
Glitazone Treatment Rescues Phenotypic Deficits in a Fly Model of Gaucher/Parkinson’s Disease

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
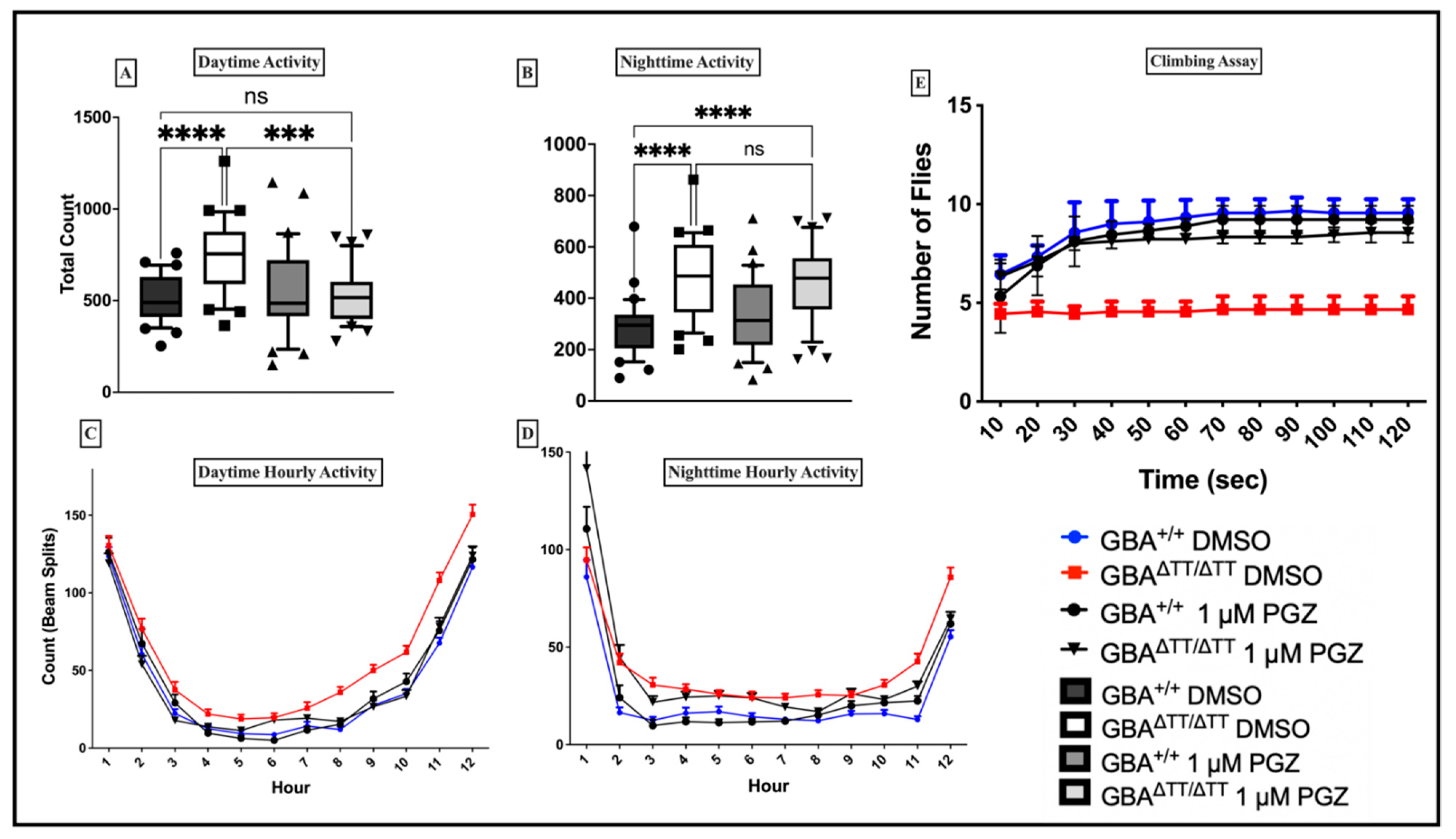
2.1. Sleep-Wake Behavior
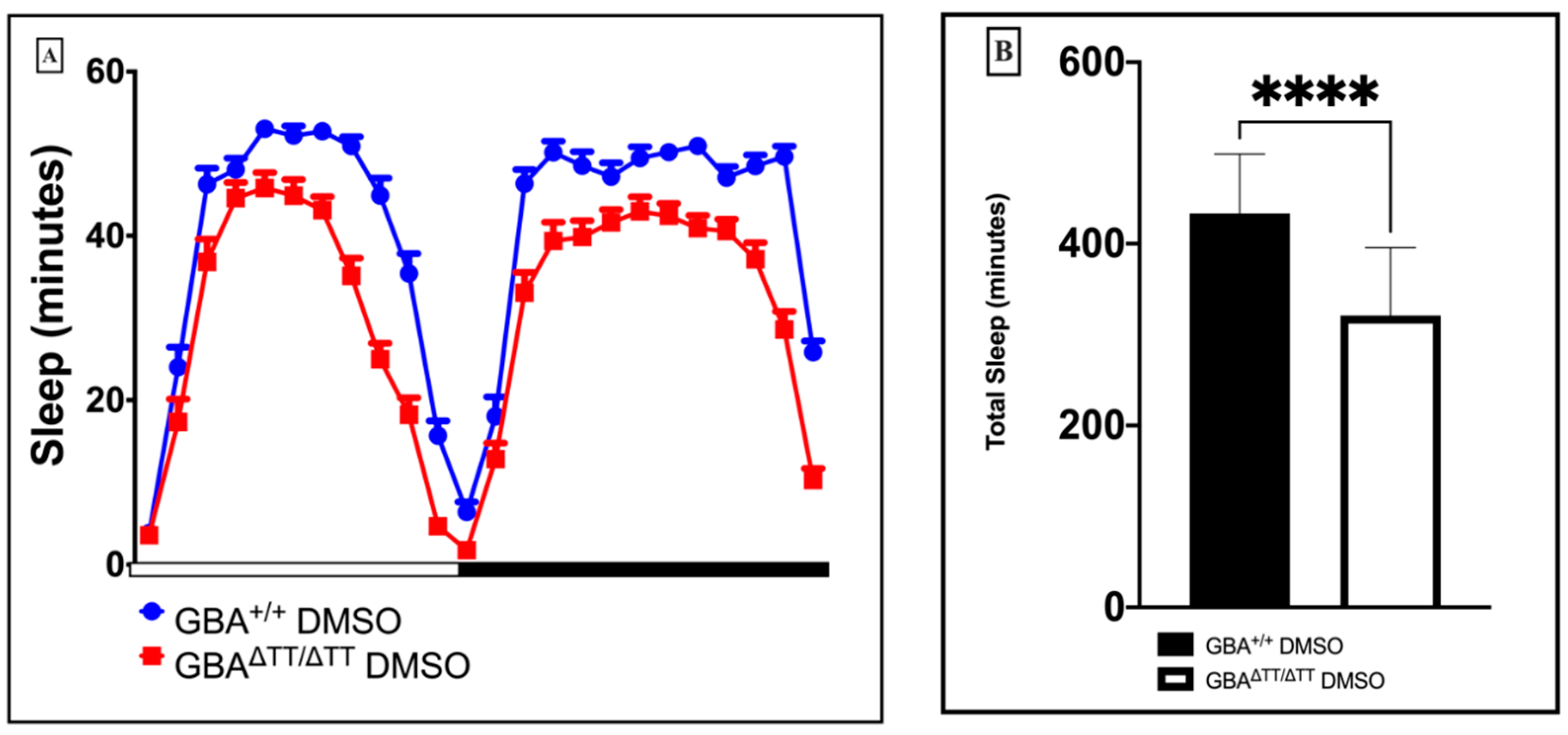
2.1.1. Changes in Sleep-Wake Behavior in GBA1ΔTT/ΔTT Mutants
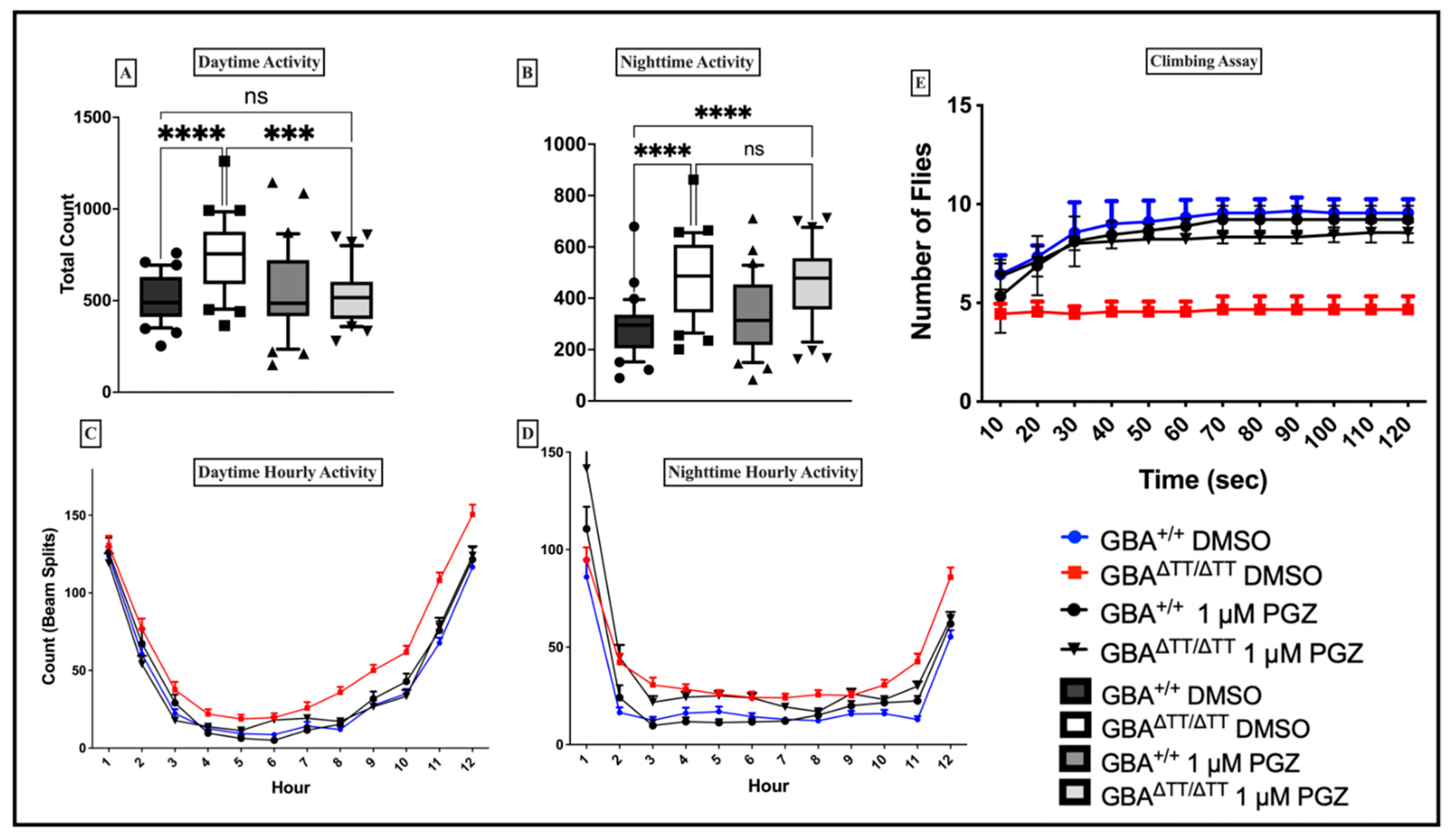
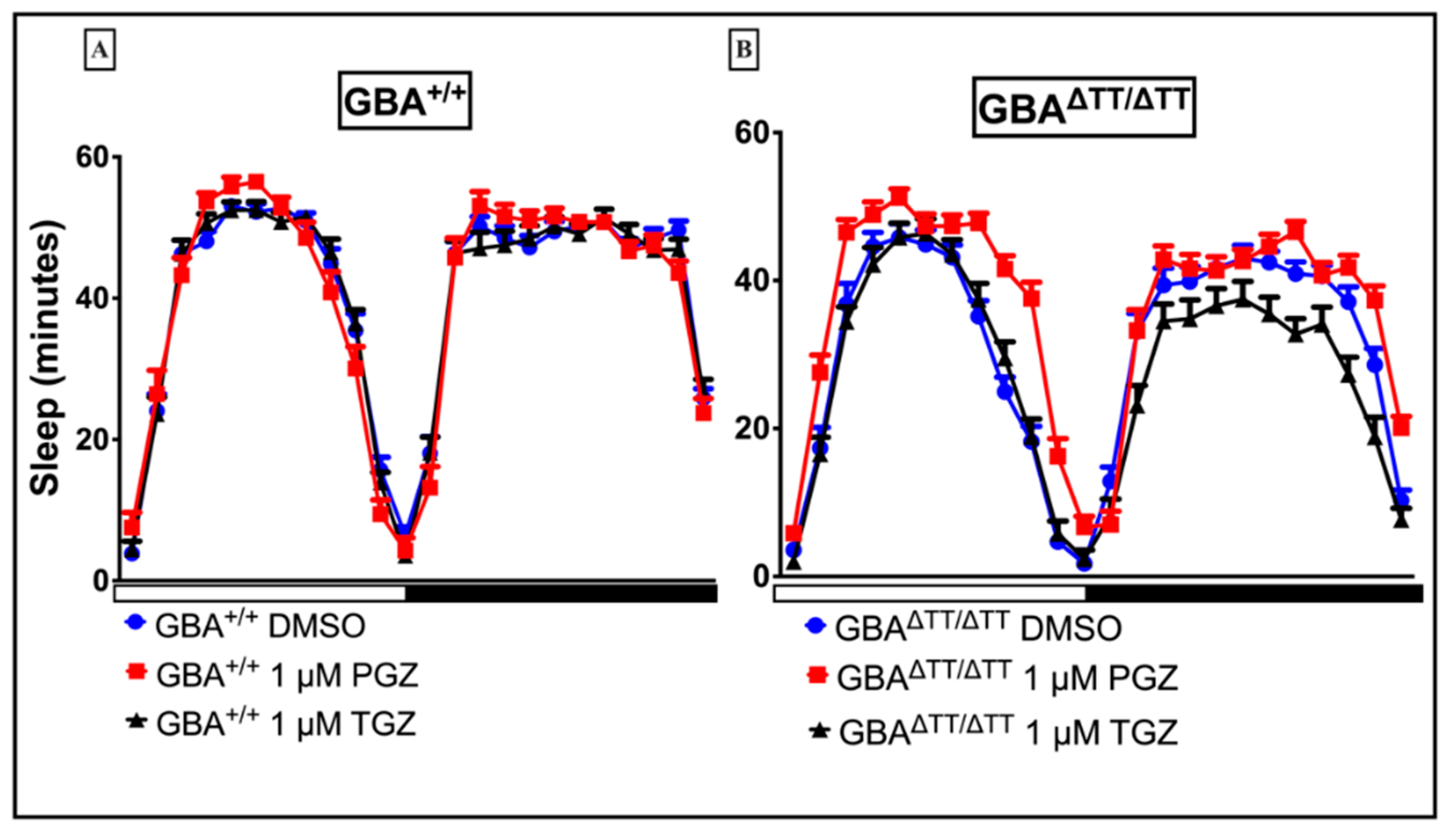
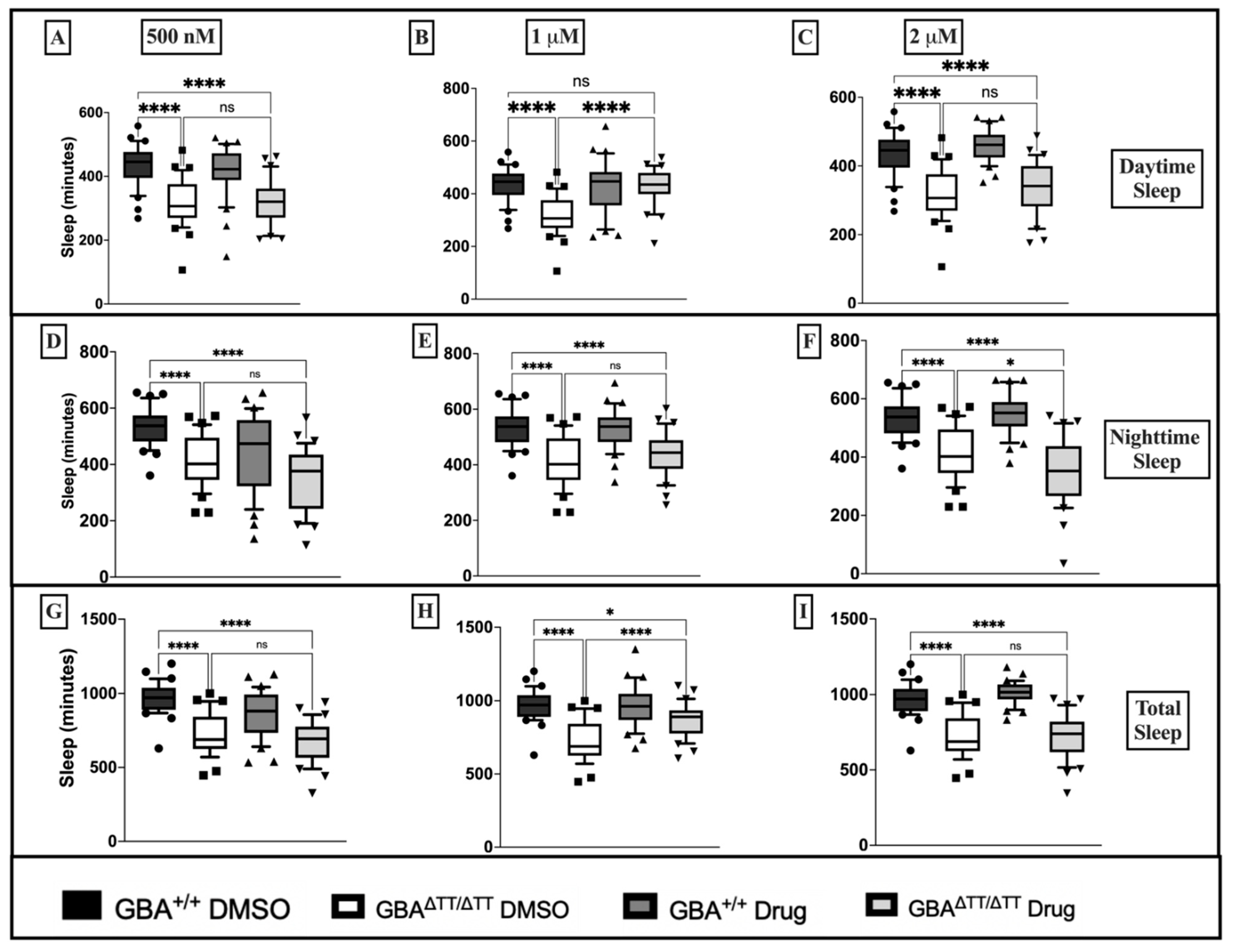
2.1.2. Thiazolidinedione (Glitazones) Screening in the GBA1 Deficiency Fly Model of PD
2.1.3. Locomotor Deficits Reduced by 1 μM Pioglitazone
2.1.4. GBA Deficiency Leads to Lysosomal-Autophagic Deficits Reduced by 1 μM Pioglitazone
3. Discussion and Conclusions
4. Materials and Methods
4.1. Drosophila Strains
4.2. Sleep/Wake Monitoring
4.3. Mobility
4.4. Negative Geotaxis Assay
4.5. Western Blots
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
References
- Billingsley, K.J.; Bandres-Ciga, S.; Saez-Atienzar, S.; Singleton, A.B. Genetic risk factors in Parkinson’s disease. Z. Zellforsch. Mikrosk. Anat. 2018, 373, 9–20. [Google Scholar] [CrossRef]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Goker-Alpan, O.; Stubblefield, B.K.; Giasson, B.I.; Sidransky, E. Glucocerebrosidase is present in α-synuclein inclusions in Lewy body disorders. Acta Neuropathol. 2010, 120, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H. Glucocerebrosidase and Parkinson disease: Recent advances. Mol. Cell. Neurosci. 2015, 66, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.-H.; Xu, K.; Sun, Y.; Liou, B.; Quinn, B.; Li, R.-H.; Xue, L.; Zhang, W.; Setchell, K.D.; Witte, D.; et al. Multiple pathogenic proteins implicated in neuronopathic Gaucher disease mice. Hum. Mol. Genet. 2014, 23, 3943–3957. [Google Scholar] [CrossRef] [Green Version]
