
Role of Endothelial Glucocorticoid Receptor in the Pathogenesis of Kidney Diseases

 , and
, and
Abstract
1. Introduction
2. Role of Endothelial Glucocorticoid Receptor in the Pathogenesis of Arterial Hypertension and Vascular Diseases
3. Role of Endothelial Glucocorticoid Receptor in the Pathogenesis of Glomerulopathies
4. Role of Endothelial Glucocorticoid Receptor in the Pathogenesis of Diabetic Kidney Disease
5. Role of Endothelial Glucocorticoid Receptor in the Pathogenesis of Chronic Kidney Disease
6. Role of Endothelial Glucocorticoid Receptor in Pharmacotherapy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ANCA | Anti-neutrophil cytoplasmic antibody |
| AP-1 | Activator protein 1 |
| AT1 | Angiotensin II receptor type 1 |
| AT-II | Angiotensin II |
| BAG-1 | BAG family molecular chaperone regulator 1 |
| bcl-2 | B-cell lymphoma 2 |
| BP | Blood pressure |
| CCL | CC chemokine ligand |
| COX-1 | Cyclooxygenase 1 |
| Dickkopf-3 | Dickkopf-related protein 3 |
| DUSP1 | Dual specificity protein phosphatase 1 |
| eNOS | Endothelial nitric oxide synthase |
| EC | Endothelial cell |
| EndMT | Endothelial to mesenchymal transition |
| ET-1 | Endothelin 1 |
| FKBP5 | FK506 binding protein 5 |
| FAO | Fatty acid oxidation |
| FPR1 | Formyl peptide receptor 1 |
| GATA | GATA transcription factor |
| GBM | Glomerular base membranę |
| GC | Glucocorticoids |
| GFR | Glomerular filtration rate |
| GR | Glucocorticoid receptor |
| GTP | Guanosine-5’-triphosphate |
| HDAC | Histone deacetylase |
| HIF1α | Hypoxia-inducible factor 1-alpha |
| Hsp | Heat shock protein |
| HUVEC | Human umbilical vein endothelial cell |
| ICAM-1 | Intracellular adhesion molecule 1 |
| IL | Interleukin |
| IL1R2 | Interleukin 1 receptor, type II |
| iNOS | Inducible nitric oxide synthase |
| LDL | Low-density lipoprotein |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MPK-1 | MAPK phosphatase-1 |
| MR | Mineralocorticoid receptor |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| NCC | Sodium chloride cotransporter |
| NLRP | Nuclear binding oligomerization domain |
| NF-κB | Nuclear factor kappa B |
| NLPR3 | NLR family pyrin domain containing 3 |
| NO | Nitric oxide |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PDGFβ | Platelet-derived growth factor beta |
| PPARα | Peroxisome proliferator-activated receptor alpha |
| ROS | Reactive oxygen species |
| SAP30 | Sin3A-associated protein |
| SDF-1 | Stromal cell-derived factor 1 |
| siRNA | Small interfering RNA |
| Snail1 | Zinc finger protein SNAI1 |
| STAT3 | Signal transducer and activator of transcription 3 |
| sVEGFR | Soluble vascular endothelial growth factor receptor |
| TLR | Toll-like receptor |
| TNF-α | Tumour necrosis factor alpha |
| TNFSF10 | Tumour necrosis factor ligand superfamily member 10 |
| V2R | Vasopressin receptor 2 |
| VCAM-1 | vascular cell adhesion 1 |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| Wnt | Wnt signalling pathway |
References
- Reichardt, S.D.; Amouret, A.; Muzzi, C.; Vettorazzi, S.; Tuckermann, J.P.; Lühder, F.; Reichardt, H.M. The Role of Glucocorticoids in Inflammatory Diseases. Cells 2021, 10, 2921. [Google Scholar] [CrossRef] [PubMed]
- Noureddine, L.M.; Trédan, O.; Hussein, N.; Badran, B.; Le Romancer, M.; Poulard, C. Glucocorticoid Receptor: A Multifaceted Actor in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 4446. [Google Scholar] [CrossRef]
- Ponticelli, C.; Locatelli, F. Glucocorticoids in the Treatment of Glomerular Diseases: Pitfalls and Pearls. Clin. J. Am. Soc. Nephrol. 2018, 13, 815–822. [Google Scholar] [CrossRef]
- Timmermans, S.; Souffriau, J.; Libert, C. A General Introduction to Glucocorticoid Biology. Front. Immunol. 2019, 10, 1545. [Google Scholar] [CrossRef]
- Whirledge, S.; DeFranco, D.B. Glucocorticoid Signaling in Health and Disease: Insights From Tissue-Specific GR Knockout Mice. Endocrinology 2018, 159, 46–64. [Google Scholar] [CrossRef]
