
The Cytokine Mediated Molecular Pathophysiology of Psoriasis and Its Clinical Implications



Abstract
1. Introduction
2. Results
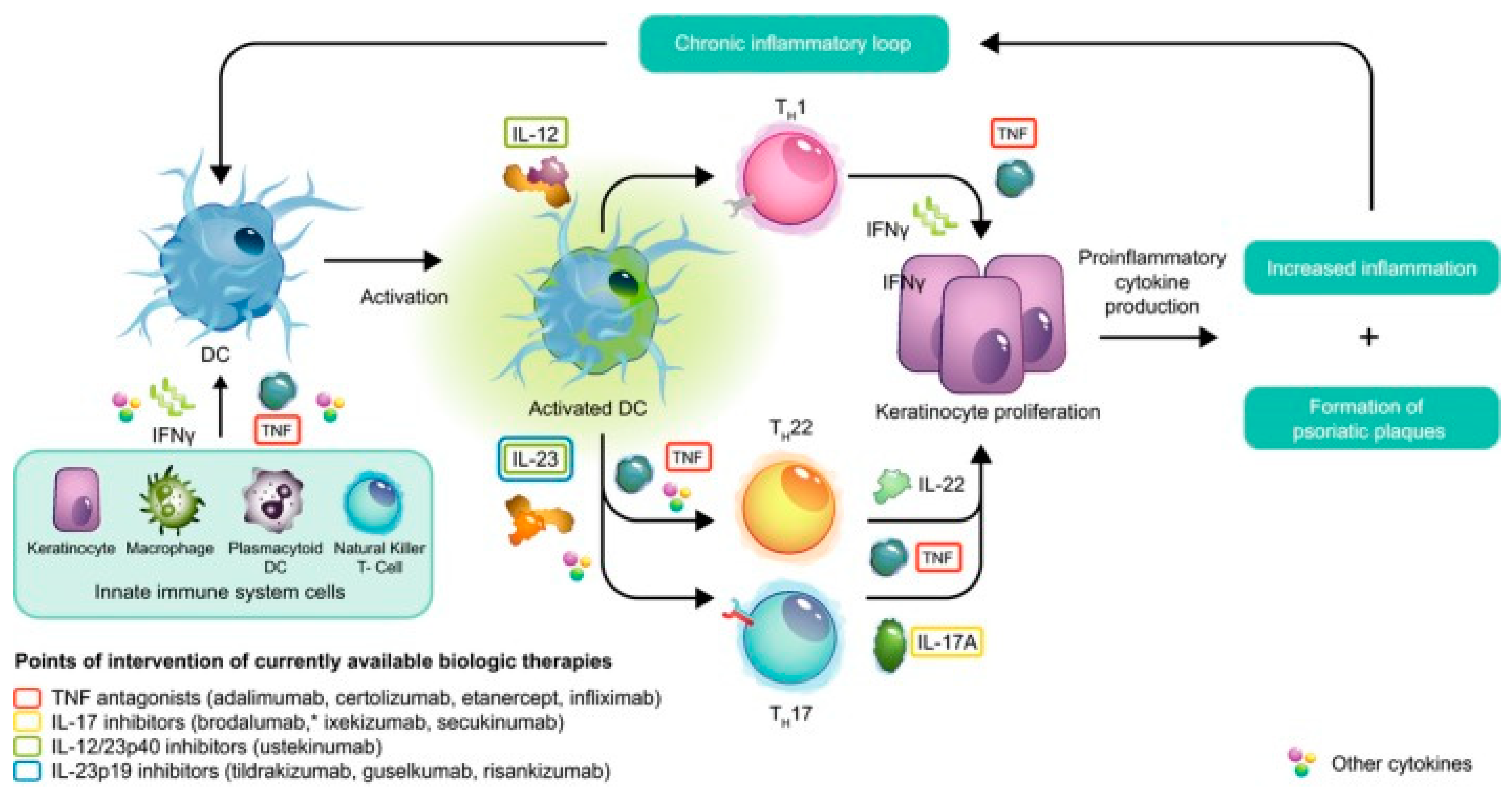
2.1. The Pathophysiology of Plaque Psoriasis
2.1.1. Pathogenesis of Pustular Psoriasis
2.1.2. The Pathophysiology of Psoriatic Arthritis
2.2. Clinical Manifestations of Psoriasis
2.2.1. Clinical Manifestations of Plaque Psoriasis
2.2.2. Clinical Manifestations of Pustular Psoriasis
2.3. Biological Therapies for Plaque Psoriasis
2.3.1. TNF-α Inhibitors
2.3.2. IL-12/IL-23 Inhibitors
2.3.3. IL-17 Inhibitors
2.3.4. IL-23/IL-39 Inhibitors
2.4. Emerging Role of JAK Inhibitors
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Rendon, A.; Schäkel, K. Psoriasis Pathogenesis and Treatment. Int. J. Mol. Sci. 2019, 20, 1475. [Google Scholar] [CrossRef]
- Armstrong, A.W.; Read, C. Pathophysiology, Clinical Presentation, and Treatment of Psoriasis: A Review. JAMA 2020, 323, 1945–1960. [Google Scholar] [CrossRef]
- Schadler, E.D.; Ortel, B.; Mehlis, S.L. Biologics for the primary care physician: Review and treatment of psoriasis. Disease-A-Month 2019, 65, 51–90. [Google Scholar] [CrossRef] [PubMed]
- Parisi, R.; Iskandar, I.Y.K.; Kontopantelis, E.; Augustin, M.; Griffiths, C.E.M.; Ashcroft, D.M. National, regional, and worldwide epidemiology of psoriasis: Systematic analysis and modelling study. BMJ 2020, 369, m1590. [Google Scholar] [CrossRef]
- Sawyer, L.M.; Malottki, K.; Sabry-Grant, C.; Yasmeen, N.; Wright, E.; Sohrt, A.; Borg, E.; Warren, R.B. Assessing the relative efficacy of interleukin-17 and interleukin-23 targeted treatments for moderate-to-severe plaque psoriasis: A systematic review and network meta-analysis of PASI response. PLoS ONE 2019, 14, e0220868. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, M.; Rullo, E.V.; Berretta, M.; Cacopardo, B.; Pellicanò, G.F.; Nunnari, G.; Guarneri, C. New generation biologics for the treatment of psoriasis and psoriatic arthritis. State of the art and considerations about the risk of infection. Dermatol. Ther. 2021, 34, e14660. [Google Scholar] [CrossRef] [PubMed]
- Badri, T.; Kumar, P.; Oakley, A.M. Plaque Psoriasis. StatPearls 2021, 19, 560–565. [Google Scholar]
- Boehncke, W.H. Systemic inflammation and cardiovascular comorbidity in psoriasis patients: Causes and consequences. Front. Immunol. 2018, 9, 579. [Google Scholar] [CrossRef] [PubMed]
- Korman, N.J. Management of psoriasis as a systemic disease: What is the evidence? Br. J. Dermatol. 2020, 182, 840. [Google Scholar] [CrossRef]
- Afonina, I.S.; Van Nuffel, E.; Beyaert, R. Immune responses and therapeutic options in psoriasis. Cell. Mol. Life Sci. 2021, 78, 2709–2727. [Google Scholar] [CrossRef]
- Petit, R.G.; Cano, A.; Ortiz, A.; Espina, M.; Prat, J.; Muñoz, M.; Severino, P.; Souto, E.B.; García, M.L.; Pujol, M.; et al. Psoriasis: From Pathogenesis to Pharmacological and Nano-Technological-Based Therapeutics. Int. J. Mol. Sci. 2021, 22, 4983. [Google Scholar] [CrossRef]
- Sato, Y.; Ogawa, E.; Okuyama, R. Role of Innate Immune Cells in Psoriasis. Int. J. Mol. Sci. 2020, 21, 6604. [Google Scholar] [CrossRef]
- Herster, F.; Bittner, Z.; Archer, N.K.; Dickhöfer, S.; Eisel, D.; Eigenbrod, T.; Knorpp, T.; Schneiderhan-Marra, N.; Löffler, M.W.; Kalbacher, H.; et al. Neutrophil extracellular trap-associated RNA and LL37 enable self-amplifying inflammation in psoriasis. Nat. Commun. 2020, 11, 105. [Google Scholar] [CrossRef]
- Amin, M.; Darji, K.; No, D.J.; Wu, J.J. Review of phase III trial data on IL-23 inhibitors tildrakizumab and guselkumab for psoriasis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1627–1632. [Google Scholar] [CrossRef]
- Cai, Y.; Shen, X.; Ding, C.; Qi, C.; Li, K.; Li, X.; Jala, V.R.; Zhang, H.G.; Wang, T.; Zheng, J.; et al. Pivotal role of dermal IL-17-producing γδ T cells in skin inflammation. Immunity 2011, 35, 596–610. [Google Scholar] [CrossRef]
- Matos, T.R.; O’Malley, J.T.; Lowry, E.L.; Hamm, D.; Kirsch, I.R.; Robins, H.S.; Kupper, T.S.; Krueger, J.G.; Clark, R.A. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing αβ T cell clones. J. Clin. Investig. 2017, 127, 4031–4041. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, M.B.M.; Munneke, J.M.; Bernink, J.H.; Spuls, P.I.; Res, P.C.M.; Te Velde, A.; Cheuk, S.; Brouwer, M.W.D.; Menting, S.P.; Eidsmo, L.; et al. Composition of innate lymphoid cell subsets in the human skin: Enrichment of NCR(+) ILC3 in lesional skin and blood of psoriasis patients. J. Investig. Dermatol. 2014, 134, 2351–2360. [Google Scholar] [CrossRef] [PubMed]
