
γ-Tocotrienol Protects against Mitochondrial Dysfunction, Energy Deficits, Morphological Damage, and Decreases in Renal Functions after Renal Ischemia

Abstract
:1. Introduction
2. Results
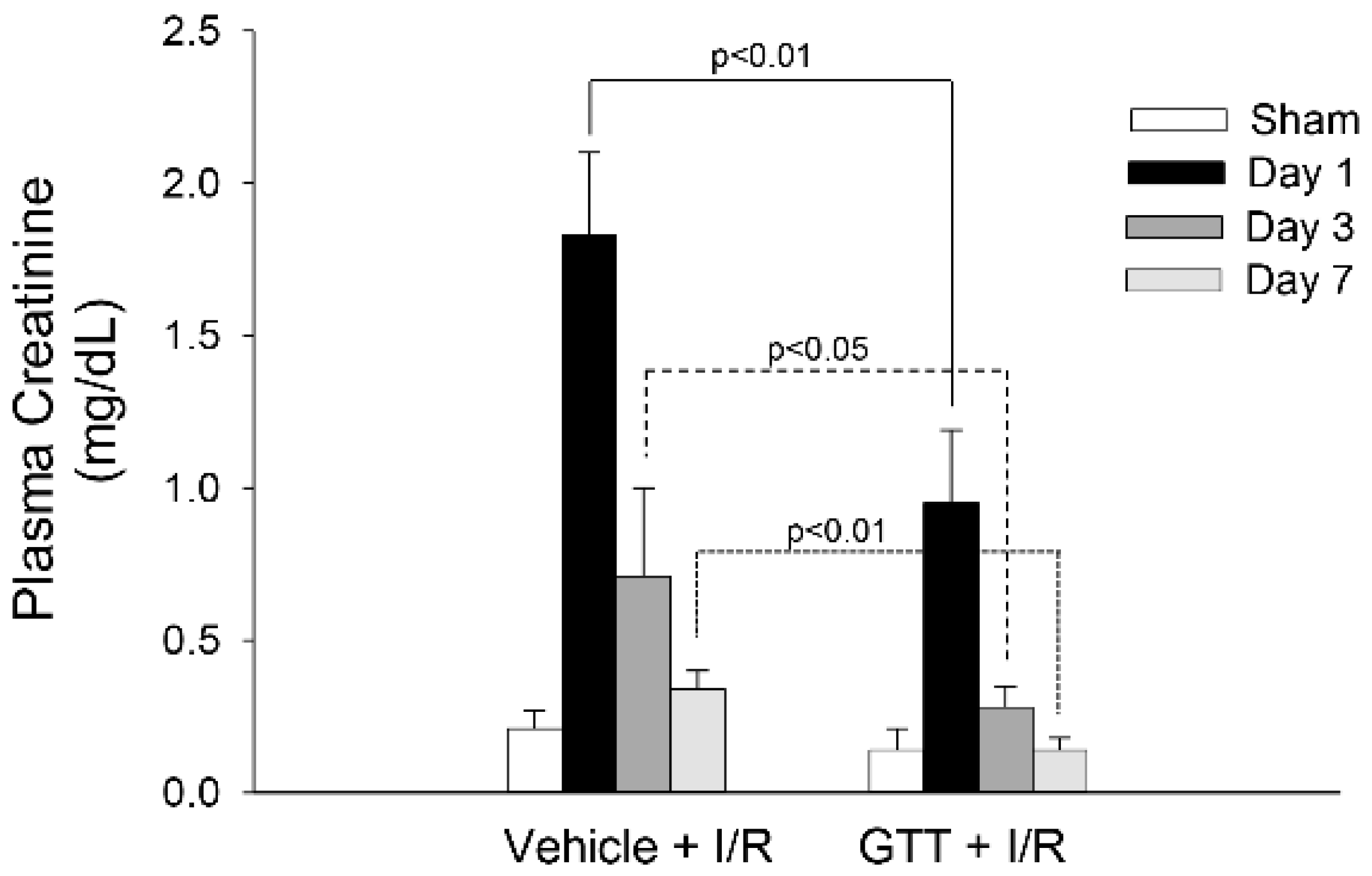
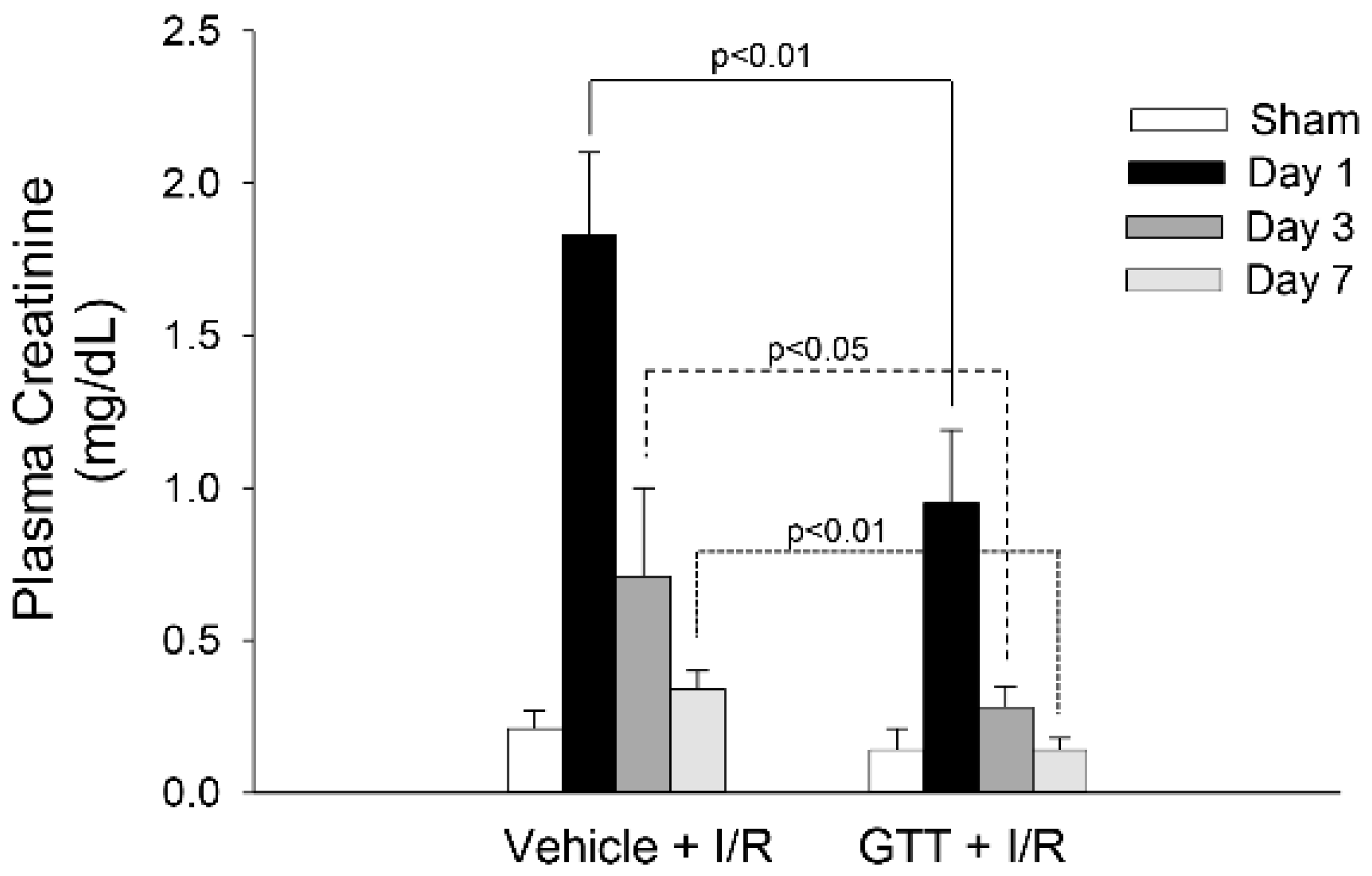
2.1. Treatment of Mice with GTT Improves Renal Function after Ischemia
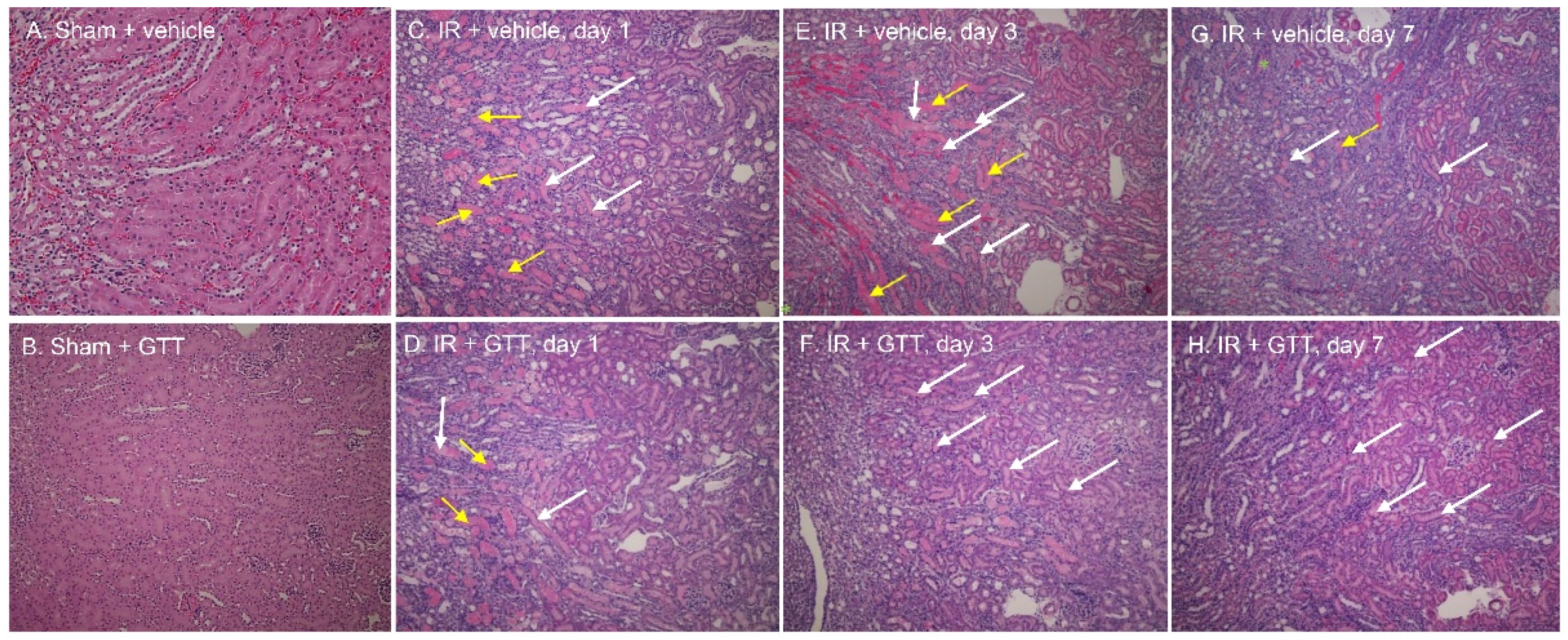
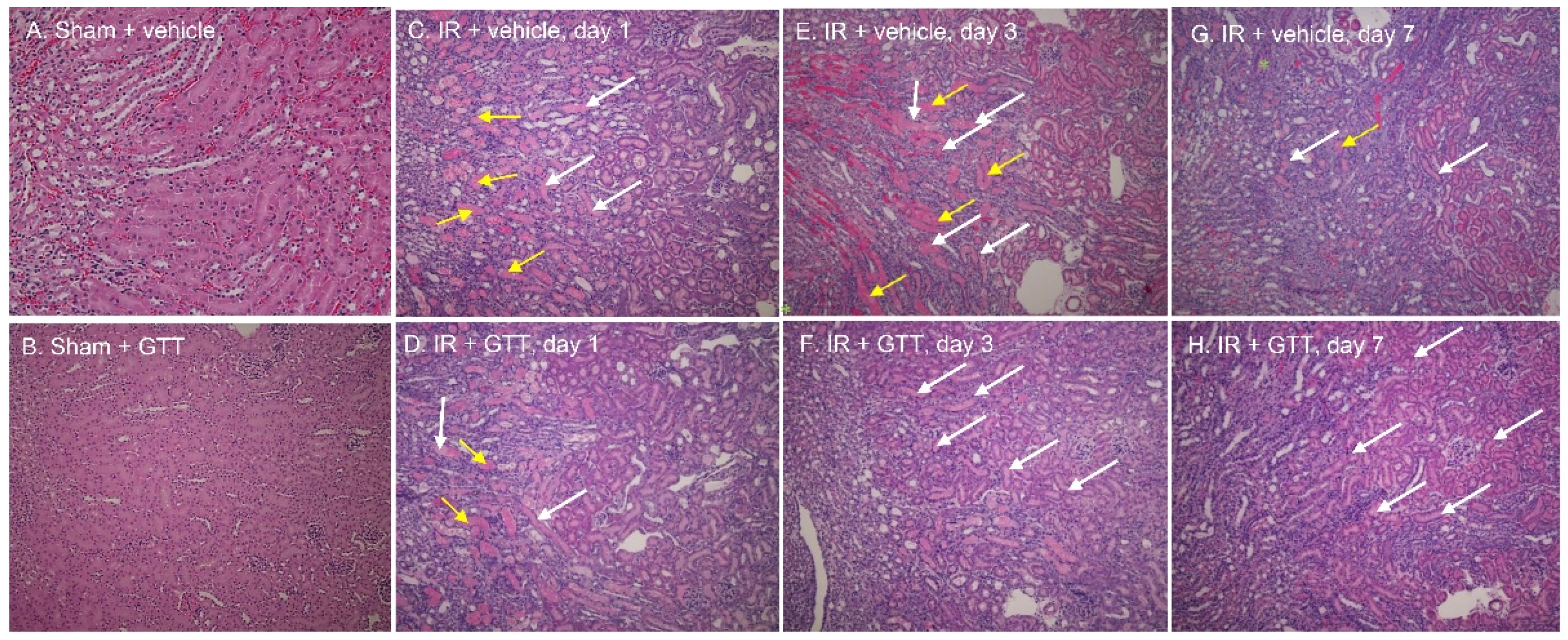
2.2. GTT Administration Improves Renal Morphology after Ischemia
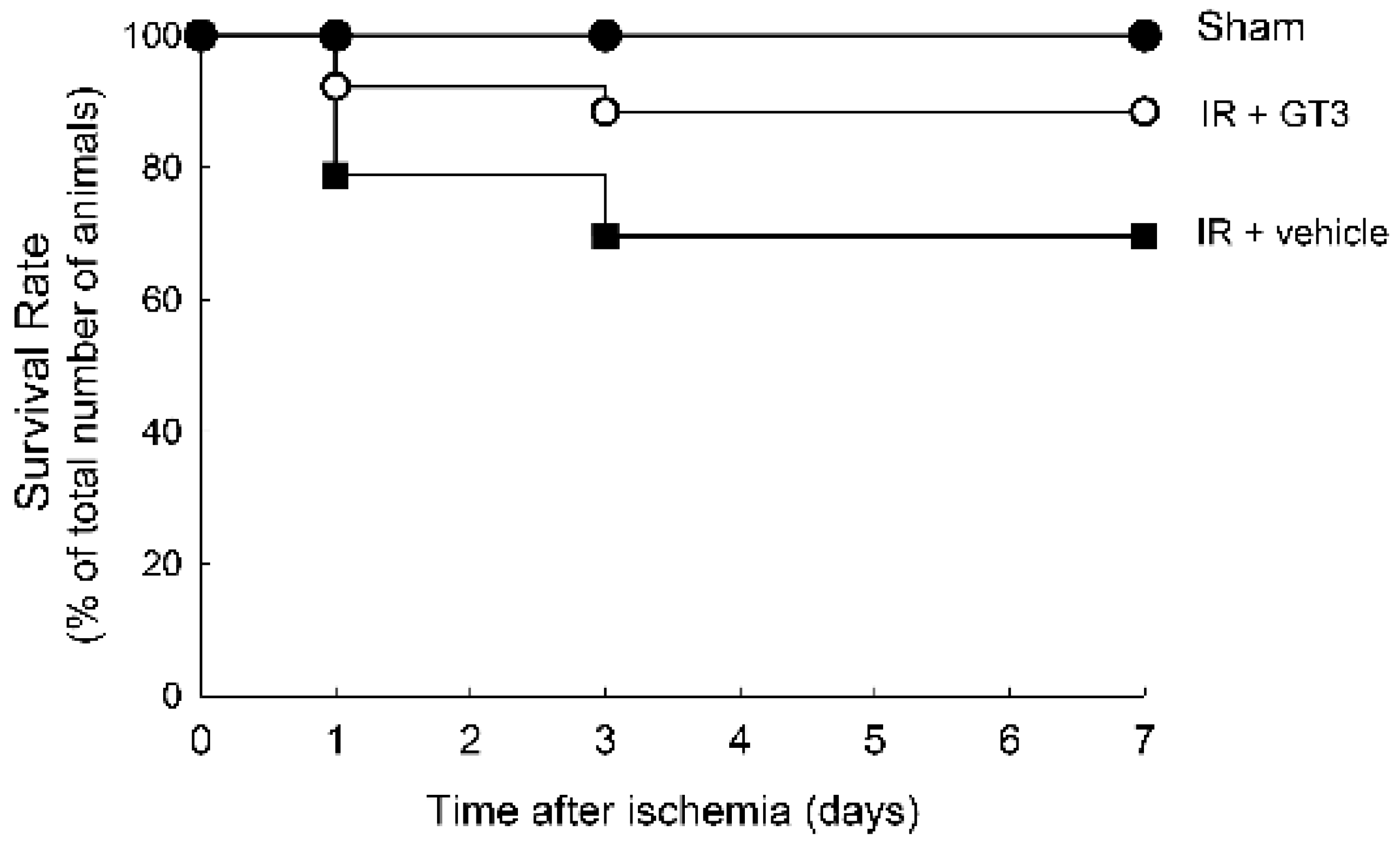
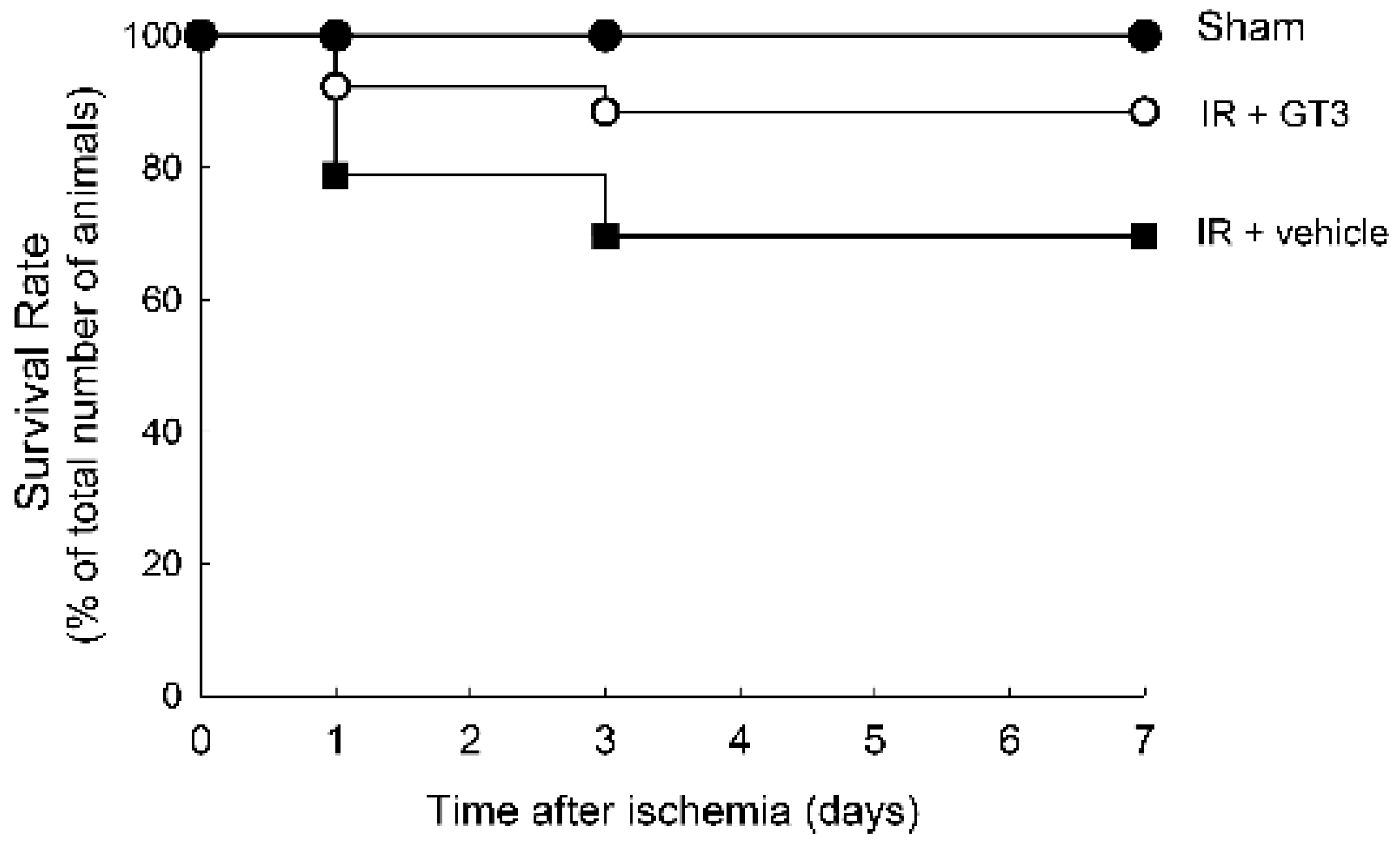
2.3. Treatment with GTT before Ischemia Improves Survival during Reperfusion
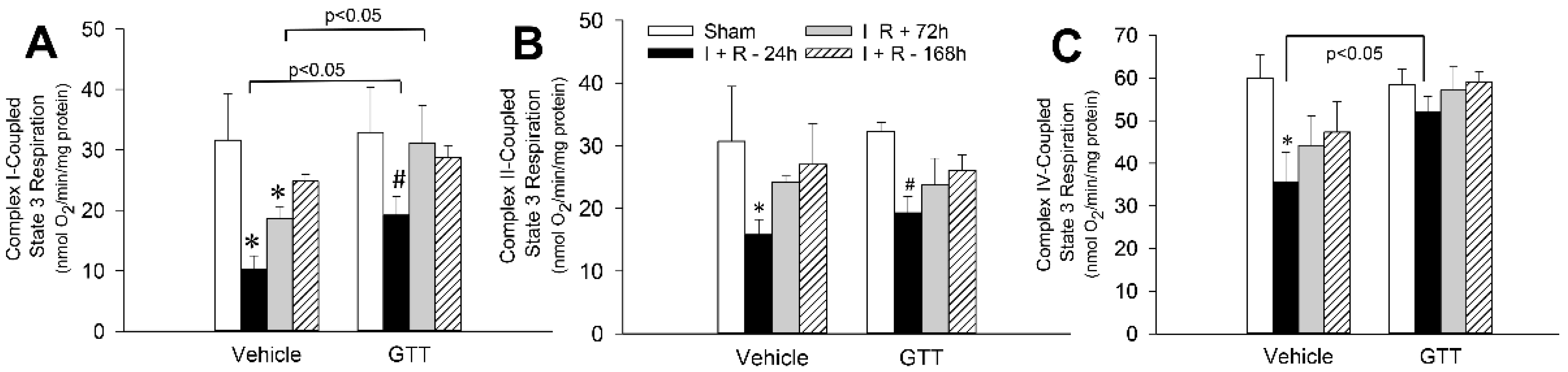
2.4. GTT Administration Promotes Mitochondrial Capacity for Maximum Respiration after Ischemia
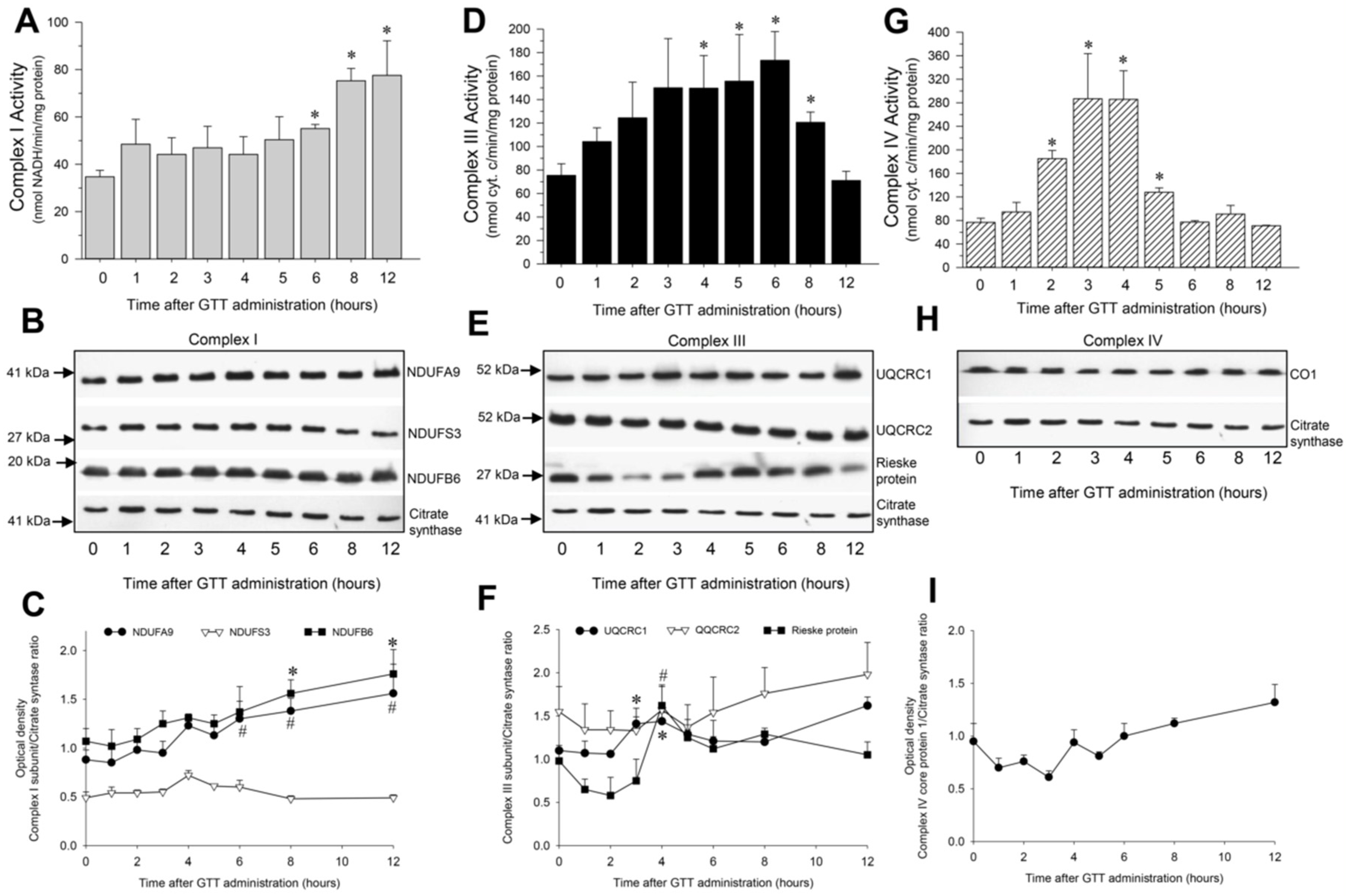
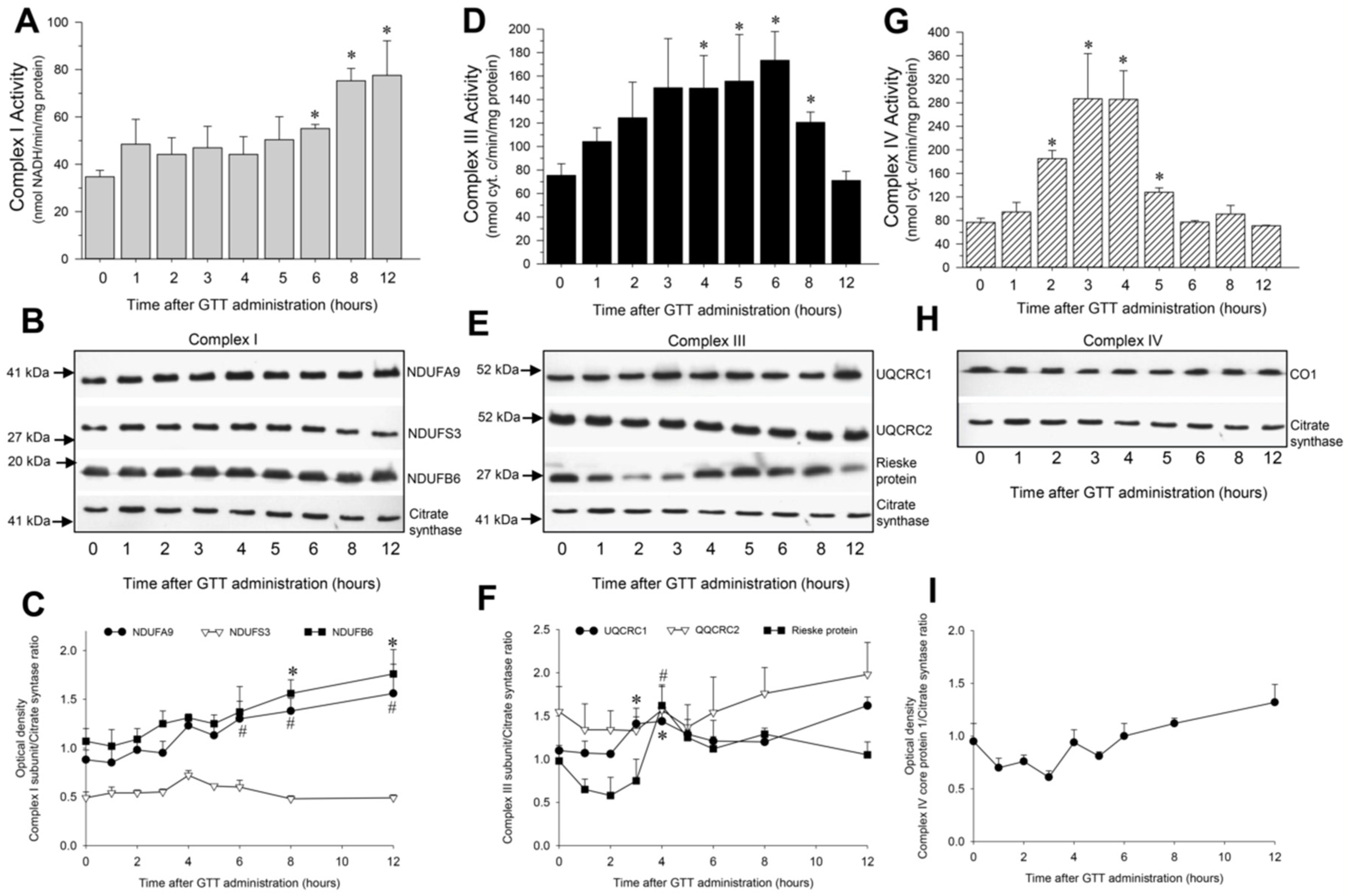
2.5. GTT Administration Increases Activities of Complexes of the Electron Chain in Cortical Mitochondria of Noninjured Kidneys
2.5.1. NADH:Ubiquinone Oxidoreductase
2.5.2. Ubiquinol:Cytochrome c Oxidoreductase
2.5.3. Cytochrome Oxidase
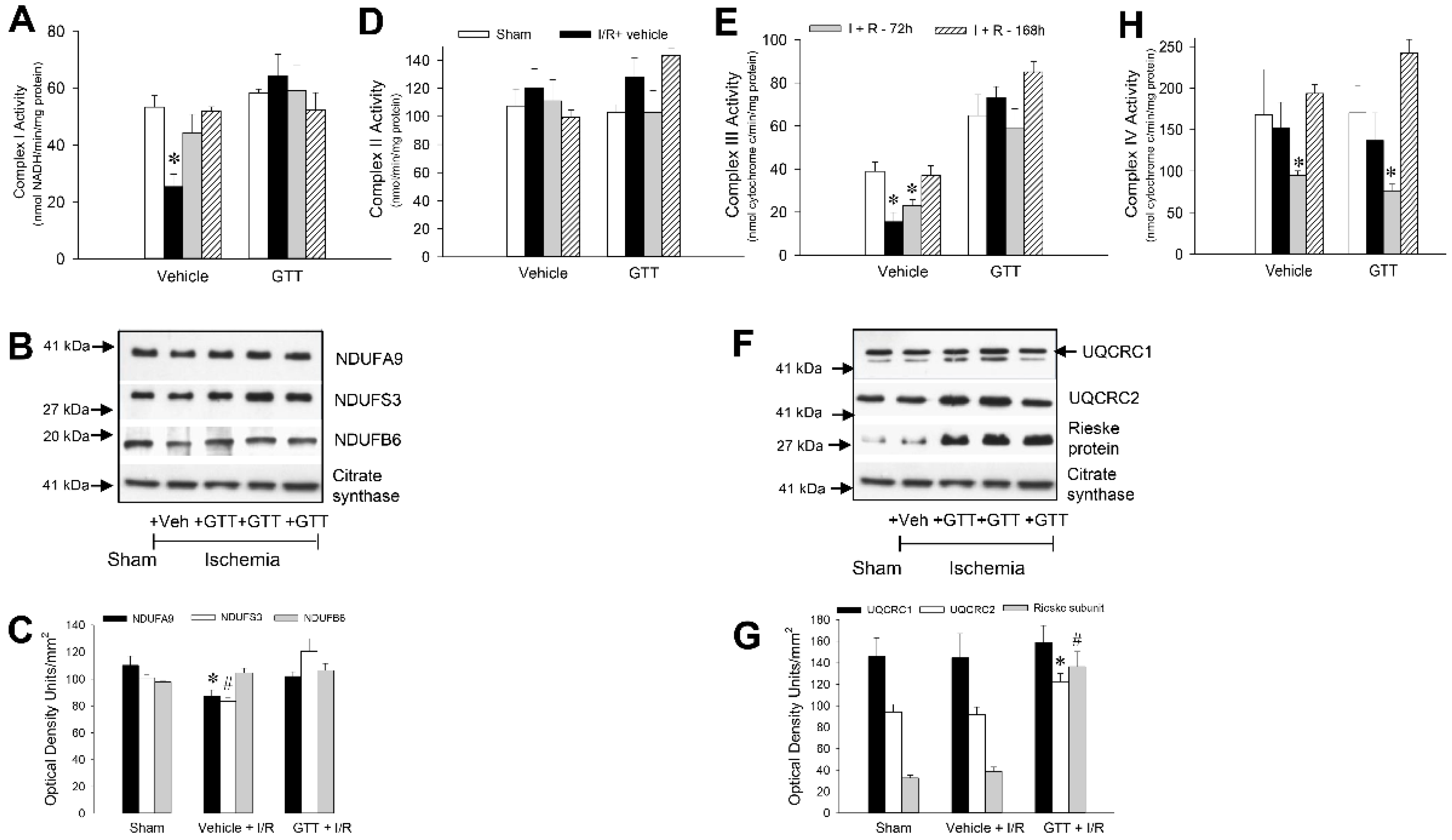
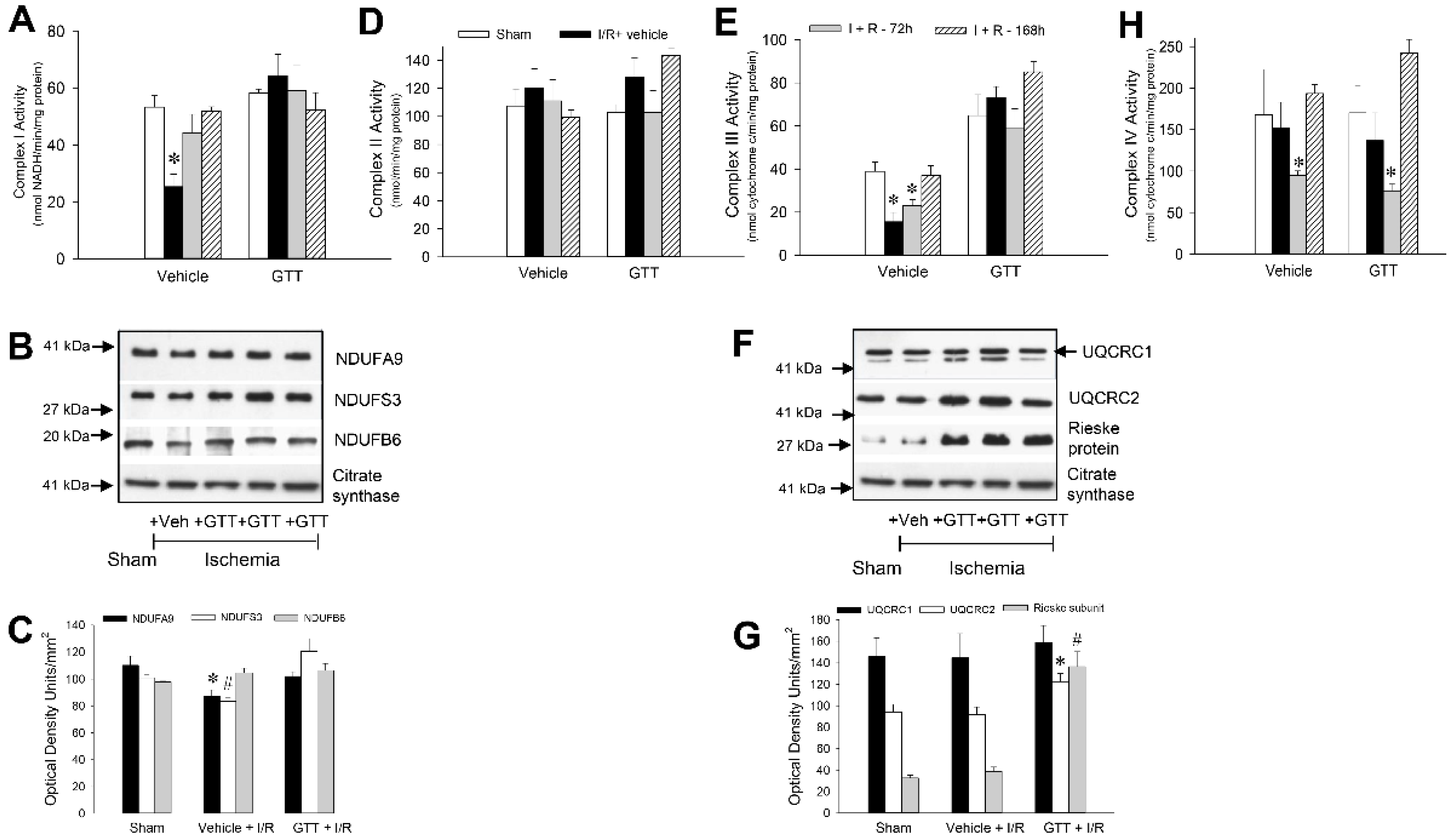
2.6. GTT Treatment Preserves Activities of Complexes I and III after Ischemia/Reperfusion Injury
2.6.1. NADH:Ubiquinone Oxidoreductase
2.6.2. Succinate:Ubiquinone Oxidoreductase
2.6.3. Ubiquinol:Cytochrome c Oxidoreductase
2.6.4. Cytochrome Oxidase
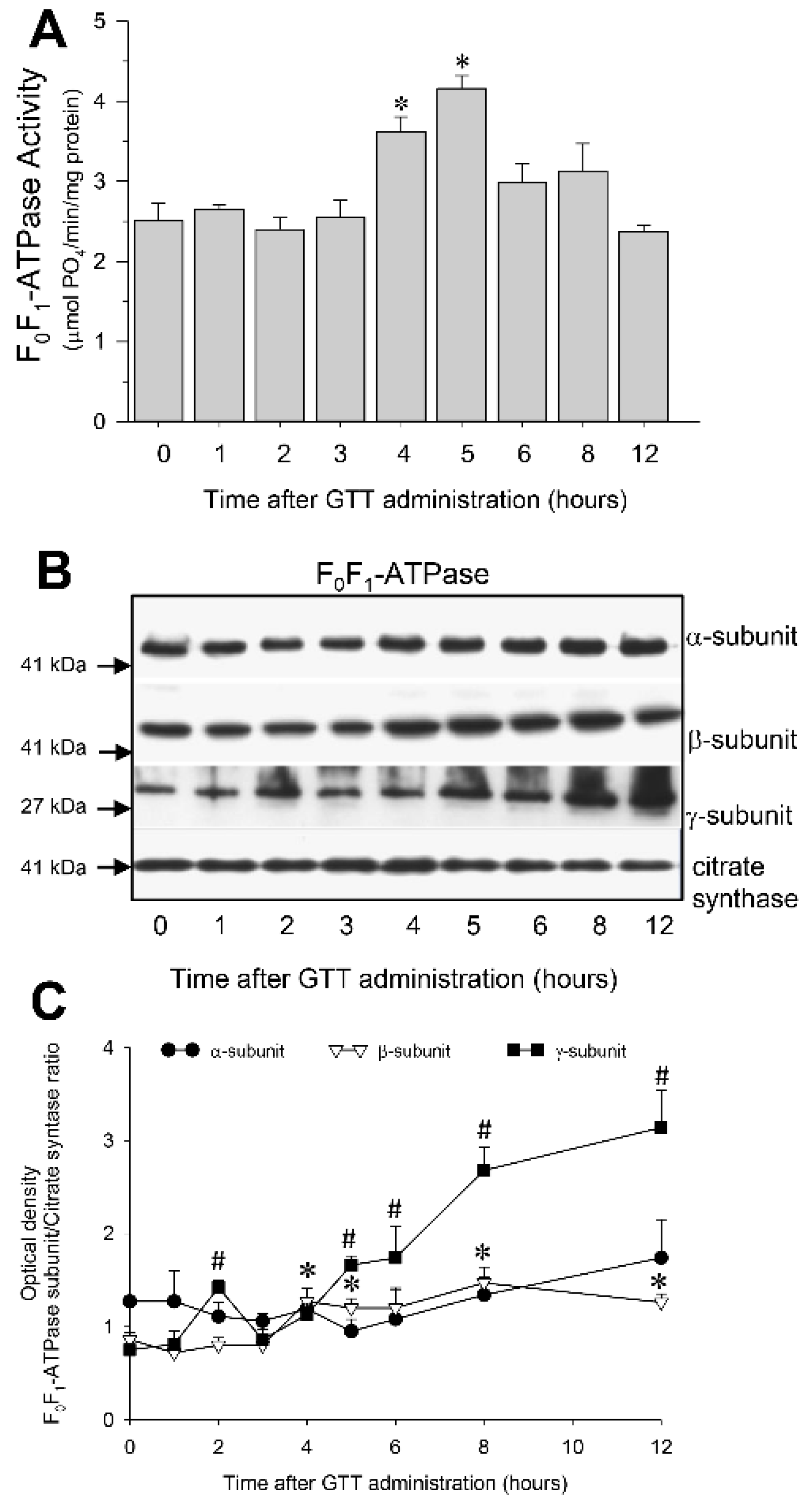
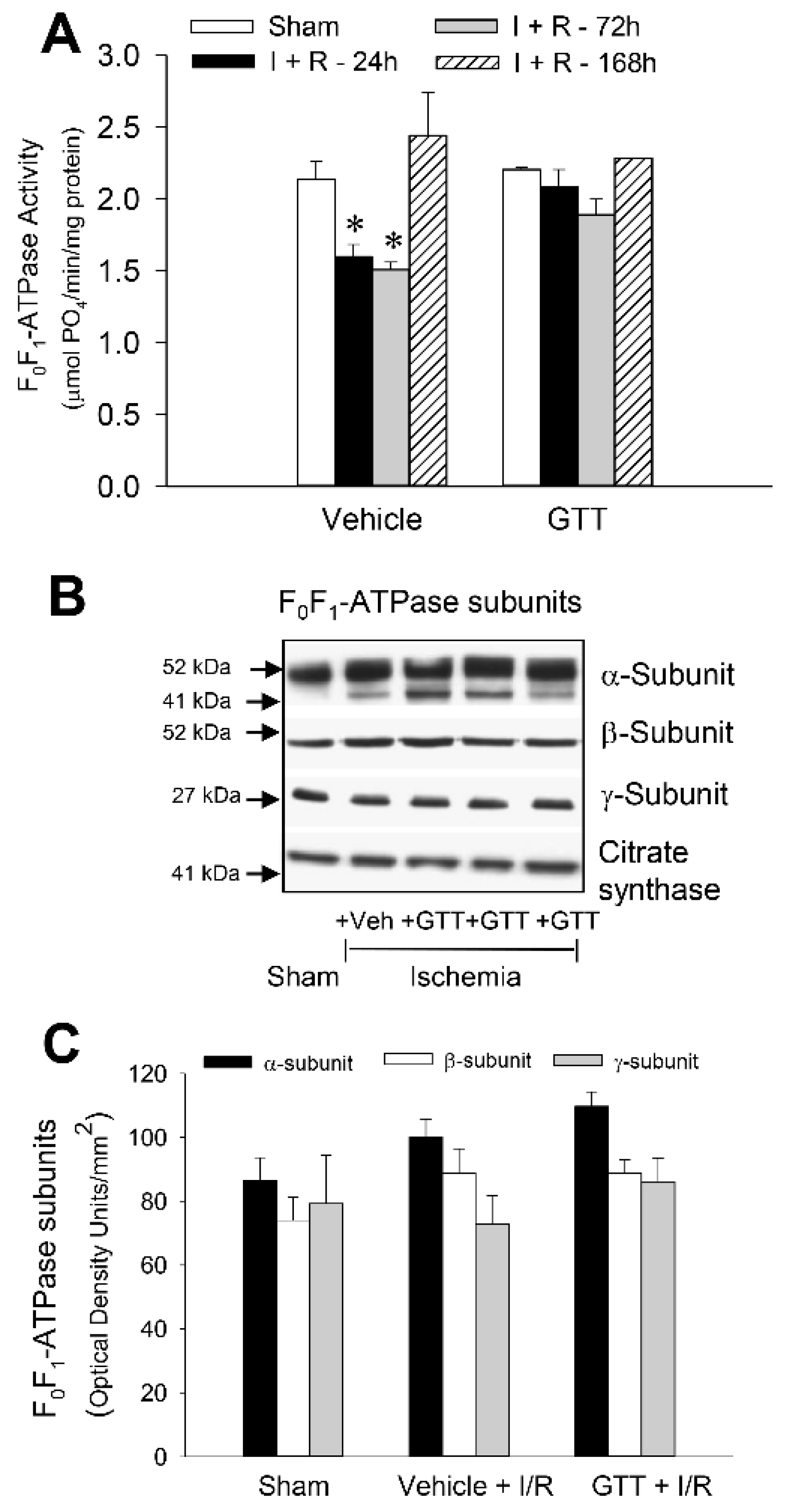
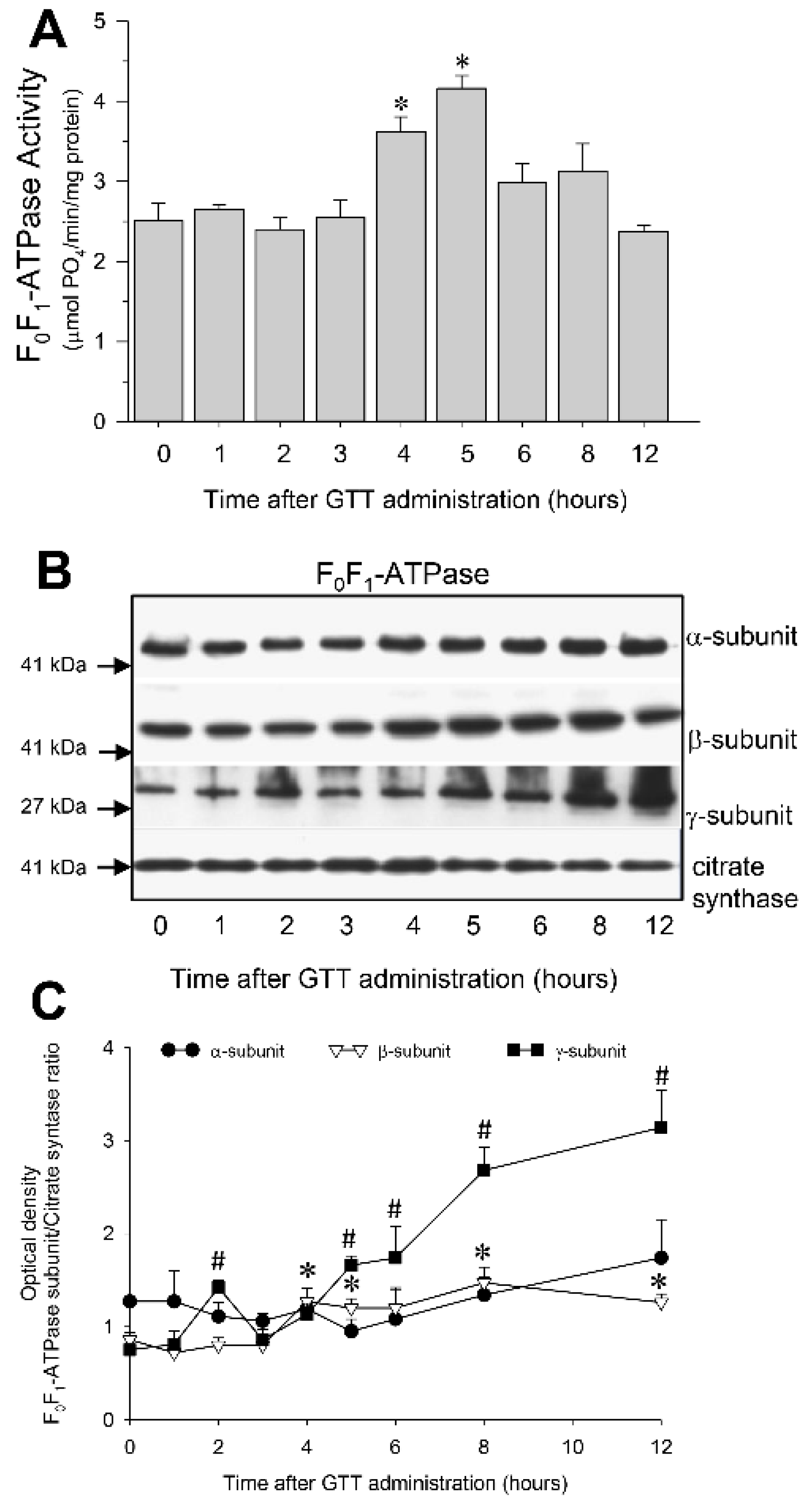
2.7. GTT Promotes Activity of ATP Synthase and Ameliorates Decreases in ATP Synthase Activity in Renal Cortical Mitochondria after Ischemia-Induced Injury
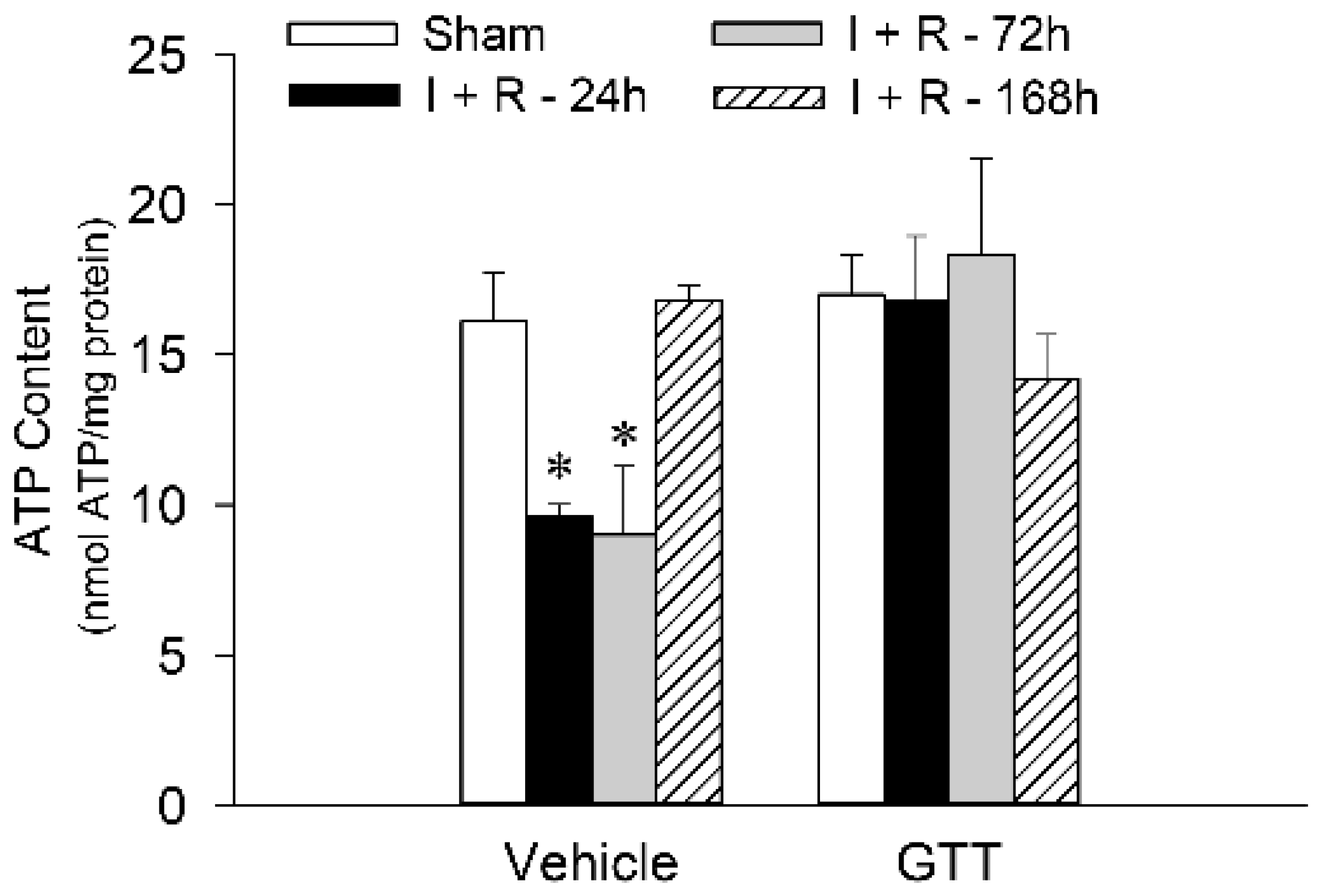
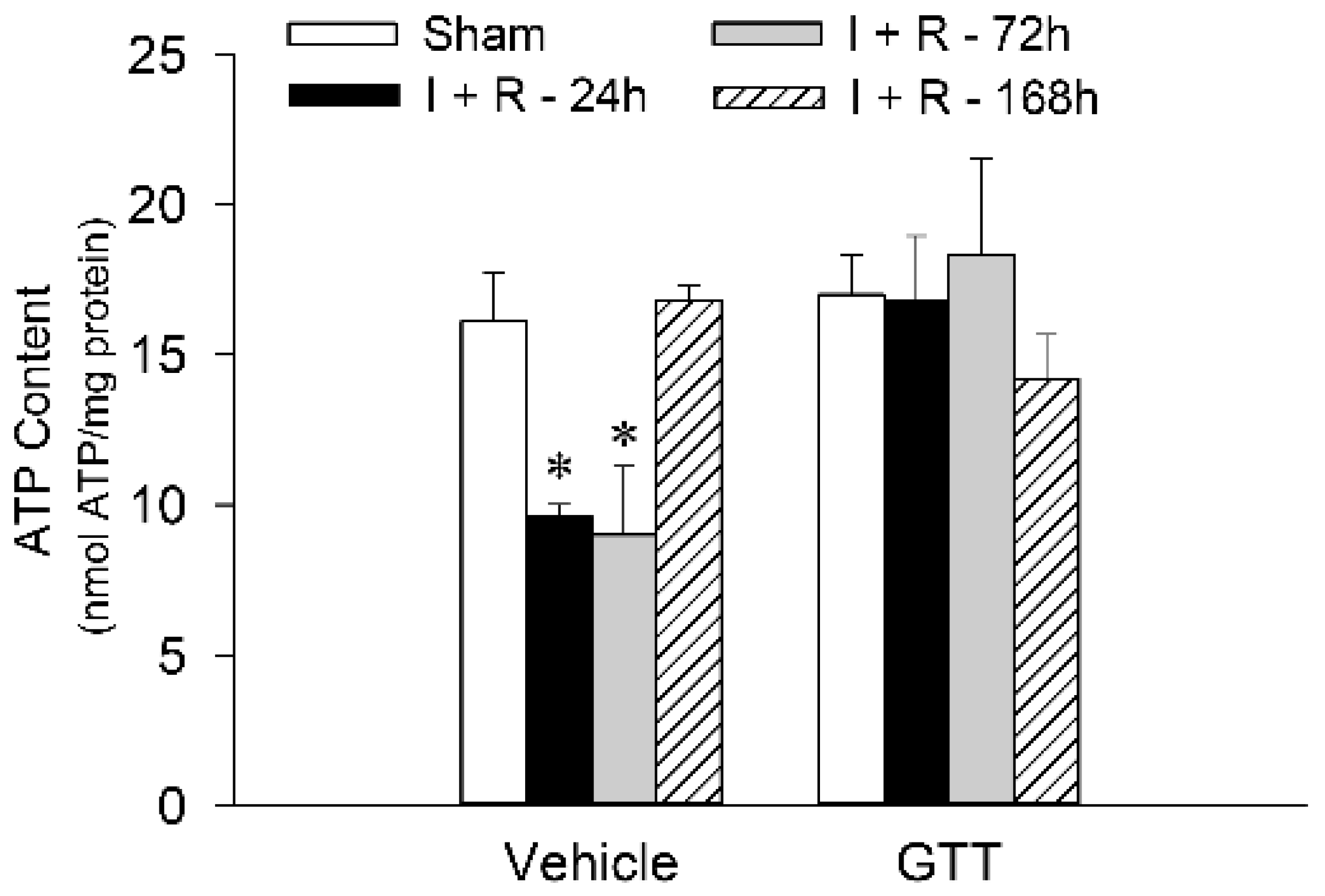
2.8. GTT Administration Maintains Renal ATP Content after Ischemic Injury
3. Discussion
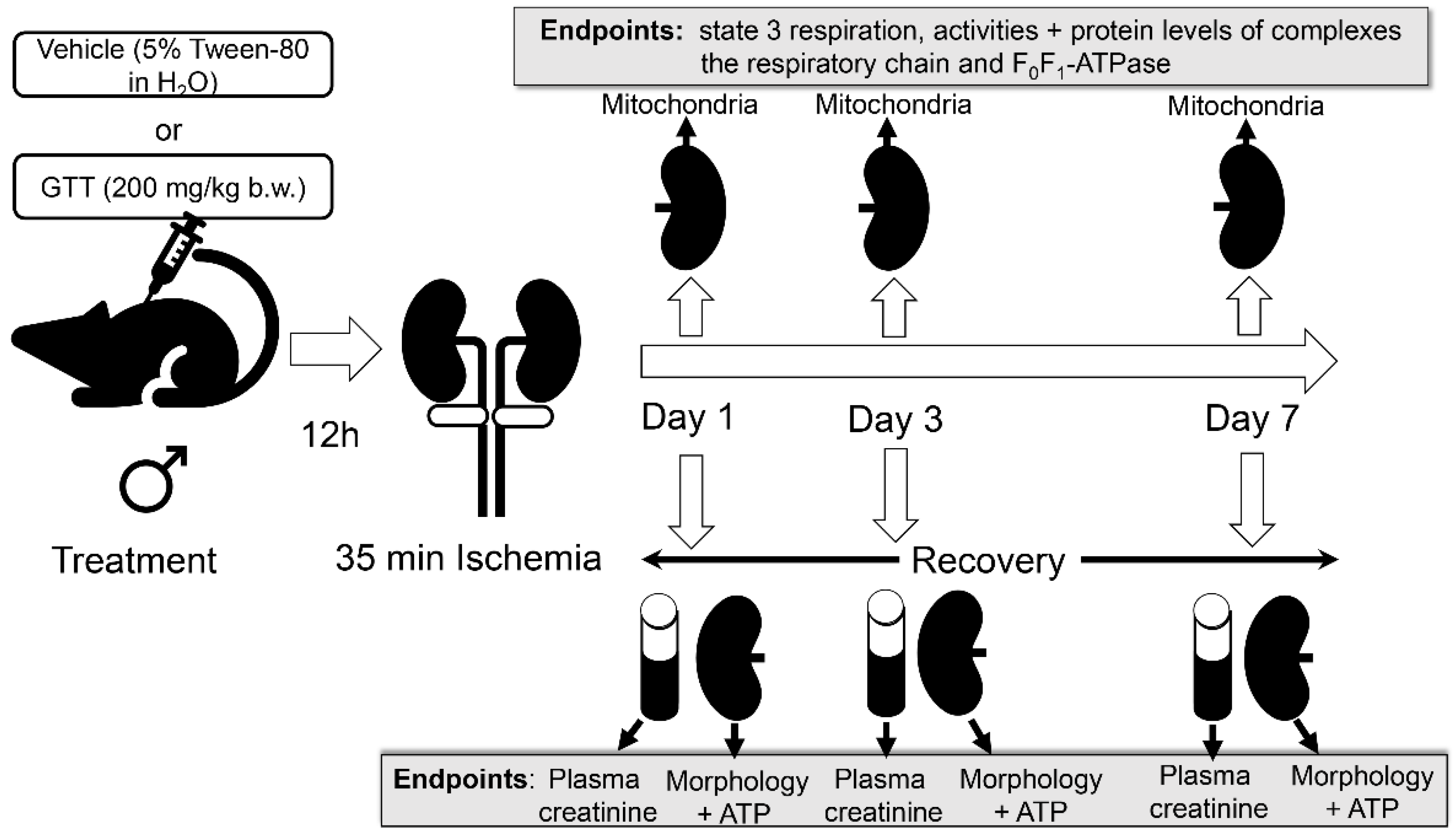
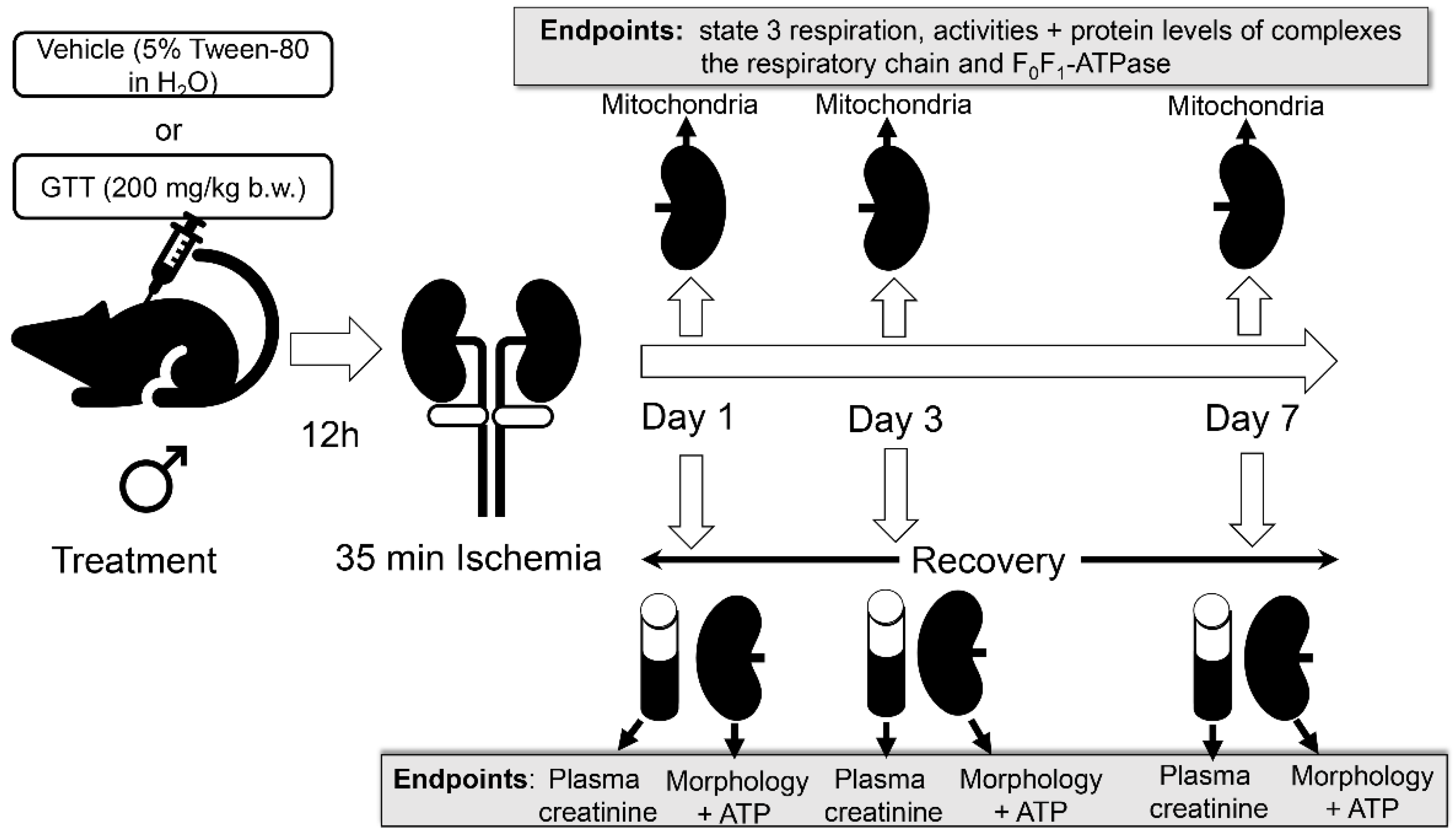
4. Materials and Methods
4.1. Animals
4.2. γ-Tocotrienol Administration
4.3. Ischemia/Reperfusion Injury
4.4. Assessment of Renal Morphology
4.5. Plasma Levels of Creatinine
4.6. Isolation of Mitochondria
4.7. State 3 Respiration
4.8. Activities of Respiratory Complexes
4.9. Activity of F0F1-ATPase
4.10. Tissue ATP Content
4.11. Immunoblotting
4.12. Protein Concentration
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brigelius-Flohe, R.; Traber, M.G. Vitamin E: Function and Metabolism. FASEB J. 1999, 13, 1145–1155. [Google Scholar] [CrossRef]