- Giandalia, A.; Giuffrida, A.E.; Gembillo, G.; Cucinotta, D.; Squadrito, G.; Santoro, D.; Russo, G.T. Gender Differences in Diabetic Kidney Disease: Focus on Hormonal, Genetic and Clinical Factors. Int. J. Mol. Sci. 2021, 22, 5808. [Google Scholar] [CrossRef]
- Vodošek Hojs, N.; Bevc, S.; Ekart, R.; Piko, N.; Petreski, T.; Hojs, R. Mineralocorticoid Receptor Antagonists in Diabetic Kidney Disease. Pharmaceuticals 2021, 14, 561. [Google Scholar] [CrossRef]
- Agrawal, S.; He, J.C.; Tharaux, P.L. Nuclear receptors in podocyte biology and glomerular disease. Nat. Rev. Nephrol. 2021, 17, 185–204. [Google Scholar] [CrossRef]
- Srivastava, S.P.; Zhou, H.; Setia, O.; Dardik, A.; Fernandez-Hernando, C.; Goodwin, J. Podocyte Glucocorticoid Receptors Are Essential for Glomerular Endothelial Cell Homeostasis in Diabetes Mellitus. J. Am. Heart Assoc. 2021, 10, e019437. [Google Scholar] [CrossRef]
- Taves, M.D.; Gomez-Sanchez, C.E.; Soma, K.K. Extra-adrenal glucocorticoids and mineralocorticoids: Evidence for local synthesis, regulation, and function. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E11–E24. [Google Scholar] [CrossRef]
- Pagotto, M.A.; Roldán, M.L.; Pagotto, R.M.; Lugano, M.C.; Pisani, G.B.; Rogic, G.; Molinas, S.M.; Trumper, L.; Pignataro, O.P.; Monasterolo, L.A. Localization and functional activity of cytochrome P450 side chain cleavage enzyme (CYP11A1) in the adult rat kidney. Mol. Cell. Endocrinol. 2011, 332, 253–260. [Google Scholar] [CrossRef]
- Gong, R.; Morris, D.J.; Brem, A.S. Variable expression of 11beta Hydroxysteroid dehydrogenase (11beta-HSD) isoforms in vascular endothelial cells. Steroids 2008, 73, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Scheschowitsch, K.; Leite, J.A.; Assreuy, J. New Insights in Glucocorticoid Receptor Signaling-More Than Just a Ligand-Binding Receptor. Front. Endocrinol. 2017, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, T.N.; Knight, J.K.; Goodwin, J.E. The Glucocorticoid Receptor in Cardiovascular Health and Disease. Cells 2019, 8, 1227. [Google Scholar] [CrossRef]
- Ji, J.Y.; Jing, H.; Diamond, S.L. Shear stress causes nuclear localization of endothelial glucocorticoid receptor and expression from the GRE promoter. Circ. Res. 2003, 92, 279–285. [Google Scholar] [CrossRef]
- Nayebosadri, A.; Ji, J.Y. Endothelial nuclear lamina is not required for glucocorticoid receptor nuclear import but does affect receptor-mediated transcription activation. Am. J. Physiol. Cell Physiol. 2013, 305, C309–C322. [Google Scholar] [CrossRef]
- Ji, J.Y.; Diamond, S.L. Exogenous nitric oxide activates the endothelial glucocorticoid receptor. Biochem. Biophys. Res. Commun. 2004, 318, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef]
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58. [Google Scholar] [CrossRef]
- Todd-Turla, K.M.; Schnermann, J.; Fejes-Tóth, G.; Naray-Fejes-Tóth, A.; Smart, A.; Killen, P.D.; Briggs, J.P. Distribution of mineralocorticoid and glucocorticoid receptor mRNA along the nephron. Am. J. Physiol. 1993, 264 Pt 2, F781–F791. [Google Scholar] [CrossRef]
- Ackermann, D.; Gresko, N.; Carrel, M.; Loffing-Cueni, D.; Habermehl, D.; Gomez-Sanchez, C.; Rossier, B.C.; Loffing, J. In vivo nuclear translocation of mineralocorticoid and glucocorticoid receptors in rat kidney: Differential effect of corticosteroids along the distal tubule. Am. J. Physiol. Ren. Physiol. 2010, 299, F1473–F1485. [Google Scholar] [CrossRef]
- Yan, K.; Kudo, A.; Hirano, H.; Watanabe, T.; Tasaka, T.; Kataoka, S.; Nakajima, N.; Nishibori, Y.; Shibata, T.; Kohsaka, T.; et al. Subcellular localization of glucocorticoid receptor protein in the human kidney glomerulus. Kidney Int. 1999, 56, 65–73. [Google Scholar] [CrossRef][Green Version]
- Fürst, R.; Schroeder, T.; Eilken, H.M.; Bubik, M.F.; Kiemer, A.K.; Zahler, S.; Vollmar, A.M. MAPK phosphatase-1 represents a novel anti-inflammatory target of glucocorticoids in the human endothelium. FASEB J. 2007, 21, 74–80. [Google Scholar] [CrossRef]