- Boniface, K.; Bernard, F.X.; Garcia, M.; Gurney, A.L.; Lecron, J.C.; Morel, F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J. Immunol. 2005, 174, 3695–3702. [Google Scholar] [CrossRef]
- Bovenschen, H.J.; Gerritsen, W.J.; van Rens, D.W.A.; Seyger, M.M.B.; de Jong, E.M.G.J.; van de Kerkhof, P.C.M. Explorative immunohistochemical study to evaluate the addition of a topical corticosteroid in the early phase of alefacept treatment for psoriasis. Arch. Dermatol. Res. 2007, 298, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Gilhar, A.; Ullmann, Y.; Kerner, H.; Assy, B.; Shalaginov, R.; Serafimovich, S.; Kalish, R.S. Psoriasis is mediated by a cutaneous defect triggered by activated immunocytes: Induction of psoriasis by cells with natural killer receptors. J. Investig. Dermatol. 2002, 119, 384–391. [Google Scholar] [CrossRef][Green Version]
- Hammarén, H.M.; Virtanen, A.T.; Raivola, J.; Silvennoinen, O. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine 2019, 118, 48–63. [Google Scholar] [CrossRef]
- Fardos, M.I.; Singh, R.; Perche, P.O.; Kelly, K.A.; Feldman, S.R. Evaluating topical JAK inhibitors as a treatment option for atopic dermatitis. Expert Rev. Clin. Immunol. 2021, 1–11. [Google Scholar] [CrossRef]
- Szilveszter, K.P.; Németh, T.; Mócsai, A. Tyrosine Kinases in Autoimmune and Inflammatory Skin Diseases. Front. Immunol. 2019, 10, 1862. [Google Scholar] [CrossRef] [PubMed]
- Raharja, A.; Mahil, S.K.; Barker, J.N. Psoriasis: A brief overview. Clin. Med. (Lond.) 2021, 21, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, M.; Puig, L.; Torres, T. JAK Inhibitors for Treatment of Psoriasis: Focus on Selective TYK2 Inhibitors. Drugs 2020, 80, 341–352. [Google Scholar] [CrossRef]
- Zhou, J.; Luo, Q.; Cheng, Y.; Wen, X.; Liu, J. An update on genetic basis of generalized pustular psoriasis (Review). Int. J. Mol. Med. 2021, 47, 118. [Google Scholar] [CrossRef]
- Mahil, S.K.; Twelves, S.; Farkas, K.; Setta-Kaffetzi, N.; Burden, A.D.; Gach, J.E.; Irvine, A.D.; Képíró, L.; Mockenhaupt, M.; Oon, H.H.; et al. AP1S3 Mutations Cause Skin Autoinflammation by Disrupting Keratinocyte Autophagy and Up-Regulating IL-36 Production. J. Investig. Dermatol. 2016, 136, 2251–2259. [Google Scholar] [CrossRef]
- Setta-Kaffetzi, N.; Simpson, M.A.; Navarini, A.A.; Patel, V.M.; Lu, H.C.; Allen, M.H.; Duckworth, M.; Bachelez, H.; Burden, A.D.; Choon, S.E.; et al. AP1S3 mutations are associated with pustular psoriasis and impaired Toll-like receptor 3 trafficking. Am. J. Hum. Genet. 2014, 94, 790–797. [Google Scholar] [CrossRef]
- Li, L.; You, J.; Fu, X.; Wang, Z.; Sun, Y.; Liu, H.; Zhang, F. Variants of CARD14 are predisposing factors for generalized pustular psoriasis (GPP) with psoriasis vulgaris but not for GPP alone in a Chinese population. Br. J. Dermatol. 2019, 180, 425–426. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, K.; Muto, M.; Akiyama, M. CARD14 c.526G>C (p.Asp176His) is a significant risk factor for generalized pustular psoriasis with psoriasis vulgaris in the Japanese cohort. J. Investig. Dermatol. 2014, 134, 1755–1757. [Google Scholar] [CrossRef]
- Sugiura, K. The genetic background of generalized pustular psoriasis: IL36RN mutations and CARD14 gain-of-function variants. J. Dermatol. Sci. 2014, 74, 187–192. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, R.; Lu, Z.; Guo, Y.; Yan, M.; Liang, J.; Huang, P.; Li, M.; Yao, Z. Clinical profiles of pediatric patients with GPP alone and with different IL36RN genotypes. J. Dermatol. Sci. 2017, 85, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Hoegler, K.M.; John, A.M.; Handler, M.Z.; Schwartz, R.A. Generalized pustular psoriasis: A review and update on treatment. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 1645–1651. [Google Scholar] [CrossRef]
- Qin, P.; Zhang, Q.; Chen, M.; Fu, X.; Wang, C.; Wang, Z.; Yu, G.; Yu, Y.; Li, X.; Sun, Y.; et al. Variant analysis of CARD14 in a Chinese Han population with psoriasis vulgaris and generalized pustular psoriasis. J. Investig. Dermatol. 2014, 134, 2994–2996. [Google Scholar] [CrossRef] [PubMed]
- Alinaghi, F.; Calov, M.; Kristensen, L.E.; Gladman, D.D.; Coates, L.C.; Jullien, D.; Gottlieb, A.B.; Gisondi, P.; Wu, J.J.; Thyssen, J.P.; et al. Prevalence of psoriatic arthritis in patients with psoriasis: A systematic review and meta-analysis of observational and clinical studies. J. Am. Acad. Dermatol. 2019, 80, 251–265.e19. [Google Scholar] [CrossRef] [PubMed]