- Sen, C.K.; Gordillo, G.M.; Khanna, S.; Roy, S. Micromanaging Vascular Biology: Tiny microRNAs Play Big Band. J. Vasc. Res. 2009, 46, 527–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berbee, M.; Fu, Q.; Boerma, M.; Kumar, K.S.; Loose, D.S.; Hauer-Jensen, M. Mechanisms Underlying the Radioprotective Properties of Gamma-Tocotrienol: Comparative Gene Expression Profiling in Tocol-Treated Endothelial Cells. Genes Nutr. 2012, 7, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Podda, M.; Weber, C.; Traber, M.G.; Packer, L. Simultaneous Determination of Tissue Tocopherols, Tocotrienols, Ubiquinols, and Ubiquinones. J. Lipid Res. 1996, 37, 893–901. [Google Scholar] [CrossRef]
- Zhou, C.; Tabb, M.M.; Sadatrafiei, A.; Grün, F.; Blumberg, B. Tocotrienols Activate the Steroid and Xenobiotic Receptor, SXR, and Selectively Regulate Expression of its Target Genes. Drug. Metab. Dispos. 2004, 32, 1075–1082. [Google Scholar] [CrossRef] [Green Version]
- Komiyama, K.; Iizuka, K.; Yamaoka, M.; Watanabe, H.; Tsuchiya, N.; Umezawa, I. Studies on the Biological Activity of Tocotrienols. Chem. Pharm. Bull. 1989, 37, 1369–1371. [Google Scholar] [CrossRef] [Green Version]
- Goh, S.H.; Hew, N.F.; Norhanom, A.W.; Yadav, M. Inhibition of Tumor Promotion by Various Palm-Oil Tocotrienols. Int. J. Cancer 1994, 57, 529–531. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Sundaram, C.; Prasad, S.; Kannappan, R. Tocotrienols, the Vitamin E of the 21st Century: Its Potential Against Cancer and Other Chronic Diseases. Biochem. Pharmacol. 2010, 80, 1613–1631. [Google Scholar] [CrossRef] [Green Version]
- Sen, C.K.; Khanna, S.; Roy, S. Tocotrienols: Vitamin E beyond Tocopherols. Life Sci. 2006, 78, 2088–2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntyre, B.S.; Briski, K.P.; Gapor, A.; Sylvester, P.W. Antiproliferative and Apoptotic Effects of Tocopherols and Tocotrienols on Preneoplastic and Neoplastic Mouse Mammary Epithelial Cells. Proc. Soc. Exp. Biol. Med. 2000, 224, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Loo, G. Disruption of Mitochondria during Tocotrienol-Induced Apoptosis in MDA-MB-231 Human Breast Cancer Cells. Biochem. Pharmacol. 2004, 67, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samant, G.V.; Sylvester, P.W. γ-Tocotrienol Inhibits ErbB3-Dependent PI3K/Akt Mitogenic Signalling in Neoplastic Mammary Epithelial Cells. Cell Prolif. 