- Cruz-Topete, D.; Cidlowski, J.A. One hormone, two actions: Anti- and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation 2015, 22, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Mehta, S.; Srivastava, S.P.; Grabinska, K.; Zhang, X.; Wong, C.; Hedayat, A.; Perrotta, P.; Fernández-Hernando, C.; Sessa, W.C.; et al. Endothelial cell-glucocorticoid receptor interactions and regulation of Wnt signaling. JCI Insight 2020, 5, e131384. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, C.; Hadoke, P.W.F.; Nixon, M. Glucocorticoids: Fuelling the Fire of Atherosclerosis or Therapeutic Extinguishers? Int. J. Mol. Sci. 2021, 22, 7622. [Google Scholar] [CrossRef]
- Logie, J.J.; Ali, S.; Marshall, K.M.; Heck, M.M.; Walker, B.R.; Hadoke, P.W. Glucocorticoid-mediated inhibition of angiogenic changes in human endothelial cells is not caused by reductions in cell proliferation or migration. PLoS ONE. 2010, 5, e14476. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, J.; Hashimoto, T.; Shoji, K.; Yoneda, K.; Tsukamoto, I.; Moriue, T.; Kubota, Y.; Kosaka, H. Dexamethasone induces caveolin-1 in vascular endothelial cells: Implications for attenuated responses to VEGF. Am. J. Physiol. Cell Physiol. 2013, 304, C790–C800. [Google Scholar] [CrossRef] [PubMed]
- Ozmen, A.; Unek, G.; Kipmen-Korgun, D.; Mendilcioglu, I.; Sanhal, C.; Sakıncı, M.; Korgun, E.T. Glucocorticoid effects on angiogenesis are associated with mTOR pathway activity. Biotech. Histochem. 2016, 91, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhou, H.; Zhang, T.; Gao, X.; Tao, B.; Xing, H.; Zhuang, Z.; Dardik, A.; Kyriakides, T.R.; Goodwin, J.E. Loss of endothelial glucocorticoid receptor promotes angiogenesis via upregulation of Wnt/β-catenin pathway. Angiogenesis 2021, 24, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.E.; Geller, D.S. Glucocorticoid-induced hypertension. Pediatr. Nephrol. 2012, 27, 1059–1066. [Google Scholar] [CrossRef]
- Huesler, C.; Lauterburg, M.; Frey, B.M.; Frey, F.J. Evidence for glucocorticoid-mediated hypertension after uninephrectomy. Physiol. Rep. 2013, 1, e00101. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Baldes, A.; Berger, S.; Grahammer, F.; Warth, R.; Goldschmidt, I.; Peters, J.; Schütz, G.; Greger, R.; Bleich, M. Induction of the epithelial Na+ channel via glucocorticoids in mineralocorticoid receptor knockout mice. Pflug. Arch. 2001, 443, 297–305. [Google Scholar] [CrossRef]
- Goodwin, J.E.; Zhang, J.; Velazquez, H.; Geller, D.S. The glucocorticoid receptor in the distal nephron is not necessary for the development or maintenance of dexamethasone-induced hypertension. Biochem. Biophys. Res. Commun. 2010, 394, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Canonica, J.; Frateschi, S.; Boscardin, E.; Ebering, A.; Sergi, C.; Jäger, Y.; Peyrollaz, T.; Mérillat, A.M.; Maillard, M.; Klusonova, P.; et al. Lack of Renal Tubular Glucocorticoid Receptor Decreases the Thiazide-Sensitive Na. Front. Physiol. 2019, 10, 989. [Google Scholar] [CrossRef]
- Zhu, X.; Huang, Y.; Li, S.; Ge, N.; Li, T.; Wang, Y.; Liu, K.; Liu, C. Glucocorticoids Reverse Diluted Hyponatremia Through Inhibiting Arginine Vasopressin Pathway in Heart Failure Rats. J. Am. Heart Assoc. 2020, 9, e014950. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, L. Glucocorticoids and vascular reactivity. Curr. Vasc. Pharmacol. 2004, 2, 1–12. [Google Scholar] [CrossRef]
- Goodwin, J.E.; Zhang, J.; Gonzalez, D.; Albinsson, S.; Geller, D.S. Knockout of the vascular endothelial glucocorticoid receptor abrogates dexamethasone-induced hypertension. J. Hypertens. 2011, 29, 1347–1356. [Google Scholar] [CrossRef]