- Roberts, R.; Wade-Martins, R.; Alegre-Abarrategui, J. Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinson’s disease brain. Brain 2015, 138, 1642–1657. [Google Scholar] [CrossRef] [Green Version]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Boot, B.; Locascio, J.J.; Ms, I.E.J.; Winder-Rhodes, S.; Eberly, S.; Elbaz, A.; Brice, A.; Ravina, B.; Van Hilten, J.J.; et al. Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann. Neurol. 2016, 80, 674–685. [Google Scholar] [CrossRef]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef]
- Bilen, J.; Bonini, N.M. Drosophila as a Model for Human Neurodegenerative Disease. Annu. Rev. Genet. 2005, 39, 153–171. [Google Scholar] [CrossRef] [Green Version]
- Ugur, B.; Chen, K.; Bellen, H.J. Drosophila tools and assays for the study of human diseases. Dis. Model. Mech. 2016, 9, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Pandey, U.B.; Nichols, C.D. Human Disease Models in Drosophila melanogaster and the Role of the Fly in Therapeutic Drug Discovery. Pharmacol. Rev. 2011, 63, 411–436. [Google Scholar] [CrossRef] [Green Version]
- The modENCODE Consortium; Roy, S.; Ernst, J.; Kharchenko, P.V.; Kheradpour, P.; Negre, N.; Eaton, M.L.; Landolin, J.M.; Bristow, C.A.; Ma, L.; et al. Identification of Functional Elements and Regulatory Circuits by Drosophila modENCODE. Science 2010, 330, 1787–1797. [Google Scholar] [CrossRef] [Green Version]