- Veale, D.J.; Fearon, U. The pathogenesis of psoriatic arthritis. Lancet 2018, 391, 2273–2284. [Google Scholar] [CrossRef]
- Rauber, S.; Luber, M.; Weber, S.; Maul, L.; Soare, A.; Wohlfahrt, T.; Lin, N.Y.; DIetel, K.; Bozec, A.; Herrmann, M.; et al. Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat. Med. 2017, 23, 938–944. [Google Scholar] [CrossRef]
- Kamata, M.; Tada, Y. Efficacy and Safety of Biologics for Psoriasis and Psoriatic Arthritis and Their Impact on Comorbidities: A Literature Review. Int. J. Mol. Sci. 2020, 21, 1690. [Google Scholar] [CrossRef]
- Watanabe, R.; Shirai, T.; Namkoong, H.; Zhang, H.; Berry, G.J.; Wallis, B.B.; Schaefgen, B.; Harrison, D.G.; Tremmel, J.A.; Giacomini, J.C.; et al. Pyruvate controls the checkpoint inhibitor PD-L1 and suppresses T cell immunity. J. Clin. Investig. 2017, 127, 2725–2738. [Google Scholar] [CrossRef]
- Sucur, A.; Jajic, Z.; Artukovic, M.; Matijasevic, M.I.; Anic, B.; Flegar, D.; Markotic, A.; Kelava, T.; Ivcevic, S.; Kovacic, N.; et al. Chemokine signals are crucial for enhanced homing and differentiation of circulating osteoclast progenitor cells. Arthritis Res. Ther. 2017, 19, 142. [Google Scholar] [CrossRef]
- Tiwari, V.; Brent, L.H. Psoriatic Arthritis; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Sherlock, J.P.; Joyce-Shaikh, B.; Turner, S.P.; Chao, C.C.; Sathe, M.; Grein, J.; Gorman, D.M.; Bowman, E.P.; McClanahan, T.K.; Yearley, J.H.; et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4-CD8- entheseal resident T cells. Nat. Med. 2012, 18, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Ritchlin, C.T.; Colbert, R.A.; Gladman, D.D. Psoriatic Arthritis. N. Engl. J. Med. 2017, 376, 957–970, Erratum in N. Engl. J. Med. 2017, 376, 2097. [Google Scholar] [CrossRef]
- Venken, K.; Jacques, P.; Mortier, C.; Labadia, M.E.; Decruy, T.; Coudenys, J.; Hoyt, K.; Wayne, A.L.; Hughes, R.; Turner, M.; et al. RORγt inhibition selectively targets IL-17 producing iNKT and γδ-T cells enriched in Spondyloarthritis patients. Nat. Commun. 2019, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Ljubenovic, M.; Lazarevic, V.; Golubovic, M.; Binic, I. Integrative Approach to Psoriasis Vulgaris. Holist. Nurs. Pract. 2018, 32, 133–139. [Google Scholar] [CrossRef]
- Tonini, A.; Gualtieri, B.; Panduri, S.; Romanelli, M.; Chiricozzi, A. A new class of biologic agents facing the therapeutic paradigm in psoriasis: Anti-IL-23 agents. Expert Opin. Biol. Ther. 2018, 18, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.; Azad, J.; Takwale, A. Management of nail psoriasis. Clin. Exp. Dermatol. 2021, 46, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Wang, H.; Bao, C.; Zhang, L.; Ruan, S.; Zhang, J.; Gong, T.; Cheng, B. Challenge of Nail Psoriasis: An Update Review. Clin. Rev. Allergy Immunol. 2021, 1, 1–26. [Google Scholar] [CrossRef]
- Micali, G.; Verzì, A.E.; Giuffrida, G.; Panebianco, E.; Musumeci, M.L.; Lacarrubba, F. Inverse psoriasis: From diagnosis to current treatment options. Clin. Cosmet. Investig. Dermatol. 2019, 12, 953–959. [Google Scholar] [CrossRef]
- Khosravi, H.; Siegel, M.P.; Van Voorhees, A.S.; Merola, J.F. Treatment of Inverse/Intertriginous Psoriasis: Updated Guidelines from the Medical Board of the National Psoriasis Foundation. J. Drugs Dermatol. 2017, 16, 760–766. [Google Scholar] [PubMed]
- Wang, W.M.; Jin, H.Z. Biologics in the treatment of pustular psoriasis. Expert Opin. Drug Saf. 2020, 19, 969–980. [Google Scholar] [CrossRef]
- Aslam, N.; Saleem, H.; Murtazaliev, S.; Quazi, S.J.; Khan, S. FDA Approved Biologics: Can Etanercept and Ustekinumab be Considered a First-Line Systemic Therapy for Pediatric/Adolescents in Moderate to Severe Psoriasis? A Systematic Review. Cureus 2020, 12, e9812. [Google Scholar] [CrossRef]
- Honma, M.; Hayashi, K. Psoriasis: Recent progress in molecular-targeted therapies. J. Dermatol. 2021, 48, 761–777. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Gooderham, M.; Green, L.; Bewley, A.; Zhang, Z.; Khanskaya, I.; Day, R.M.; Goncalves, J.; Shah, K.; Piguet, V.; et al. The efficacy and safety of apremilast, etanercept and placebo in patients with moderate-to-severe plaque psoriasis: 52-week results from a phase IIIb, randomized, placebo-controlled trial (LIBERATE). J. Eur. Acad. Dermatol. Venereol. 2017, 31, 507–517. [Google Scholar] [CrossRef]
- Highlights of Prescribing Information. Available online: www.fda.gov/medwatch (accessed on 11 August 2021).