2006, 39, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.S.; Sethi, G.; Krishnan, K.; Aggarwal, B.B. Gamma-Tocotrienol Inhibits Nuclear Factor-kappaB Signaling Pathway through Inhibition of Receptor-Interacting Protein and TAK1 Leading to Suppression of Antiapoptotic Gene Products and Potentiation of Apoptosis. J. Biol. Chem. 2007, 282, 809–820. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Xu, W.; Liu, H.; Liu, J.; Wang, Q.; Zhou, J.; Dong, F.; Chen, B. Gamma-Tocotrienol Induces Mitochondria-Mediated Apoptosis in Human Gastric Adenocarcinoma SGC-7901 Cells. J. Nutr. Biochem. 2009, 20, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Huang, G.; Ng, L. γ-Tocotrienol Induced Cell Cycle Arrest and Apoptosis Via Activating the Bax-Mediated Mitochondrial and AMPK Signaling Pathways in 3T3-L1 Adipocytes. Food Chem. Toxicol. 2013, 59, 501–513. [Google Scholar] [CrossRef]
- Wang, H.; Luo, J.; Tian, W.; Yan, W.; Ge, S.; Zhang, Y.; Sun, W. γ-Tocotrienol Inhibits Oxidative Phosphorylation and Triggers Apoptosis by Inhibiting Mitochondrial Complex I Subunit NDUFB8 and Complex II Subunit SDHB. Toxicology 2019, 417, 42–53. [Google Scholar] [CrossRef]
- Palozza, P.; Verdecchia, S.; Avanzi, L.; Vertuani, S.; Serini, S.; Iannone, A.; Manfredini, S. Comparative Antioxidant Activity of Tocotrienols and the Novel Chromanyl-Polyisoprenyl Molecule FeAox-6 in Isolated Membranes and Intact Cells. Mol. Cell. Biochem. 2006, 287, 21–32. [Google Scholar] [CrossRef]
- Lekli, I.; Ray, D.; Mukherjee, S.; Gurusamy, N.; Ahsan, M.K.; Juhasz, B.; Bak, I.; Tosaki, A.; Gherghiceanu, M.; Popescu, L.M.; et al. Co-Ordinated Autophagy with Resveratrol and Gamma-Tocotrienol Confers Synergetic Cardioprotection. J. Cell. Mol. Med. 2010, 14, 2506–2518. [Google Scholar] [CrossRef] [Green Version]
- Das, M.; Das, S.; Wang, P.; Powell, S.R.; Das, D.K. Caveolin and Proteasome in Tocotrienol Mediated Myocardial Protection. Cell. Physiol. Biochem. 2008, 22, 287–294. [Google Scholar] [CrossRef]
- Das, S.; Lekli, I.; Das, M.; Szabo, G.; Varadi, J.; Juhasz, B.; Bak, I.; Nesaretam, K.; Tosaki, A.; Powell, S.R.; et al. Cardioprotection with Palm Oil Tocotrienols: Comparision of Different Isomers. Am. J. Physiol.-Heart Circ. Physiol. 2008, 294, H970–H978. [Google Scholar] [CrossRef]
- Sen, C.K.; Khanna, S.; Roy, S.; Packer, L. Molecular Basis of Vitamin E Action. Tocotrienol Potently Inhibits Glutamate-Induced pp60(c-Src) Kinase Activation and Death of HT4 Neuronal Cells. J. Biol. Chem. 2000, 275, 13049–13055. [Google Scholar] [CrossRef] [Green Version]