- Chintapalli, V.R.; Wang, J.; Dow, J. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat. Genet. 2007, 39, 715–720. [Google Scholar] [CrossRef]
- Davis, M.Y.; Trinh, K.; Thomas, R.E.; Yu, S.; Germanos, A.A.; Whitley, B.N.; Sardi, S.P.; Montine, T.J.; Pallanck, L.J. Glucocerebrosidase Deficiency in Drosophila Results in α-Synuclein-Independent Protein Aggregation and Neurodegeneration. PLoS Genet. 2016, 12, e1005944. [Google Scholar] [CrossRef] [Green Version]
- Kinghorn, K.J.; Grönke, S.; Castillo-Quan, J.I.; Woodling, N.S.; Li, L.; Sirka, E.; Gegg, M.; Mills, K.; Hardy, J.; Bjedov, I.; et al. A Drosophila Model of Neuronopathic Gaucher Disease Demonstrates Lysosomal-Autophagic Defects and Altered mTOR Signalling and Is Functionally Rescued by Rapamycin. J. Neurosci. 2016, 36, 11654–11670. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, H.; Suzuki, T.; Ito, K.; Takahara, T.; Goto-Inoue, N.; Setou, M.; Sakata, K.; Ishida, N. Minos-insertion mutant of the Drosophila GBA gene homologue showed abnormal phenotypes of climbing ability, sleep and life span with accumulation of hydroxy-glucocerebroside. Gene 2017, 614, 49–55. [Google Scholar] [CrossRef]
- Maor, G.; Cabasso, O.; Krivoruk, O.; Rodriguez, J.; Steller, H.; Segal, D.; Horowitz, M. The contribution of mutantGBAto the development of Parkinson disease in Drosophila. Hum. Mol. Genet. 2016, 25, 2712–2727. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-C.; Wu, J.-S.; Tsai, H.-D.; Huang, C.-Y.; Chen, J.-J.; Sun, G.Y.; Lin, T.-N. Peroxisome Proliferator-Activated Receptor Gamma (PPAR-γ) and Neurodegenerative Disorders. Mol. Neurobiol. 2012, 46, 114–124. [Google Scholar] [CrossRef]