- Iuchi, T.; Akaike, M.; Mitsui, T.; Ohshima, Y.; Shintani, Y.; Azuma, H.; Matsumoto, T. Glucocorticoid excess induces superoxide production in vascular endothelial cells and elicits vascular endothelial dysfunction. Circ. Res. 2003, 92, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.E.; Feng, Y.; Velazquez, H.; Sessa, W.C. Endothelial glucocorticoid receptor is required for protection against sepsis. Proc. Natl. Acad. Sci. USA 2013, 110, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.E.; Feng, Y.; Velazquez, H.; Zhou, H.; Sessa, W.C. Loss of the endothelial glucocorticoid receptor prevents the therapeutic protection afforded by dexamethasone after LPS. PLoS ONE 2014, 9, e108126. [Google Scholar] [CrossRef] [PubMed]
- Hafezi-Moghadam, A.; Simoncini, T.; Yang, Z.; Limbourg, F.P.; Plumier, J.C.; Rebsamen, M.C.; Hsieh, C.M.; Chui, D.S.; Thomas, K.L.; Prorock, A.J.; et al. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat. Med. 2002, 8, 473–479. [Google Scholar] [CrossRef]
- Goodwin, J.E.; Zhang, X.; Rotllan, N.; Feng, Y.; Zhou, H.; Fernández-Hernando, C.; Yu, J.; Sessa, W.C. Endothelial glucocorticoid receptor suppresses atherogenesis--brief report. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 779–782. [Google Scholar] [CrossRef]
- Goodwin, J.E. Role of the glucocorticoid receptor in glomerular disease. Am. J. Physiol. Renal Physiol. 2019, 317, F133–F136. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Tian, X.; Tufro, A.; Moeckel, G.; Ishibe, S.; Goodwin, J. Loss of the podocyte glucocorticoid receptor exacerbates proteinuria after injury. Sci. Rep. 2017, 7, 9833. [Google Scholar] [CrossRef]
- Kuppe, C.; van Roeyen, C.; Leuchtle, K.; Kabgani, N.; Vogt, M.; Van Zandvoort, M.; Smeets, B.; Floege, J.; Gröne, H.J.; Moeller, M.J. Investigations of Glucocorticoid Action in GN. J. Am. Soc. Nephrol. 2017, 28, 1408–1420. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Schreiber, A.; Eulenberg-Gustavus, C.; Scheidereit, C.; Kamps, J.; Kettritz, R. Endothelial NF-κB Blockade Abrogates ANCA-Induced GN. J. Am. Soc. Nephrol. 2017, 28, 3191–3204. [Google Scholar] [CrossRef]
- Bartlett, C.S.; Jeansson, M.; Quaggin, S.E. Vascular Growth Factors and Glomerular Disease. Annu. Rev. Physiol. 2016, 78, 437–461. [Google Scholar] [CrossRef]
- Veron, D.; Reidy, K.; Marlier, A.; Bertuccio, C.; Villegas, G.; Jimenez, J.; Kashgarian, M.; Tufro, A. Induction of podocyte VEGF164 overexpression at different stages of development causes congenital nephrosis or steroid-resistant nephrotic syndrome. Am. J. Pathol. 2010, 177, 2225–2233. [Google Scholar] [CrossRef]
- Wasilewska, A.; Zoch-Zwierz, W. Glucocorticoid receptor and vascular endothelial growth factor in nephrotic syndrome. Acta Paediatr. 2006, 95, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Ballermann, B.J. Contribution of the endothelium to the glomerular permselectivity barrier in health and disease. Nephron Physiol. 2007, 106, 19–25. [Google Scholar] [CrossRef]
- Eremina, V.; Jefferson, J.A.; Kowalewska, J.; Hochster, H.; Haas, M.; Weisstuch, J.; Richardson, C.; Kopp, J.B.; Kabir, M.G.; Backx, P.H.; et al. VEGF inhibition and renal thrombotic microangiopathy. N. Engl. J. Med. 2008, 358, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Betzen, C.; Plotnicki, K.; Fathalizadeh, F.; Pappan, K.; Fleming, T.; Bielaszewska, M.; Karch, H.; Tönshoff, B.; Rafat, N. Shiga Toxin 2a-Induced Endothelial Injury in Hemolytic Uremic Syndrome: A Metabolomic Analysis. J. Infect. Dis. 2016, 213, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tan, Y.; Qu, Z.; Yu, F. Renal microvascular lesions in lupus nephritis. Ren. Fail. 2020, 42, 19–29. [Google Scholar] [CrossRef]
- Amaral, L.M.; Wallace, K.; Owens, M.; LaMarca, B. Pathophysiology and Current Clinical Management of Preeclampsia. Curr. Hypertens. Rep. 2017, 19, 61. [Google Scholar] [CrossRef]
- Messmer, U.K.; Winkel, G.; Briner, V.A.; Pfeilschifter, J. Glucocorticoids potently block tumour necrosis factor-alpha- and lipopolysaccharide-induced apoptotic cell death in bovine glomerular endothelial cells upstream of caspase 3 activation. Br. J. Pharmacol. 1999, 127, 1633–1640. [Google Scholar] [CrossRef]