- Mazlan, M.; Sue Mian, T.; Mat Top, G.; Zurinah Wan Ngah, W. Comparative Effects of α-Tocopherol and γ-Tocotrienol Against Hydrogen Peroxide Induced Apoptosis on Primary-Cultured Astrocytes. J. Neurol. Sci. 2006, 243, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.P.; Kulkarni, S.; Hieber, K.; Toles, R.; Romanyukha, L.; Kao, T.C.; Hauer-Jensen, M.; Kumar, K.S. Gamma-Tocotrienol, a Tocol Antioxidant as a Potent Radioprotector. Int. J. Radiat. Biol. 2009, 85, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.; Ghosh, S.P.; Satyamitra, M.; Mog, S.; Hieber, K.; Romanyukha, L.; Gambles, K.; Toles, R.; Kao, T.C.; Hauer-Jensen, M.; et al. γ-Tocotrienol Protects Hematopoietic Stem and Progenitor Cells in Mice After Total-Body Irradiation. Radiat. Res. 2010, 173, 738–747. [Google Scholar] [CrossRef]
- Peh, H.Y.; Tan, W.S.D.; Chan, T.; Pow, C.W.; Foster, P.S.; Wong, W.S.F. Vitamin E Isoform γ-Tocotrienol Protects Against Emphysema in Cigarette Smoke-Induced COPD. Free Rad. Biol. Med. 2017, 110, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Makpol, S.; Abdul Rahim, N.; Hui, C.K.; Ngah, W.Z. Inhibition of Mitochondrial Cytochrome c Release and Suppression of Caspases by γ-Tocotrienol Prevent Apoptosis and Delay Aging in Stress-Induced Premature Senescence of Skin Fibroblasts. Oxid. Med. Cell Longev. 2012, 785743, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Elson, C.E. Suppression of Mevalonate Pathway Activities by Dietary Isoprenoids: Protective Roles in Cancer and Cardiovascular Disease. J. Nutr. 1995, 125, 1666S–1672S. [Google Scholar]
- Elson, C.E.; Qureshi, A.A. Coupling the Cholesterol- and Tumor-Suppressive Actions of Palm Oil to the Impact of its Minor Constituents on 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Activity. Prostaglandins Leukot. Essent. Fatty Acids 1995, 52, 205–207. [Google Scholar] [CrossRef]
- Khor, H.T.; Chieng, D.Y.; Ong, K.K. Tocotrienols Inhibit Liver HMG CoA Reductase Activity in the Guinea. Pig. Nutr. Res. 1995, 15, 537–544. [Google Scholar] [CrossRef]
- Song, B.; DeBose-Boyd, R.A. Insig-Dependent Ubiquitination and Degradation of 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Stimulated by δ- and γ-Tocotrienols. J. Biol. Chem. 2006, 281, 25054–25061. [Google Scholar] [CrossRef] [Green Version]