- Fda. HUMIRA® (Adalimumab) Injection, for Subcutaneous Use. Available online: www.fda.gov/medwatch (accessed on 25 October 2021).
- Conrad, C.; Gilliet, M. Psoriasis: From Pathogenesis to Targeted Therapies. Clin. Rev. Allergy Immunol. 2018, 54, 102–113. [Google Scholar] [CrossRef]
- Bagel, J.; Tyring, S.; Rice, K.C.; Collier, D.H.; Kricorian, G.; Chung, J.; Iles, J.; Stolshek, B.S.; Kaliyaperumal, A.; Papp, K.A. Open-label study of etanercept treatment in patients with moderate-to-severe plaque psoriasis who lost a satisfactory response to adalimumab. Br. J. Dermatol. 2017, 177, 411–418. [Google Scholar] [CrossRef]
- Gottlieb, A.B.; Blauvelt, A.; Thaçi, D.; Leonardi, C.L.; Poulin, Y.; Drew, J.; Peterson, L.; Arendt, C.; Burge, D.; Reich, K. Certolizumab pegol for the treatment of chronic plaque psoriasis: Results through 48 weeks from 2 phase 3, multicenter, randomized, double-blinded, placebo-controlled studies (CIMPASI-1 and CIMPASI-2). J. Am. Acad. Dermatol. 2018, 79, 302–314.e6. [Google Scholar] [CrossRef] [PubMed]
- De Vries, A.C.Q.; Thio, H.B.; de Kort, W.J.A.; Opmeer, B.C.; van der Stok, H.M.; de Jong, E.M.G.J.; Horvath, B.; Busschbach, J.J.V.; Nijsten, T.E.C.; Spuls, P.I. A prospective randomized controlled trial comparing infliximab and etanercept in patients with moderate-to-severe chronic plaque-type psoriasis: The Psoriasis Infliximab vs. Etanercept Comparison Evaluation (PIECE) study. Br. J. Dermatol. 2017, 176, 624–633. [Google Scholar] [CrossRef]
- Armstrong, A.W.; Soliman, A.M.; Betts, K.A.; Wang, Y.; Gao, Y.; Puig, L.; Augustin, M. Comparative Efficacy and Relative Ranking of Biologics and Oral Therapies for Moderate-to-Severe Plaque Psoriasis: A Network Meta-analysis. Dermatol. Ther. (Heidelb) 2021, 11, 885–905. [Google Scholar] [CrossRef]
- Kimball, A.B.; Rothman, K.J.; Kricorian, G.; Pariser, D.; Yamauchi, P.S.; Menter, A.; Teller, C.F.; Aras, G.; Accortt, N.A.; Hooper, M.; et al. OBSERVE-5: Observational postmarketing safety surveillance registry of etanercept for the treatment of psoriasis final 5-year results. J. Am. Acad. Dermatol. 2015, 72, 115. [Google Scholar] [CrossRef] [PubMed]
- Menter, A.; Thaçi, D.; Wu, J.J.; Abramovits, W.; Kerdel, F.; Arikan, D.; Guo, D.; Ganguli, A.; Bereswill, M.; Camez, A.; et al. Long-Term Safety and Effectiveness of Adalimumab for Moderate to Severe Psoriasis: Results from 7-Year Interim Analysis of the ESPRIT Registry. Dermatol. Ther. (Heidelb) 2017, 7, 365–381. [Google Scholar] [CrossRef]
- Kalb, R.E.; Fiorentino, D.F.; Lebwohl, M.G.; Toole, J.; Poulin, Y.; Cohen, A.D.; Goyal, K.; Fakharzadeh, S.; Calabro, S.; Chevrier, M.; et al. Risk of Serious Infection With Biologic and Systemic Treatment of Psoriasis: Results From the Psoriasis Longitudinal Assessment and Registry (PSOLAR). JAMA Dermatol. 2015, 151, 961–969. [Google Scholar] [CrossRef]
- Fiorentino, D.; Ho, V.; Lebwohl, M.G.; Leite, L.; Hopkins, L.; Galindo, C.; Goyal, K.; Langholff, W.; Fakharzadeh, S.; Srivastava, B.; et al. Risk of malignancy with systemic psoriasis treatment in the Psoriasis Longitudinal Assessment Registry. J. Am. Acad. Dermatol. 2017, 77, 845–854.e5. [Google Scholar] [CrossRef]
- Gerriets, V.; Bansal, P.; Goyal, A.; Khaddour, K. Tumor Necrosis Factor Inhibitors; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Brownstone, N.D.; Hong, J.; Mosca, M.; Hadeler, E.; Liao, W.; Bhutani, T.; Koo, J. Biologic Treatments of Psoriasis: An Update for the Clinician. Biologics 2021, 15, 39–51. [Google Scholar] [CrossRef]
- Pan, A.; Gerriets, V. Etanercept; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Fatima, R.; Bittar, K.; Aziz, M. Infliximab; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR (accessed on 14 November 2021).