- Hsiao, P.-J.; Chiou, H.-Y.C.; Jiang, H.J.; Lee, M.-Y.; Hsieh, T.-J.; Kuo, K.-K. Pioglitazone Enhances Cytosolic Lipolysis, β-oxidation and Autophagy to Ameliorate Hepatic Steatosis. Sci. Rep. 2017, 7, 9030. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, M.; Costa, L.; Palermo, R.; Giovenco, A.; Vacca, A.; Gulino, A. Protective effect of pioglitazone, a PPARγ ligand, in a 3 nitropropionic acid model of Huntington’s disease. Brain Res. Bull. 2011, 85, 231–237. [Google Scholar] [CrossRef]
- Ridder, D.A.; Schwaninger, M. In search of the neuroprotective mechanism of thiazolidinediones in Parkinson’s disease. Exp. Neurol. 2012, 238, 133–137. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, W.; Li, G.; Chen, J.; Guan, X.; Chen, X.; Guan, Z. Neuroprotective Effect and Mechanism of Thiazolidinedione on Dopaminergic Neurons In Vivo and In Vitro in Parkinson’s Disease. PPAR Res. 2017, 2017, 4089214. [Google Scholar] [CrossRef] [Green Version]
- Prabitha, P.; Justin, A.; Kumar, T.D.A.; Chinaswamy, M.; Kumar, B.R.P. Glitazones Activate PGC-1α Signaling via PPAR-γ: A Promising Strategy for Antiparkinsonism Therapeutics. ACS Chem. Neurosci. 2021, 12, 2261–2272. [Google Scholar] [CrossRef]
- Kumar, A.P.; Prabitha, P.; Kumar, B.R.P.; Jeyarani, V.; Dhanabal, S.P.; Justin, A. Glitazones, PPAR-γ and Neuroprotection. Mini Rev. Med. Chem. 2021, 21, 1457–1464. [Google Scholar] [CrossRef]