- Messmer, U.K.; Winkel, G.; Briner, V.A.; Pfeilschifter, J. Suppression of apoptosis by glucocorticoids in glomerular endothelial cells: Effects on proapoptotic pathways. Br. J. Pharmacol. 2000, 129, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Newton, C.J.; Ran, G.; Xie, Y.X.; Bilko, D.; Burgoyne, C.H.; Adams, I.; Abidia, A.; McCollum, P.T.; Atkin, S.L. Statin-induced apoptosis of vascular endothelial cells is blocked by dexamethasone. J. Endocrinol. 2002, 174, 7–16. [Google Scholar] [CrossRef]
- Koenen, P.; Barczyk, K.; Wolf, M.; Roth, J.; Viemann, D. Endothelial cells present an innate resistance to glucocorticoid treatment: Implications for therapy of primary vasculitis. Ann. Rheum. Dis. 2012, 71, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Pelaia, G.; Cuda, G.; Vatrella, A.; Grembiale, R.D.; De Sarro, G.; Maselli, R.; Costanzo, F.S.; Avvedimento, V.E.; Rotiroti, D.; Marsico, S.A. Effects of glucocorticoids on activation of c-jun N-terminal, extracellular signal-regulated, and p38 MAP kinases in human pulmonary endothelial cells. Biochem. Pharmacol. 2001, 62, 1719–1724. [Google Scholar] [CrossRef]
- Xu, X.; Otsuki, M.; Saito, H.; Sumitani, S.; Yamamoto, H.; Asanuma, N.; Kouhara, H.; Kasayama, S. PPARalpha and GR differentially down-regulate the expression of nuclear factor-kappaB-responsive genes in vascular endothelial cells. Endocrinology 2001, 142, 3332–3339. [Google Scholar] [CrossRef][Green Version]
- Gembillo, G.; Siligato, R.; Amatruda, M.; Conti, G.; Santoro, D. Vitamin D and Glomerulonephritis. Medicina 2021, 57, 186. [Google Scholar] [CrossRef]
- Badal, S.S.; Danesh, F.R. New insights into molecular mechanisms of diabetic kidney disease. Am. J. Kidney Dis. 2014, 63 (Suppl. S2), S63–S83. [Google Scholar] [CrossRef]
- Santoro, D.; Torreggiani, M.; Pellicanò, V.; Cernaro, V.; Messina, R.M.; Longhitano, E.; Siligato, R.; Gembillo, G.; Esposito, C.; Piccoli, G.B. Kidney Biopsy in Type 2 Diabetic Patients: Critical Reflections on Present Indications and Diagnostic Alternatives. Int. J. Mol. Sci. 2021, 22, 5425. [Google Scholar] [CrossRef]
- Selby, N.M.; Taal, M.W. An updated overview of diabetic nephropathy: Diagnosis, prognosis, treatment goals and latest guidelines. Diabetes Obes. Metab. 2020, 22 (Suppl. S1), 3–15. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.R.; Chu, Y.M.; Wei, L.; Tu, C.; Han, Y.Y. Antiangiogenic therapy in diabetic nephropathy: A double-edged sword (Review). Mol. Med. Rep. 2021, 23, 260. [Google Scholar] [CrossRef]
- Sison, K.; Eremina, V.; Baelde, H.; Min, W.; Hirashima, M.; Fantus, I.G.; Quaggin, S.E. Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J. Am. Soc. Nephrol. 2010, 21, 1691–1701. [Google Scholar] [CrossRef]
- Majumder, S.; Advani, A. VEGF and the diabetic kidney: More than too much of a good thing. J. Diabetes Complicat. 2017, 31, 273–279. [Google Scholar] [CrossRef]
- Veron, D.; Reidy, K.J.; Bertuccio, C.; Teichman, J.; Villegas, G.; Jimenez, J.; Shen, W.; Kopp, J.B.; Thomas, D.B.; Tufro, A. Overexpression of VEGF-A in podocytes of adult mice causes glomerular disease. Kidney Int. 2010, 77, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Oltean, S.; Qiu, Y.; Ferguson, J.K.; Stevens, M.; Neal, C.; Russell, A.; Kaura, A.; Arkill, K.P.; Harris, K.; Symonds, C.; et al. Vascular Endothelial Growth Factor-A165b Is Protective and Restores Endothelial Glycocalyx in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2015, 26, 1889–1904. [Google Scholar] [CrossRef] [PubMed]