- Fda. STELARA® (Ustekinumab) Injection, for Subcutaneous or Intravenous. Available online: www.fda.gov/medwatch (accessed on 25 October 2021).
- Rønholt, K.; Iversen, L. Old and New Biological Therapies for Psoriasis. Int. J. Mol. Sci. 2017, 18, 2297. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/nepexto (accessed on 14 November 2021).
- Tsai, T.F.; Ho, J.C.; Song, M.; Szapary, P.; Guzzo, C.; Shen, Y.K.; Li, S.; Kim, K.J.; Kim, T.Y.; Choi, J.H.; et al. Efficacy and safety of ustekinumab for the treatment of moderate-to-severe psoriasis: A phase III, randomized, placebo-controlled trial in Taiwanese and Korean patients (PEARL). J. Dermatol. Sci. 2011, 63, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.B.; Strober, B.; Lebwohl, M.; Augustin, M.; Blauvelt, A.; Poulin, Y.; Papp, K.A.; Sofen, H.; Puig, L.; Foley, P.; et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): Results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet 2018, 392, 650–661. [Google Scholar] [CrossRef]
- Lebwohl, M.; Strober, B.; Menter, A.; Gordon, K.; Weglowska, J.; Puig, L.; Papp, K.; Spelman, L.; Toth, D.; Kerdel, F.; et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N. Engl. J. Med. 2015, 373, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Bagel, J.; Blauvelt, A.; Nia, J.; Hashim, P.; Patekar, M.; de Vera, A.; Ahmad, K.; Paguet, B.; Xia, S.; Muscianisi, E.; et al. Secukinumab maintains superiority over ustekinumab in clearing skin and improving quality of life in patients with moderate to severe plaque psoriasis: 52-week results from a double-blind phase 3b trial (CLARITY). J. Eur. Acad. Dermatol. Venereol. 2021, 35, 135–142. [Google Scholar] [CrossRef]
- Egeberg, A.; Ottosen, M.B.; Gniadecki, R.; Broesby-Olsen, S.; Dam, T.N.; Bryld, L.E.; Rasmussen, M.K.; Skov, L. Safety, efficacy and drug survival of biologics and biosimilars for moderate-to-severe plaque psoriasis. Br. J. Dermatol. 2018, 178, 509–519. [Google Scholar] [CrossRef]
- Colquhoun, M.; Kemp, A.K. Ustekinumab; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Krueger, J.G.; Wharton, K.A.; Schlitt, T.; Suprun, M.; Torene, R.I.; Jiang, X.; Wang, C.Q.; Fuentes-Duculan, J.; Hartmann, N.; Peters, T.; et al. IL-17A inhibition by secukinumab induces early clinical, histopathologic, and molecular resolution of psoriasis. J. Allergy Clin. Immunol. 2019, 144, 750–763. [Google Scholar] [CrossRef] [PubMed]
- Fda. TALTZ (Ixekizumab) Injection, for Subcutaneous Use. Available online: www.fda.gov/medwatch (accessed on 25 October 2021).
- Griffiths, C.E.M.; Reich, K.; Lebwohl, M.; Van De Kerkhof, P.; Paul, C.; Menter, A.; Cameron, G.S.; Erickson, J.; Zhang, L.; Secrest, R.J.; et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): Results from two phase 3 randomised trials. Lancet 2015, 386, 541–551. [Google Scholar] [CrossRef]
- Gordon, K.B.; Foley, P.; Krueger, J.G.; Pinter, A.; Reich, K.; Vender, R.; Vanvoorden, V.; Madden, C.; White, K.; Cioffi, C.; et al. Bimekizumab efficacy and safety in moderate to severe plaque psoriasis (BE READY): A multicentre, double-blind, placebo-controlled, randomised withdrawal phase 3 trial. Lancet 2021, 397, 475–486. [Google Scholar] [CrossRef]
- Adams, R.; Maroof, A.; Baker, T.; Lawson, A.D.G.; Oliver, R.; Paveley, R.; Rapecki, S.; Shaw, S.; Vajjah, P.; West, S.; et al. Bimekizumab, a Novel Humanized IgG1 Antibody That Neutralizes Both IL-17A and IL-17F. Front. Immunol. 2020, 11, 1894. [Google Scholar] [CrossRef] [PubMed]
- Ali, Z.; Matthews, R.; Al-Janabi, A.; Warren, R.B. Bimekizumab: A dual IL-17A and IL-17F inhibitor for the treatment of psoriasis and psoriatic arthritis. Expert Rev. Clin. Immunol. 2021, 17, 1073–1081. [Google Scholar] [CrossRef]
- Mease, P.J.; Helliwell, P.S.; Hjuler, K.F.; Raymond, K.; Mcinnes, I. Brodalumab in psoriatic arthritis: Results from the randomised phase III AMVISION-1 and AMVISION-2 trials. Ann. Rheum. Dis. 2021, 80, 185–193. [Google Scholar] [CrossRef]
- Blauvelt, A.; Reich, K.; Tsai, T.F.; Tyring, S.; Vanaclocha, F.; Kingo, K.; Ziv, M.; Pinter, A.; Vender, R.; Hugot, S.; et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate-to-severe plaque psoriasis up to 1 year: Results from the CLEAR study. J. Am. Acad. Dermatol. 2017, 76, 60–69.e9. [Google Scholar] [CrossRef]
- Papp, K.A.; Reich, K.; Paul, C.; Blauvelt, A.; Baran, W.; Bolduc, C.; Toth, D.; Langley, R.G.; Cather, J.; Gottlieb, A.B.; et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br. J. Dermatol. 2016, 175, 273–286. [Google Scholar] [CrossRef]
- Reich, K.; Papp, K.A.; Blauvelt, A.; Langley, R.G.; Armstrong, A.; Warren, R.B.; Gordon, K.B.; Merola, J.F.; Okubo, Y.; Madden, C.; et al. Bimekizumab versus ustekinumab for the treatment of moderate to severe plaque psoriasis (BE VIVID): Efficacy and safety from a 52-week, multicentre, double-blind, active comparator and placebo controlled phase 3 trial. Lancet 2021, 397, 487–498. [Google Scholar] [CrossRef]
- Warren, R.B.; Blauvelt, A.; Bagel, J.; Papp, K.A.; Yamauchi, P.; Armstrong, A.; Langley, R.G.; Vanvoorden, V.; De Cuyper, D.; Cioffi, C.; et al. Bimekizumab versus Adalimumab in Plaque Psoriasis. N. Engl. J. Med. 2021, 385, 130–141. [Google Scholar] [CrossRef]