- Corona, J.C.; Duchen, M.R. PPARγ and PGC-1α as Therapeutic Targets in Parkinson’s. Neurochem. Res. 2015, 40, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Jiang, S.; Yang, Z.; Hu, W.; Wang, Z.; Li, T.; Yang, Y. PGC-1α sparks the fire of neuroprotection against neurodegenerative disorders. Ageing Res. Rev. 2018, 44, 8–21. [Google Scholar] [CrossRef]
- Swanson, C.R.; Joers, V.; Bondarenko, V.; Brunner, K.; Simmons, H.A.; Ziegler, T.E.; Kemnitz, J.W.; Johnson, J.A.; Emborg, M.E. The PPAR-γ agonist pioglitazone modulates inflammation and induces neuroprotection in parkinsonian monkeys. J. Neuroinflamm. 2011, 8, 91. [Google Scholar] [CrossRef] [Green Version]
- Brauer, R.; Bhaskaran, K.; Chaturvedi, N.; Dexter, D.T.; Smeeth, L.; Douglas, I. Glitazone Treatment and Incidence of Parkinson’s Disease among People with Diabetes: A Retrospective Cohort Study. PLoS Med. 2015, 12, e1001854. [Google Scholar] [CrossRef] [Green Version]
- Brakedal, B.; Flønes, I.; Reiter, S.F.; Torkildsen, Ø.; Dölle, C.; Assmus, J.; Haugarvoll, K.; Tzoulis, C. Glitazone use associated with reduced risk of Parkinson’s disease. Mov. Disord. 2017, 32, 1594–1599. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.-L.; Tseng, Y.-F.; Chao, J.C.-J.; Hsu, C.-Y. Association of thiazolidinedione with a lower risk of Parkinson’s disease in a population with newly-diagnosed diabetes mellitus. Ann. Med. 2018, 50, 430–436. [Google Scholar] [CrossRef]
- Chang, Y.; Yen, S.; Wu, W.; Lin, K. Pioglitazone and statins lower incidence of Parkinson disease in patients with diabetes mellitus. Eur. J. Neurol. 2021, 28, 430–437. [Google Scholar] [CrossRef]