- Veron, D.; Aggarwal, P.K.; Velazquez, H.; Kashgarian, M.; Moeckel, G.; Tufro, A. Podocyte-specific VEGF-a gain of function induces nodular glomerulosclerosis in eNOS null mice. J. Am. Soc. Nephrol. 2014, 25, 1814–1824. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Wang, H.; Fan, X.; Paueksakon, P.; Harris, R.C. Improvement of endothelial nitric oxide synthase activity retards the progression of diabetic nephropathy in db/db mice. Kidney Int. 2012, 82, 1176–1183. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yuen, D.A.; Stead, B.E.; Zhang, Y.; White, K.E.; Kabir, M.G.; Thai, K.; Advani, S.L.; Connelly, K.A.; Takano, T.; Zhu, L.; et al. eNOS deficiency predisposes podocytes to injury in diabetes. J. Am. Soc. Nephrol. 2012, 23, 1810–1823. [Google Scholar] [CrossRef]
- Siddiqi, F.S.; Advani, A. Endothelial-podocyte crosstalk: The missing link between endothelial dysfunction and albuminuria in diabetes. Diabetes 2013, 62, 3647–3655. [Google Scholar] [CrossRef]
- He, W.; Dai, C.; Li, Y.; Zeng, G.; Monga, S.P.; Liu, Y. Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J. Am. Soc. Nephrol. 2009, 20, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Gruenwald, A.; Suh, J.H.; Miner, J.H.; Barisoni-Thomas, L.; Taketo, M.M.; Faul, C.; Millar, S.E.; Holzman, L.B.; Susztak, K. Wnt/β-catenin pathway in podocytes integrates cell adhesion, differentiation, and survival. J. Biol. Chem. 2011, 286, 26003–26015. [Google Scholar] [CrossRef]
- Li, L.; Chen, L.; Zang, J.; Tang, X.; Liu, Y.; Zhang, J.; Bai, L.; Yin, Q.; Lu, Y.; Cheng, J.; et al. C3a and C5a receptor antagonists ameliorate endothelial-myofibroblast transition via the Wnt/β-catenin signaling pathway in diabetic kidney disease. Metabolism 2015, 64, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.P.; Zhou, H.; Setia, O.; Liu, B.; Kanasaki, K.; Koya, D.; Dardik, A.; Fernandez-Hernando, C.; Goodwin, J. Loss of endothelial glucocorticoid receptor accelerates diabetic nephropathy. Nat. Commun. 2021, 12, 2368. [Google Scholar] [CrossRef]
- Xiong, J.; Kawagishi, H.; Yan, Y.; Liu, J.; Wells, Q.S.; Edmunds, L.R.; Fergusson, M.M.; Yu, Z.X.; Rovira, I.I.; Brittain, E.L.; et al. A Metabolic Basis for Endothelial-to-Mesenchymal Transition. Mol. Cell 2018, 69, 689–698.e7. [Google Scholar] [CrossRef]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef]
- Bábíčková, J.; Klinkhammer, B.M.; Buhl, E.M.; Djudjaj, S.; Hoss, M.; Heymann, F.; Tacke, F.; Floege, J.; Becker, J.U.; Boor, P. Regardless of etiology, progressive renal disease causes ultrastructural and functional alterations of peritubular capillaries. Kidney Int. 2017, 91, 70–85. [Google Scholar] [CrossRef]
- Lin, S.L.; Chang, F.C.; Schrimpf, C.; Chen, Y.T.; Wu, C.F.; Wu, V.C.; Chiang, W.C.; Kuhnert, F.; Kuo, C.J.; Chen, Y.M.; et al. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am. J. Pathol. 2011, 178, 911–923. [Google Scholar] [CrossRef]
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508. [Google Scholar] [CrossRef]
- Takagaki, Y.; Lee, S.M.; Dongqing, Z.; Kitada, M.; Kanasaki, K.; Koya, D. Endothelial autophagy deficiency induces IL6—dependent endothelial mesenchymal transition and organ fibrosis. Autophagy 2020, 16, 1905–1914. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.A.; Modarage, K.; Goggolidou, P. The Role of Wnt Signalling in Chronic Kidney Disease (CKD). Genes 2020, 11, 496. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Zhou, D.; Tan, R.J.; Fu, H.; Zhou, L.; Hou, F.F.; Liu, Y. Sustained Activation of Wnt/β-Catenin Signaling Drives AKI to CKD Progression. J. Am. Soc. Nephrol. 2016, 27, 1727–1740. [Google Scholar] [CrossRef]
- Zhou, D.; Tan, R.J.; Fu, H.; Liu, Y. Wnt/β-catenin signaling in kidney injury and repair: A double-edged sword. Lab. Invest. 2016, 96, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Castañeda, J.R.; Rodelo-Haad, C.; Pendon-Ruiz de Mier, M.V.; Martin-Malo, A.; Santamaria, R.; Rodriguez, M. Klotho/FGF23 and Wnt Signaling as Important Players in the Comorbidities Associated with Chronic Kidney Disease. Toxins 2020, 12, 185. [Google Scholar] [CrossRef]