- Reich, K.; Warren, R.B.; Lebwohl, M.; Gooderham, M.; Strober, B.; Langley, R.G.; Paul, C.; De Cuyper, D.; Vanvoorden, V.; Madden, C.; et al. Bimekizumab versus Secukinumab in Plaque Psoriasis. N. Engl. J. Med. 2021, 385, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Li, G.G.; Liu, Q.; Niu, X.; Li, R.; Ma, H. Short-Term Efficacy and Safety of IL-17, IL-12/23, and IL-23 Inhibitors Brodalumab, Secukinumab, Ixekizumab, Ustekinumab, Guselkumab, Tildrakizumab, and Risankizumab for the Treatment of Moderate to Severe Plaque Psoriasis: A Systematic Review and Network Meta-Analysis of Randomized Controlled Trials. J. Immunol. Res. 2019, 2019, 2546161. [Google Scholar] [CrossRef]
- Preuss, C.V.; Quick, J. Ixekizumab; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Aboobacker, S.; Kurn, H.; Al Aboud, A.M. Secukinumab; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Golbari, N.M.; Basehore, B.M.; Zito, P.M. Brodalumab; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Lu, Z.; Xu, K.; Wang, X.; Li, Y.; Li, M. Interleukin 39: A new member of interleukin 12 family. Cent. J. Immunol. 2020, 45, 214. [Google Scholar] [CrossRef]
- Kulig, P.; Musiol, S.; Freiberger, S.N.; Schreiner, B.; Gyülveszi, G.; Russo, G.; Pantelyushin, S.; Kishihara, K.; Alessandrini, F.; Kündig, T.; et al. IL-12 protects from psoriasiform skin inflammation. Nat. Commun. 2016, 7, 13466. [Google Scholar] [CrossRef]
- Reich, K.; Rich, P.; Maari, C.; Bissonnette, R.; Leonardi, C.; Menter, A.; Igarashi, A.; Klekotka, P.; Patel, D.; Li, J.; et al. Efficacy and safety of mirikizumab (LY3074828) in the treatment of moderate-to-severe plaque psoriasis: Results from a randomized phase II study. Br. J. Dermatol. 2019, 181, 88–95. [Google Scholar] [CrossRef]
- Yang, K.; Oak, A.S.W.; Elewski, B.E. Use of IL-23 Inhibitors for the Treatment of Plaque Psoriasis and Psoriatic Arthritis: A Comprehensive Review. Am. J. Clin. Dermatol. 2021, 22, 173–192. [Google Scholar] [CrossRef] [PubMed]
- Foley, P.; Gordon, K.; Griffiths, C.E.M.; Wasfi, Y.; Randazzo, B.; Song, M.; Li, S.; Shen, Y.K.; Blauvelt, A. Efficacy of guselkumab compared with adalimumab and placebo for psoriasis in specific body regions a secondary analysis of 2 randomized clinical trials. JAMA Dermatol. 2018, 154, 676–683. [Google Scholar] [CrossRef]
- Reich, K.; Armstrong, A.W.; Foley, P.; Song, M.; Wasfi, Y.; Randazzo, B.; Li, S.; Shen, Y.K.; Gordon, K.B. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: Results from the phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trial. J. Am. Acad. Dermatol. 2017, 76, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Leonardi, C.; Elewski, B.; Crowley, J.J.; Guenther, L.C.; Gooderham, M.; Langley, R.G.; Vender, R.; Pinter, A.; Griffiths, C.E.M.; et al. IXORA-R Study Group. A head-to-head comparison of ixekizumab vs. guselkumab in patients with moderate-to-severe plaque psoriasis: 24-week efficacy and safety results from a randomized, double-blinded trial. Br. J. Dermatol. 2021, 184, 1047–1058. [Google Scholar] [CrossRef]
- Diels, J.; Thilakarathne, P.; Cameron, C.; McElligott, S.; Schubert, A.; Puig, L. Adjusted treatment COMPArisons between guSelkumab and uStekinumab for treatment of moderate-to-severe plaque psoriasis: The COMPASS analysis. Br. J. Dermatol. 2020, 183, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Armstrong, A.W.; Langley, R.G.; Flavin, S.; Randazzo, B.; Li, S.; Hsu, M.-C.; Branigan, P.; Blauvelt, A. Guselkumab versus secukinumab for the treatment of moderate-to-severe psoriasis (ECLIPSE): Results from a phase 3, randomised controlled trial. Lancet 2019, 394, 831–839. [Google Scholar] [CrossRef]
- Reich, K.; Papp, K.A.; Blauvelt, A.; Tyring, S.K.; Sinclair, R.; Thaçi, D.; Nograles, K.; Mehta, A.; Cichanowitz, N.; Li, Q.; et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): Results from two randomised controlled, phase 3 trials. Lancet 2017, 390, 276–288. [Google Scholar] [CrossRef]
- Reich, K.; Gooderham, M.; Thaçi, D.; Crowley, J.J.; Ryan, C.; Krueger, J.G.; Tsai, T.F.; Flack, M.; Gu, Y.; Williams, D.A.; et al. Risankizumab compared with adalimumab in patients with moderate-to-severe plaque psoriasis (IMMvent): A randomised, double-blind, active-comparator-controlled phase 3 trial. Lancet 2019, 394, 576–586. [Google Scholar] [CrossRef]
- Warren, R.B.; Blauvelt, A.; Poulin, Y.; Beeck, S.; Kelly, M.; Wu, T.; Geng, Z.; Paul, C. Efficacy and safety of risankizumab vs. secukinumab in patients with moderate-to-severe plaque psoriasis (IMMerge): Results from a phase III, randomized, open-label, efficacy-assessor-blinded clinical trial. Br. J. Dermatol. 2021, 184, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Słuczanowska-Głąbowska, S.; Ziegler-Krawczyk, A.; Szumilas, K.; Pawlik, A. Role of Janus Kinase Inhibitors in Therapy of Psoriasis. J. Clin. Med. 2021, 10, 4307. [Google Scholar] [CrossRef]
- Bellinato, F.; Gisondi, P.; Girolomoni, G. Latest Advances for the Treatment of Chronic Plaque Psoriasis with Biologics and Oral Small Molecules. Biologics 2021, 15, 247. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.; Gordon, K.; Thaçi, D.; Morita, A.; Gooderham, M.; Foley, P.; Girgis, I.G.; Kundu, S.; Banerjee, S. Phase 2 Trial of Selective Tyrosine Kinase 2 Inhibition in Psoriasis. N. Engl. J. Med. 2018, 379, 1313–1321. [Google Scholar] [CrossRef]