- Cirelli, C. The genetic and molecular regulation of sleep: From fruit flies to humans. Nat. Rev. Neurosci. 2009, 10, 549–560. [Google Scholar] [CrossRef] [Green Version]
- Videnovic, A.; Golombek, D. Circadian and sleep disorders in Parkinson’s disease. Exp. Neurol. 2013, 243, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Gerstner, J.R.; Vanderheyden, W.M.; Shaw, P.J.; Landry, C.F.; Yin, J.C.P. Fatty-Acid Binding Proteins Modulate Sleep and Enhance Long-Term Memory Consolidation in Drosophila. PLoS ONE 2011, 6, e15890. [Google Scholar] [CrossRef] [Green Version]
- Gerstner, J.R.; Lenz, O.; Vanderheyden, W.M.; Chan, M.T.; Pfeiffenberger, C.; Pack, A. Amyloid-β induces sleep fragmentation that is rescued by fatty acid binding proteins in Drosophila. J. Neurosci. Res. 2017, 95, 1548–1564. [Google Scholar] [CrossRef]
- Joardar, A.; Menzl, J.; Podolsky, T.C.; Manzo, E.; Estes, P.S.; Ashford, S.; Zarnescu, D.C. PPAR gamma activation is neuroprotective in a Drosophila model of ALS based on TDP-43. Hum. Mol. Genet. 2015, 24, 1741–1754. [Google Scholar] [CrossRef] [Green Version]
- Madabattula, S.T.; Strautman, J.C.; Bysice, A.M.; O’Sullivan, J.A.; Androschuk, A.; Rosenfelt, C.; Doucet, K.; Rouleau, G.; Bolduc, F. Quantitative Analysis of Climbing Defects in a Drosophila Model of Neurodegenerative Disorders. J. Vis. Exp. 2015, 10, e52741. [Google Scholar] [CrossRef]
- Ali, Y.O.; Escala, W.; Ruan, K.; Zhai, R. Assaying Locomotor, Learning, and Memory Deficits in Drosophila Models of Neurodegeneration. J. Vis. Exp. 2011, 10, e2504. [Google Scholar] [CrossRef] [Green Version]