- Foulquier, S.; Daskalopoulos, E.P.; Lluri, G.; Hermans, K.C.M.; Deb, A.; Blankesteijn, W.M. WNT Signaling in Cardiac and Vascular Disease. Pharmacol. Rev. 2018, 70, 68–141. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, C.; Hong, X.; Miao, J.; Liao, Y.; Hou, F.F.; Zhou, L.; Liu, Y. Wnt/β-catenin signaling mediates both heart and kidney injury in type 2 cardiorenal syndrome. Kidney Int. 2019, 95, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Lipphardt, M.; Dihazi, H.; Jeon, N.L.; Dadafarin, S.; Ratliff, B.B.; Rowe, D.W.; Müller, G.A.; Goligorsky, M.S. Dickkopf-3 in aberrant endothelial secretome triggers renal fibroblast activation and endothelial-mesenchymal transition. Nephrol. Dial. Transplant. 2019, 34, 49–62. [Google Scholar] [CrossRef]
- Kawazoe, M.; Kaneko, K.; Nanki, T. Glucocorticoid therapy suppresses Wnt signaling by reducing the ratio of serum Wnt3a to Wnt inhibitors, sFRP-1 and Wif-1. Clin. Rheumatol. 2021, 40, 2947–2954. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, K.; Patocs, A. Glucocorticoids Influencing Wnt/β-Catenin Pathway; Multiple Sites, Heterogeneous Effects. Molecules 2020, 25, 1489. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Sugiyama, S.; Yamamoto, E.; Matsuzawa, Y.; Akiyama, E.; Kusaka, H.; Fujisue, K.; Kurokawa, H.; Matsubara, J.; Sugamura, K.; et al. Endothelial function and cardiovascular events in chronic kidney disease. Int. J. Cardiol. 2014, 173, 481–486. [Google Scholar] [CrossRef]
- Wang, X.Y.; Chen, X.L.; Wang, L.; Chen, H.W. High-dose glucocorticoids increases the expression of mineralocorticoid receptor in vascular endothelial cells. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4314–4323. [Google Scholar]
- Van der Goes, M.C.; Jacobs, J.W.; Bijlsma, J.W. The value of glucocorticoid co-therapy in different rheumatic diseases—positive and adverse effects. Arthritis Res. Ther. 2014, 16 (Suppl. S2), S2. [Google Scholar] [CrossRef]
- Pujades-Rodriguez, M.; Morgan, A.W.; Cubbon, R.M.; Wu, J. Dose-dependent oral glucocorticoid cardiovascular risks in people with immune-mediated inflammatory diseases: A population-based cohort study. PLoS Med. 2020, 17, e1003432. [Google Scholar] [CrossRef]
- Crawford, A.A.; Bankier, S.; Altmaier, E.; Barnes, C.L.K.; Clark, D.W.; Ermel, R.; Friedrich, N.; van der Harst, P.; Joshi, P.K.; Karhunen, V.; et al. Variation in the SERPINA6/SERPINA1 locus alters morning plasma cortisol, hepatic corticosteroid binding globulin expression, gene expression in peripheral tissues, and risk of cardiovascular disease. J. Hum. Genet. 2021, 66, 625–636. [Google Scholar] [CrossRef]
- Zhu, D.; Rashdan, N.A.; Chapman, K.E.; Hadoke, P.W.; MacRae, V.E. A novel role for the mineralocorticoid receptor in glucocorticoid driven vascular calcification. Vascul. Pharmacol. 2016, 86, 87–93. [Google Scholar] [CrossRef]
- Moss, M.E.; Lu, Q.; Iyer, S.L.; Engelbertsen, D.; Marzolla, V.; Caprio, M.; Lichtman, A.H.; Jaffe, I.Z. Endothelial Mineralocorticoid Receptors Contribute to Vascular Inflammation in Atherosclerosis in a Sex-Specific Manner. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1588–1601. [Google Scholar] [CrossRef]
- Lv, J.; Zhang, H.; Wong, M.G.; Jardine, M.J.; Hladunewich, M.; Jha, V.; Monaghan, H.; Zhao, M.; Barbour, S.; Reich, H.; et al. Effect of Oral Methylprednisolone on Clinical Outcomes in Patients With IgA Nephropathy: The TESTING Randomized Clinical Trial. JAMA 2017, 318, 432–442. [Google Scholar] [CrossRef]
- Rauen, T.; Eitner, F.; Fitzner, C.; Sommerer, C.; Zeier, M.; Otte, B.; Panzer, U.; Peters, H.; Benck, U.; Mertens, P.R.; et al. Intensive Supportive Care plus Immunosuppression in IgA Nephropathy. N. Engl. J. Med. 2015, 373, 2225–2236. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yu, Q.; Guo, W.; Hao, Y.; Sun, W.; Cheng, L. Effect of glucocorticoids on the function of microvascular endothelial cells in the human femoral head bone. Adv. Clin. Exp. Med. 2020, 29, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Mata-Greenwood, E.; Stewart, J.M.; Steinhorn, R.H.; Pearce, W.J. Role of BCL2-associated athanogene 1 in differential sensitivity of human endothelial cells to glucocorticoids. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1046–1055. [Google Scholar] [CrossRef]