- Papp, K.A.; Menter, M.A.; Abe, M.; Elewski, B.; Feldman, S.R.; Gottlieb, A.B.; Langley, R.; Luger, T.; Thaci, D.; Buonanno, M.; et al. OPT Pivotal 1 and OPT Pivotal 2 investigators. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: Results from two randomized, placebo-controlled, phase III trials. Br. J. Dermatol. 2015, 173, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Tsai, T.F.; Lee, M.G.; Zheng, M.; Wang, G.; Jin, H.Z.; Gu, J.; Li, R.Y.; Liu, Q.Z.; Chen, J.; et al. The efficacy and safety of tofacitinib in Asian patients with moderate to severe chronic plaque psoriasis: A Phase 3, randomized, double-blind, placebo-controlled study. J. Dermatol. Sci. 2017, 88, 36–45. [Google Scholar] [CrossRef]
- Safety Study of Tofacitinib Versus Tumor Necrosis Factor (TNF) Inhibitor in Subjects with Rheumatoid Arthritis—Study Results ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02092467 (accessed on 25 October 2021).
- Sbidian, E.; Chaimani, A.; Garcia-Doval, I.; Doney, L.; Dressler, C.; Hua, C.; Hughes, C.; Naldi, L.; Afach, S.; Le Cleach, L. Systemic pharmacological treatments for chronic plaque psoriasis: A network meta-analysis. Cochrane Database Syst. Rev. 2017, 12. [Google Scholar] [CrossRef]
- Yeung, J.; Gooderham, M.J.; Grewal, P.; Hong, C.-H.; Lansang, P.; Papp, K.A.; Poulin, Y.; Turchin, I.; Vender, R. Management of Plaque Psoriasis With Biologic Therapies in Women of Child-Bearing Potential Consensus Paper. J. Cutan. Med. Surg. 2020, 24, 3S–14S. [Google Scholar] [CrossRef]
- Poulin, Y.; Langley, R.; Teixeira, H.D.; Martel, M.J.; Cheung, S. Biologics in the treatment of psoriasis: Clinical and economic overview. J. Cutan. Med. Surg. 2009, 13 (Suppl. S2), S49–S57. [Google Scholar] [CrossRef] [PubMed]
- Stein, K.R.; Pearce, D.J.; Feldman, S.R. The impact of biologics on the quality of life of psoriasis patients and the economics of psoriasis care. Semin. Cutan. Med. Surg. 2005, 24, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Küster, D.; Nast, A.; Gerdes, S.; Weberschock, T.; Wozel, G.; Gutknecht, M.; Schmitt, J. Cost-effectiveness of systemic treatments for moderate-to-severe psoriasis in the German health care setting. Arch. Dermatol. Res. 2016, 308, 249–261. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| TNF-α Inhibitors | |||||||
|---|---|---|---|---|---|---|---|
| Name | Description | Stage | Route | Dosage | Adverse Effects | Contraindications | Black Box Warnings |
| Etanercept [2,53,54,55,56,62,66,67,68] | Recombinant fusion protein that inhibits the binding of TNF-α to TNF receptors 1 and 2 | FDA approved | Subcutaneous Injection | Induction: 50 mg twice/week for first 12 weeks Maintenance: 50 mg every week | Nasopharyngitis, upper respiratory tract infections, injection site reactions | Active tuberculosis, hepatitis B, hepatitis C, advanced congestive heart failure, demyelinating diseases, sepsis (new) | Risk of serious infections and malignancy |
| Infliximab [2,53,54,55,56,64,66,67,69] | Chimeric monoclonal IgG1 antibody | FDA approved | Intravenous Injection | Induction: 5 mg/kg at weeks 0, 2, and 6 Maintenance: 5 mg/kg every 8 weeks | Nasopharyngitis, upper respiratory tract infections, injection site reactions | Higher doses in moderate to severe heart failure (NYHA class III or IV), current severe infection, active infection (new) | Risk of serious infection and malignancy |
| Adalimumab [2,53,54,55,56,63,69] | Fully human monoclonal IgG1 antibody | FDA approved | Subcutaneous Injection | Induction: 80 mg initially and 40 mg at week 1 Maintenance: 40 mg every other week | Nasopharyngitis, upper respiratory infections, injection site reactions | Active infection, sepsis, class III or IV congestive heart failure | Risk of serious infections and malignancy |
| Certolizumab pegol [2,53,54,55,56,57,66,67] | PEGylated TNF-alpha antibody fragment | FDA approved | Subcutaneous Injection | <90 kg: Induction: 400 mg at weeks 0, 2, and 4 Maintenance: 200 mg every 2 weeks >90 kg: 400 mg every 2 weeks | Nasopharyngitis, upper respiratory tract infections, injection site reactions | Active infection, sepsis, class III or IV congestive heart failure | Risk of serious infections and malignancy |
| Name | Stage in the United States | Stage in Japan | Stage in Europe |
|---|---|---|---|
| Etanercept [53,67,70] | FDA approved | Not approved | Recommended by EMA |
| Infliximab [53,67,70] | FDA approved | Approved | Recommended by EMA |
| Adalimumab [53,67,70] | FDA approved | Approved | Recommended by EMA |
| Certolizumab pegol [53,67,70] | FDA approved | Approved | Recommended by EMA |
| Ustekinumab [53,67,70] | FDA approved | Approved | Recommended by EMA |
| Secukinumab [53,67,70] | FDA approved | Approved | Recommended by EMA |
| Ixekizumab [53,67,70] | FDA approved | Approved | Recommended by EMA |
| Brodalumab [53,67,70] | FDA approved | Approved | Recommended by EMA |
| Bimekizumab [53,67,70] | Phase III | Phase III | Recommended by EMA |
| Guselkumab [53,67,70] | FDA approved | Approved | Recommended by EMA |