- Lynch-Day, M.A.; Mao, K.; Wang, K.; Zhao, M.; Klionsky, D.J. The Role of Autophagy in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009357. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Yang, C.; Iyaswamy, A.; Krishnamoorthi, S.; Sreenivasmurthy, S.G.; Liu, J.; Wang, Z.; Tong, B.C.-K.; Song, J.; Lu, J.; et al. Balancing mTOR Signaling and Autophagy in the Treatment of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 728. [Google Scholar] [CrossRef] [Green Version]
- Cerri, S.; Blandini, F. Role of Autophagy in Parkinson’s Disease. Curr. Med. Chem. 2019, 26, 3702–3718. [Google Scholar] [CrossRef]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s Disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef]
- Cheung, Z.H.; Ip, N.Y. The emerging role of autophagy in Parkinson’s disease. Mol. Brain 2009, 2, 29. [Google Scholar] [CrossRef] [Green Version]
- DeVorkin, L.; Gorski, S.M. Monitoring Autophagic Flux Using Ref(2)P, the Drosophila p62 Ortholog. Cold Spring Harb. Protoc. 2014, 2014, 959–966. [Google Scholar] [CrossRef]
- Chen, W.; Xi, X.; Zhang, S.; Zou, C.; Kuang, R.; Ye, Z.; Huang, Y.; Hu, H. Pioglitazone Protects Against Renal Ischemia-Reperfusion Injury via the AMP-Activated Protein Kinase-Regulated Autophagy Pathway. Front. Pharmacol. 2018, 9, 851. [Google Scholar] [CrossRef] [Green Version]
- McFarthing, K.; Buff, S.; Rafaloff, G.; Dominey, T.; Wyse, R.K.; Stott, S.R. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2020. J. Parkinson’s Dis. 2020, 10, 757–774. [Google Scholar] [CrossRef]