- Berbee, M.; Fu, Q.; Boerma, M.; Wang, J.; Kumar, K.S.; Hauer-Jensen, M. γ-Tocotrienol Ameliorates Intestinal Radiation Injury and Reduces Vascular Oxidative Stress after Total-Body Irradiation by an HMG-CoA Reductase-Dependent Mechanism. Radiat. Res. 2009, 171, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Suman, S.; Datta, K.; Chakraborty, K.; Kulkarni, S.S.; Doiron, K.; Fornace, A.J.; Sree Kumar, K.; Hauer-Jensen, M.; Ghosh, S.P. Gamma Tocotrienol, a Potent Radioprotector, Preferentially Upregulates Expression of Anti-Apoptotic Genes to Promote Intestinal Cell Survival. Food Chem. Toxicol. 2013, 60, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.R.; Siddiqui, S.; Parveen, K.; Javed, S.; Diwakar, S.; Siddiqui, W.A. Nephroprotective Action of Tocotrienol-Rich Fraction (TRF) from Palm Oil Against Potassium Dichromate (K2Cr2O7)-Induced Acute Renal Injury in Rats. Chem. Biol. Interact. 2010, 186, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Nowak, G.; Bakajsova, D.; Hayes, C.; Hauer-Jensen, M.; Compadre, C.M. γ-Tocotrienol Protects against Mitochondrial Dysfunction and Renal Cell Death. J. Pharmacol. Exp. Ther. 2012, 340, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Kribben, A.; Edelstein, C.L.; Schrier, R.W. Pathophysiology of Acute Renal Failure. J. Nephrol. 1999, 12 (Suppl. S2), S142–S151. [Google Scholar]
- Lieberthal, W.; Nigam, S.K. Acute Renal Failure. I. Relative Importance of Proximal Vs. Distal Tubular Injury. Am. J. Physiol. Renal Physiol. 1998, 275, F623–F632. [Google Scholar] [CrossRef]
- Hall, A.M.; Unwin, R.J. The Not so ‘Mighty Chondrion’: Emergence of Renal Diseases due to Mitochondrial Dysfunction. Nephron Physiol 2007, 105, 1–10. [Google Scholar] [CrossRef]
- Shah, S.J.; Sylvester, P.W. Tocotrienol-Induced Cytotoxicity is Unrelated to Mitochondrial Stress Apoptotic Signaling in Neoplastic Mammary Epithelial Cells. Biochem. Cell Biol. 2005, 83, 86–95. [Google Scholar] [CrossRef]
- Aggarwal, V.; Kashyap, D.; Sak, K.; Tuli, H.S.; Jain, A.; Chaudhary, A.; Garg, V.K.; Sethi, G.; Yerer, M.B. Molecular Mechanisms of Action of Tocotrienols in Cancer: Recent Trends and Advancements. Int. J. Mol. Sci. 2019, 20, 656. [Google Scholar] [CrossRef] [Green Version]
- Hagl, S.; Kocher, A.; Schiborr, C.; Eckert, S.H.; Ciobanu, I.; Birringer, M.; El-Askary, H.; Helal, A.; Khayyal, M.T.; Frank, J.; et al. Rice Bran Extract Protects from Mitochondrial Dysfunction in Guinea Pig Brains. Pharmacol. Res. 2013, 76, 17–27. [Google Scholar] [CrossRef]
- Schloesser, A.; Esatbeyoglu, T.; Piegholdt, S.; Dose, J.; Ikuta, N.; Okamoto, H.; Ishida, Y.; Terao, K.; Matsugo, S.; Rimbach, G. Dietary Tocotrienol/γ-Cyclodextrin Complex Increases Mitochondrial Membrane Potential and ATP Concentrations in the Brains of Aged Mice. Oxid. Med. Cell Longev. 2015, 2015, 789710. [Google Scholar]