| Tildrakizumab [53,67,70] | FDA approved | Approved | Recommended by EMA |
| Risankizumab [53,67,70] | FDA approved | Approved | Recommended by EMA |
| Mirikizumab [53,67,70] | Phase III | Phase III |
| IL-12/IL-23 Inhibitor | |||||||
|---|---|---|---|---|---|---|---|
| Name | Description | Stage | Route | Dosage | Adverse Effects | Contraindications | Black Box Warnings |
| Ustekinumab [2,55,56,63,71,78,79] | Monoclonal antibody against p40 which is a subunit of IL-12 and Il-23 | FDA approved | Subcutaneous Injection | <100 kg—Induction: 45 mg initially and at 4 weeks; Maintenance: 45 mg every 12 weeks >100 kg—Induction: 90 mg initially and at 4 weeks; Maintenance: 90 mg every 12 weeks | Headache, nasopharyngitis, upper respiratory tract infections, fatigue, pruritus | Active Infection | None |
| IL-17 Inhibitors | |||||||
|---|---|---|---|---|---|---|---|
| Name | Description | Stage | Route | Dosage | Adverse Effects | Contraindications | Black Box Warnings |
| Secukinumab [55,57,67,80,81,93,94] | Monoclonal antibody that blocks IL-17A | FDA approved | Subcutaneous Injection | Induction: 300 mg at weeks 0, 1, 2, 3, 4 Maintenance: 300 mg every 4 weeks | Headache, nasopharyngitis, upper respiratory infections, mucocutaneous candidiasis | PUVA sessions, premalignancy, demyelinating disease, optic neuritis, multiple sclerosis, congestive heart failure, fever, jaundice, markedly elevated liver enzymes | None |
| Ixekizumab [55,57,67,80,81,93,94] | Monoclonal antibody that blocks IL-17A | FDA approved | Subcutaneous Injection | Induction: 160 mg initially and 80 mg at weeks 2, 4, 6, 8, 10, 12 Maintenance: 80 mg every 4 weeks | Headache, nasopharyngitis, upper respiratory tract infections, mucocutaneous candidiasis, injection site reactions | Hypersensitivity to ixekizumab | None |
| Brodalumab [55,67,95] | Monoclonal antibody that blocks IL-17 receptor type A | FDA approved | Subcutaneous Injection | Induction: 210 mg initially and weeks 1 and 2 Maintenance: 210 mg every 2 weeks | Headache, nasopharyngitis, upper respiratory tract infections, mucocutaneous candidiasis, injection site reactions | Crohn’s disease, non-resolving infection | Suicidal ideation and behavior |
| Bimekizumab [55,67] | Monoclonal antibody inhibiting IL-17A and IL-17F | Phase III (Clinical trials) | - | - | Nasopharyngitis, upper respiratory infections, oral candidiasis | - | - |
| IL-23/39 Inhibitors | |||||||
|---|---|---|---|---|---|---|---|
| Name | Description | Stage | Route | Dosage | Adverse Effects | Contraindications | Black Box Warnings |
| Guselkumab [55,56,67] | Fully human monoclonal antibody against p19 | FDA approved | Subcutaneous Injection | Induction: 100 mg initially and week 4 Maintenance: 100 mg every 12 weeks | Upper respiratory infections, nasopharyngitis, injection site reactions | Hypersensitivity to guselkumab | None |
| Tildrakizumab [55,56,67] | Fully human monoclonal antibody against p19 | FDA approved | Subcutaneous Injection | Induction: 100 mg initially and week 4 Maintenance: 100 mg every 12 weeks | Upper respiratory infections, nasopharyngitis, injection site reactions | None Hypersensitivity to tildrakizumab | None |
| Risankizumab [55,56,67] | Fully human monoclonal antibody against p19 | FDA approved (new) | Subcutaneous Injection | Induction: 150 mg initially and week 4 Maintenance: 150 mg every 12 weeks | Upper respiratory infections, injection site reactions, headache | None | None |
| Mirikizumab [55,56,67] | Monoclonal antibody against p19 (IL-23) | Phase III (Clinical trials) | - | - | Upper respiratory infections, injection-site pain, hypertension, diarrhea | - | - |
| JAK Inhibitors | |||||
|---|---|---|---|---|---|
| Name | Description | Stage | Adverse Effects | Contraindications | Black Box Warnings |
| Tofacitinib [55,108,111,112,113] | JAK1 and JAK3 inhibitor | Phase III | Upper respiratory infections, nasopharyngitis, | None | Infection, malignancy, thrombosis |
| Deucravacitinib [109,110] | TYK2 Inhibitor | Phase III | Nasopharyngitis, headache, diarrhea, nausea, upper respiratory tract infections | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, R.; Koppu, S.; Perche, P.O.; Feldman, S.R. The Cytokine Mediated Molecular Pathophysiology of Psoriasis and Its Clinical Implications. Int. J. Mol. Sci. 2021, 22, 12793. https://doi.org/10.3390/ijms222312793
Singh R, Koppu S, Perche PO, Feldman SR. The Cytokine Mediated Molecular Pathophysiology of Psoriasis and Its Clinical Implications. International Journal of Molecular Sciences. 2021; 22(23):12793. https://doi.org/10.3390/ijms222312793
Chicago/Turabian StyleSingh, Rohan, Sindhuja Koppu, Patrick O. Perche, and Steven R. Feldman. 2021. "The Cytokine Mediated Molecular Pathophysiology of Psoriasis and Its Clinical Implications" International Journal of Molecular Sciences 22, no. 23: 12793. https://doi.org/10.3390/ijms222312793
APA StyleSingh, R., Koppu, S., Perche, P. O., & Feldman, S. R. (2021). The Cytokine Mediated Molecular Pathophysiology of Psoriasis and Its Clinical Implications. International Journal of Molecular Sciences, 22(23), 12793. https://doi.org/10.3390/ijms222312793