- Church, F. Treatment Options for Motor and Non-Motor Symptoms of Parkinson’s Disease. Biomolecules 2021, 11, 612. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J.; Cirelli, C.; Greenspan, R.J.; Tononi, G. Correlates of Sleep and Waking in Drosophila melanogaster. Science 2000, 287, 1834–1837. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shola-Dare, O.; Bailess, S.; Flores, C.C.; Vanderheyden, W.M.; Gerstner, J.R. Glitazone Treatment Rescues Phenotypic Deficits in a Fly Model of Gaucher/Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 12740. https://doi.org/10.3390/ijms222312740
Shola-Dare O, Bailess S, Flores CC, Vanderheyden WM, Gerstner JR. Glitazone Treatment Rescues Phenotypic Deficits in a Fly Model of Gaucher/Parkinson’s Disease. International Journal of Molecular Sciences. 2021; 22(23):12740. https://doi.org/10.3390/ijms222312740
Chicago/Turabian StyleShola-Dare, Oluwanifemi, Shelby Bailess, Carlos C. Flores, William M. Vanderheyden, and Jason R. Gerstner. 2021. "Glitazone Treatment Rescues Phenotypic Deficits in a Fly Model of Gaucher/Parkinson’s Disease" International Journal of Molecular Sciences 22, no. 23: 12740. https://doi.org/10.3390/ijms222312740
APA StyleShola-Dare, O., Bailess, S., Flores, C. C., Vanderheyden, W. M., & Gerstner, J. R. (2021). Glitazone Treatment Rescues Phenotypic Deficits in a Fly Model of Gaucher/Parkinson’s Disease. International Journal of Molecular Sciences, 22(23), 12740. https://doi.org/10.3390/ijms222312740