- Leshinsky-Silver, E.; Lev, D.; Tzofi-Berman, Z.; Cohen, S.; Saada, A.; Yanoov-Sharav, M.; Gilad, E.; Lerman-Sagie, T. Fulminant Neurological Deterioration in a Neonate with Leigh Syndrome due to a Maternally Transmitted Missense Mutation in the Mitochondrial ND3 Gene. Biochem. Biophys. Res. Commun. 2005, 334, 582–587. [Google Scholar] [CrossRef]
- Andrews, B.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Assembly Factors for the Membrane Arm of Human Complex I. Proc. Natl. Acad. Sci. USA 2013, 110, 18934–18939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Bosch, B.J.C.; Gerards, M.; Sluiter, W.; Stegmann, A.P.A.; Jongen, E.L.C.; Hellebrekers, D.M.E.I.; Oegema, R.; Lambrichs, E.H.; Prokisch, H.; Danhauser, K.; et al. Defective NDUFA9 as a Novel Cause of Neonatally Fatal Complex I Disease. J. Med. Genet. 2011, 49, 10–15. [Google Scholar] [CrossRef]
- Loublier, S.; Bayot, A.; Rak, M.; El-Khoury, R.; Bénit, P.; Rustin, P. The NDUFB6 Subunit of the Mitochondrial Respiratory Chain Complex I is required for Electron Transfer Activity: A Proof of Principle Study on Stable and Controlled RNA Interference in Human Cell Lines. Biochem. Biophys. Res. Commun. 2011, 414, 367–372. [Google Scholar] [CrossRef]
- Wirth, C.; Brandt, U.; Hunte, C.; Zickermann, V. Structure and Function of Mitochondrial Complex I. Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1857, 902–914. [Google Scholar] [CrossRef]
- Shan, W.; Li, J.; Xu, W.; Li, H.; Zuo, Z. Critical Role of UQCRC1 in Embryo Survival, Brain Ischemic Tolerance and Normal Cognition in Mice. Cell. Mol. Life Sci. 2019, 76, 1381–1396. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Zhang, W.; Liu, Y. CYC1, SDHA, UQCRC1, UQCRQ, and SDHB might be Important Biomarkers in Kidney Transplant Rejection. Clin. Chim. Acta 2020, 507, 132–138. [Google Scholar] [CrossRef]
- Zeng, J.; Tao, J.; Xia, L.; Zeng, Z.; Chen, J.; Wang, Z.; Meng, J.; Liu, L. Melatonin Inhibits Vascular Endothelial Cell Pyroptosis by Improving Mitochondrial Function Via Up-Regulation and Demethylation of UQCRC1. Biochem. Cell Biol. 2021, 99, 339–347. [Google Scholar] [CrossRef]
- Yi, T.; Wu, X.; Li, H. Ubiquinol-Cytochrome c Reductase Core Protein 1 Overexpression Protects H9c2 Cardiac Cells Against Mimic ischemia/reperfusion Injury through PI3K/Akt/GSK-3β Pathway. Biochem. Biophys. Res. Commun. 2020, 529, 904–909. [Google Scholar] [CrossRef]