- Mata-Greenwood, E.; Jackson, P.N.; Pearce, W.J.; Zhang, L. Endothelial glucocorticoid receptor promoter methylation according to dexamethasone sensitivity. J. Mol. Endocrinol. 2015, 55, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Asgeirsdóttir, S.A.; Zwiers, P.J.; Morselt, H.W.; Moorlag, H.E.; Bakker, H.I.; Heeringa, P.; Kok, J.W.; Kallenberg, C.G.; Molema, G.; Kamps, J.A. Inhibition of proinflammatory genes in anti-GBM glomerulonephritis by targeted dexamethasone-loaded AbEsel liposomes. Am. J. Physiol. Ren. Physiol. 2008, 294, F554–F561. [Google Scholar] [CrossRef]
- Van Alem, C.M.A.; Boonstra, M.; Prins, J.; Bezhaeva, T.; van Essen, M.F.; Ruben, J.M.; Vahrmeijer, A.L.; van der Veer, E.P.; de Fijter, J.W.; Reinders, M.E.; et al. Local delivery of liposomal prednisolone leads to an anti-inflammatory profile in renal ischaemia-reperfusion injury in the rat. Nephrol. Dial. Transplant. 2018, 33, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.T.; Lv, L.L.; Wang, B.; Cao, J.Y.; Feng, Y.; Li, Z.L.; Wu, M.; Wang, F.M.; Wen, Y.; Zhou, L.T.; et al. Employing Macrophage-Derived Microvesicle for Kidney-Targeted Delivery of Dexamethasone: An Efficient Therapeutic Strategy against Renal Inflammation and Fibrosis. Theranostics 2019, 9, 4740–4755. [Google Scholar] [CrossRef]
- Bruni, R.; Possenti, P.; Bordignon, C.; Li, M.; Ordanini, S.; Messa, P.; Rastaldi, M.P.; Cellesi, F. Ultrasmall polymeric nanocarriers for drug delivery to podocytes in kidney glomerulus. J. Control. Release 2017, 255, 94–107. [Google Scholar] [CrossRef] [PubMed]
- De Bosscher, K. Selective Glucocorticoid Receptor modulators. J. Steroid Biochem. Mol. Biol. 2010, 120, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Sundahl, N.; Bridelance, J.; Libert, C.; De Bosscher, K.; Beck, I.M. Selective glucocorticoid receptor modulation: New directions with non-steroidal scaffolds. Pharmacol. Ther. 2015, 152, 28–41. [Google Scholar] [CrossRef] [PubMed]



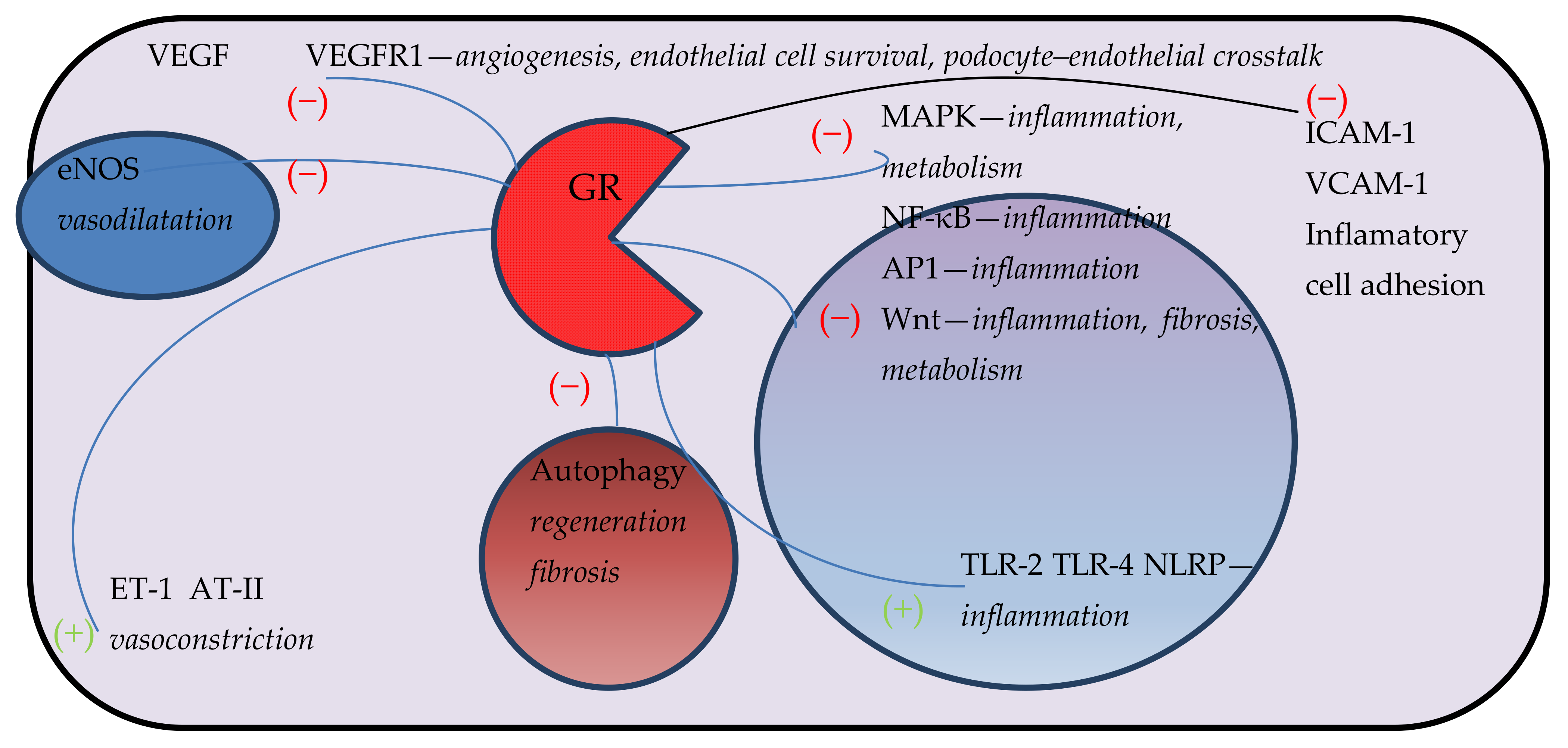{kind=link}
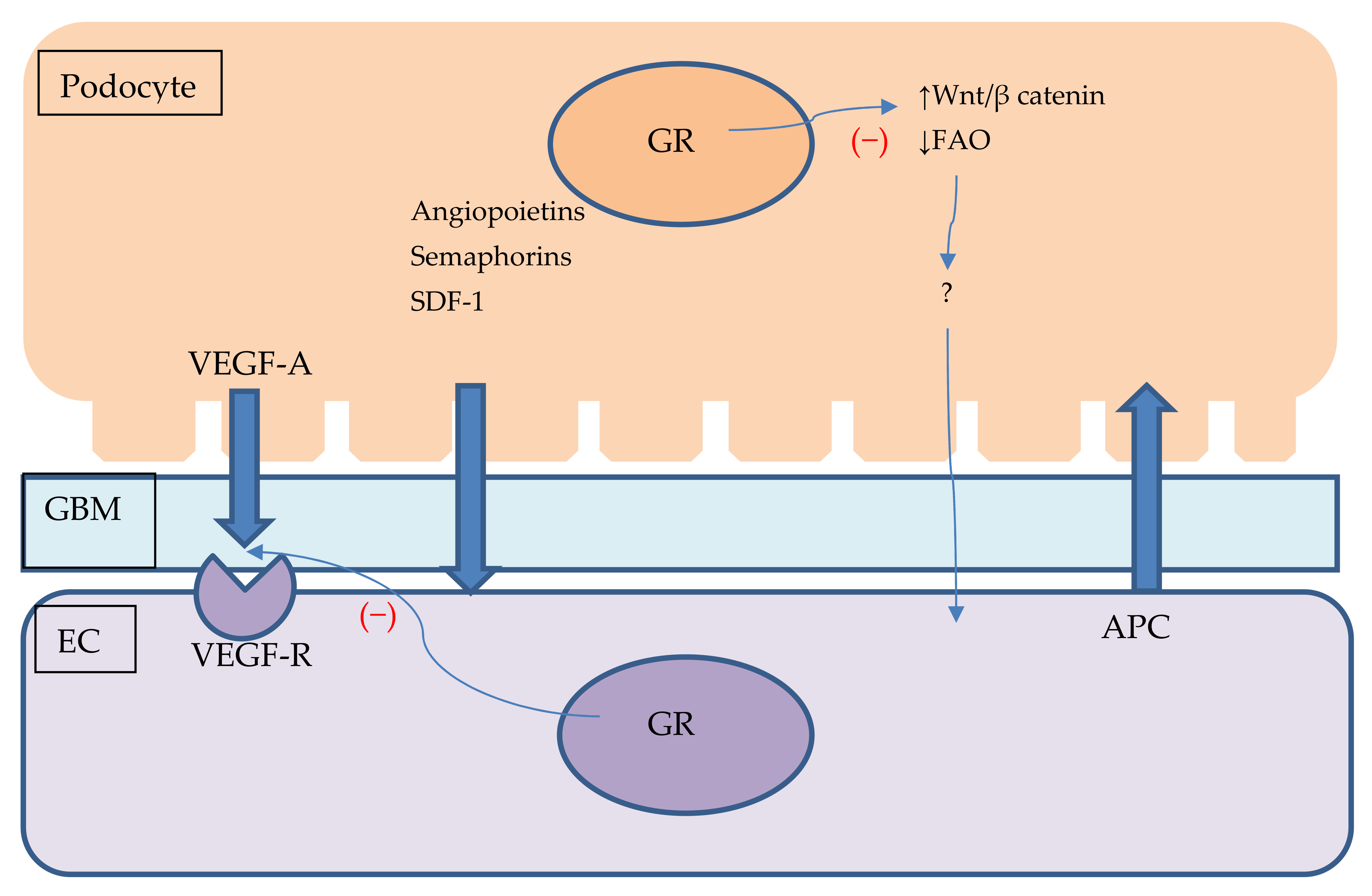{kind=link}
| Endothelial Glucocorticoid Receptor in the Pathogenesis of Particular Diseases | Pathomechanism |
|---|---|
| Hypertension and vascular diseases |
|
| Cardio-vascular diseases |
|
| Diabetic nephropathy |
|
| Chronic kidney disease |
|
| Glomerulonephritis | Pathogenesis of Selected Glomerulonephritis |
|---|---|
| Focal segmental glomerulosclerosis (FSGS) |
|
| ANCA-associated vasculitis |
|
| Minimal change disease |
|
| Lupus nephritis |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Przybyciński, J.; Drożdżal, S.; Domański, L.; Dziedziejko, V.; Pawlik, A. Role of Endothelial Glucocorticoid Receptor in the Pathogenesis of Kidney Diseases. Int. J. Mol. Sci. 2021, 22, 13295. https://doi.org/10.3390/ijms222413295
Przybyciński J, Drożdżal S, Domański L, Dziedziejko V, Pawlik A. Role of Endothelial Glucocorticoid Receptor in the Pathogenesis of Kidney Diseases. International Journal of Molecular Sciences. 2021; 22(24):13295. https://doi.org/10.3390/ijms222413295
Chicago/Turabian StylePrzybyciński, Jarosław, Sylwester Drożdżal, Leszek Domański, Violetta Dziedziejko, and Andrzej Pawlik. 2021. "Role of Endothelial Glucocorticoid Receptor in the Pathogenesis of Kidney Diseases" International Journal of Molecular Sciences 22, no. 24: 13295. https://doi.org/10.3390/ijms222413295
APA StylePrzybyciński, J., Drożdżal, S., Domański, L., Dziedziejko, V., & Pawlik, A. (2021). Role of Endothelial Glucocorticoid Receptor in the Pathogenesis of Kidney Diseases. International Journal of Molecular Sciences, 22(24), 13295. https://doi.org/10.3390/ijms222413295