- Gaignard, P.; Eyer, D.; Lebigot, E.; Oliveira, C.; Therond, P.; Boutron, A.; Slama, A. UQCRC2 Mutation in a Patient with Mitochondrial Complex III Deficiency Causing Recurrent Liver Failure, Lactic Acidosis and Hypoglycemia. J. Hum. Genet. 2017, 62, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial Complex III Rieske Fe-S Protein Processing and Assembly. Cell Cycle 2018, 17, 681–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, V.; Losonczy, G.; Heemann, U.; Vannay, Á.; Fekete, A.; Reusz, G.; Tulassay, T.; Szabó, A.J. Sexual Dimorphism in Renal Ischemia-Reperfusion Injury in Rats: Possible Role of Endothelin. Kidney Int. 2002, 62, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- Park, K.M.; Kim, J.I.; Ahn, Y.; Bonventre, A.J.; Bonventre, J.V. Testosterone is Responsible for Enhanced Susceptibility of Males to Ischemic Renal Injury. J. Biol. Chem. 2004, 279, 52282–52292. [Google Scholar] [CrossRef] [Green Version]
- Megyesi, J.; Andrade, L.; Vieira, J.M.J.; Safirstein, R.L.; Price, P.M. Positive Effect of the Induction of p21WAF1/CIP1 on the Course of Ischemic Acute Renal Failure. Kidney Int. 2001, 60, 2164–2172. [Google Scholar] [CrossRef] [Green Version]
- Nowak, G.; Takacsova-Bakajsova, D.; Megyesi, J. Deletion of Protein Kinase C-ε Attenuates Mitochondrial Dysfunction and Ameliorates Ischemic Renal Injury. Am. J. Physiol. Renal Physiol. 2017, 312, F109–F120. [Google Scholar] [CrossRef] [PubMed]
- Nowak, G.; Megyesi, J. Protein Kinase Cα Mediates Recovery of Renal and Mitochondrial Functions Following Acute Injury. FEBS J. 2020, 287, 1830–1849. [Google Scholar] [CrossRef]
- Nowak, G.; Megyesi, J.; Craigen, W.J. Deletion of VDAC1 Hinders Recovery of Mitochondrial and Renal Functions after Acute Kidney Injury. Biomolecules 2020, 10, 585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaik, Z.P.; Fifer, E.K.; Nowak, G. Akt Activation Improves Oxidative Phosphorylation in Renal Proximal Tubular Cells Following Nephrotoxicant Injury. Am. J. Physiol. Renal Physiol. 2008, 294, F423–F432. [Google Scholar] [CrossRef] [Green Version]
- Law, R.H.; Manon, S.; Devenish, R.J.; Nagley, P. ATP Synthase from Saccharomyces Cerevisiae. Methods Enzymol. 1995, 260, 133–163. [Google Scholar]
- Nowak, G.; Bakajsova, D. Protein Kinase C-α Interaction with F0F1-ATPase Promotes F0F1-ATPase Activity and Reduces Energy Deficits in Injured Renal Cells. J. Biol. Chem. 2015, 290, 7054–7066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, G. Protein Kinase C-α and ERK1/2 Mediate Mitochondrial Dysfunction, Decreases in Active Na± Transport, and Cisplatin-Induced Apoptosis in Renal Cells. J. Biol. Chem. 2002, 277, 43377–43388. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tubular Necrosis | Brush Border Loss | Cast Formation | Tubular Dilation | Inflammatory Cells | Interstitial Edema | Degeneration | Regeneration | RBC Extravasation | |
|---|---|---|---|---|---|---|---|---|---|
| Sham | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.5 ± 0.3 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 1.0 ± 0.0 |
| Vehicle + Ischemia/Reperfusion | |||||||||
| Day 1 | 3.7 ± 0.2 | 2.9 ± 0.5 | 3.2 ± 0.5 | 2.5 ± 0.4 | 2.5 ± 0.3 | 0.0 ± 0.0 | 1.7 ± 0.3 | 0.0 ± 0.0 | 2.3 ± 0.7 |
| Day 3 | 2.9 ± 0.7 | 2.3 ± 0.7 | 3.3 ± 0.8 | 2.4 ± 0.9 | 0.9 ± 0.7 | 1.0 ± 0.4 | 1.0 ± 0.6 | 1.8 ± 0.8 | 3.3 ± 0.5 |
| Day 7 | 3.3 ± 0.2 | 2.0 ± 0.6 | 3.3 ± 0.3 | 2.4 ± 0.9 | 0.9 ± 0.7 | 1.7 ± 0.7 | 1.0 ± 0.6 | 2.3 ± 0.2 | 2.3 ± 0.9 |
| γ-Tocotrienol + Ischemia/Reperfusion | |||||||||
| Day 1 | 2.6 ± 0.3 * | 2.2 ± 0.4 | 3.0 ± 0.4 | 2.2 ± 0.2 | 1.0 ± 0.0 * | 0.4 ± 0.2 * | 0.8 ± 0.6 | 2.2 ± 0.6 * | 1.9 ± 0.6 |
| Day 3 | 1.9 ± 0.2 | 1.1 ± 0.1 | 2.6 ± 0.2 | 2.1 ± 0.1 | 1.0 ± 0.0 | 0.5 ± 0.3 | 0.3 ± 0.1 | 3.1 ± 0.5 | 1.5 ± 0.2 * |
| Day 7 | 1.2 ± 0.2 * | 1.3 ± 0.5 | 1.6 ± 0.5 * | 1.0 ± 0.4 * | 1.4 ± 0.5 | 0.8 ± 0.3 | 0.4 ± 0.3 | 3.8 ± 0.3 * | 0.6 ± 0.5 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nowak, G.; Megyesi, J. γ-Tocotrienol Protects against Mitochondrial Dysfunction, Energy Deficits, Morphological Damage, and Decreases in Renal Functions after Renal Ischemia. Int. J. Mol. Sci. 2021, 22, 12674. https://doi.org/10.3390/ijms222312674
Nowak G, Megyesi J. γ-Tocotrienol Protects against Mitochondrial Dysfunction, Energy Deficits, Morphological Damage, and Decreases in Renal Functions after Renal Ischemia. International Journal of Molecular Sciences. 2021; 22(23):12674. https://doi.org/10.3390/ijms222312674
Chicago/Turabian StyleNowak, Grazyna, and Judit Megyesi. 2021. "γ-Tocotrienol Protects against Mitochondrial Dysfunction, Energy Deficits, Morphological Damage, and Decreases in Renal Functions after Renal Ischemia" International Journal of Molecular Sciences 22, no. 23: 12674. https://doi.org/10.3390/ijms222312674
APA StyleNowak, G., & Megyesi, J. (2021). γ-Tocotrienol Protects against Mitochondrial Dysfunction, Energy Deficits, Morphological Damage, and Decreases in Renal Functions after Renal Ischemia. International Journal of Molecular Sciences, 22(23), 12674. https://doi.org/10.3390/ijms222312674