
Repurposing Riociguat to Target a Novel Paracrine Nitric Oxide-TRPC6 Pathway to Prevent Podocyte Injury



Abstract
:1. Introduction
2. Results
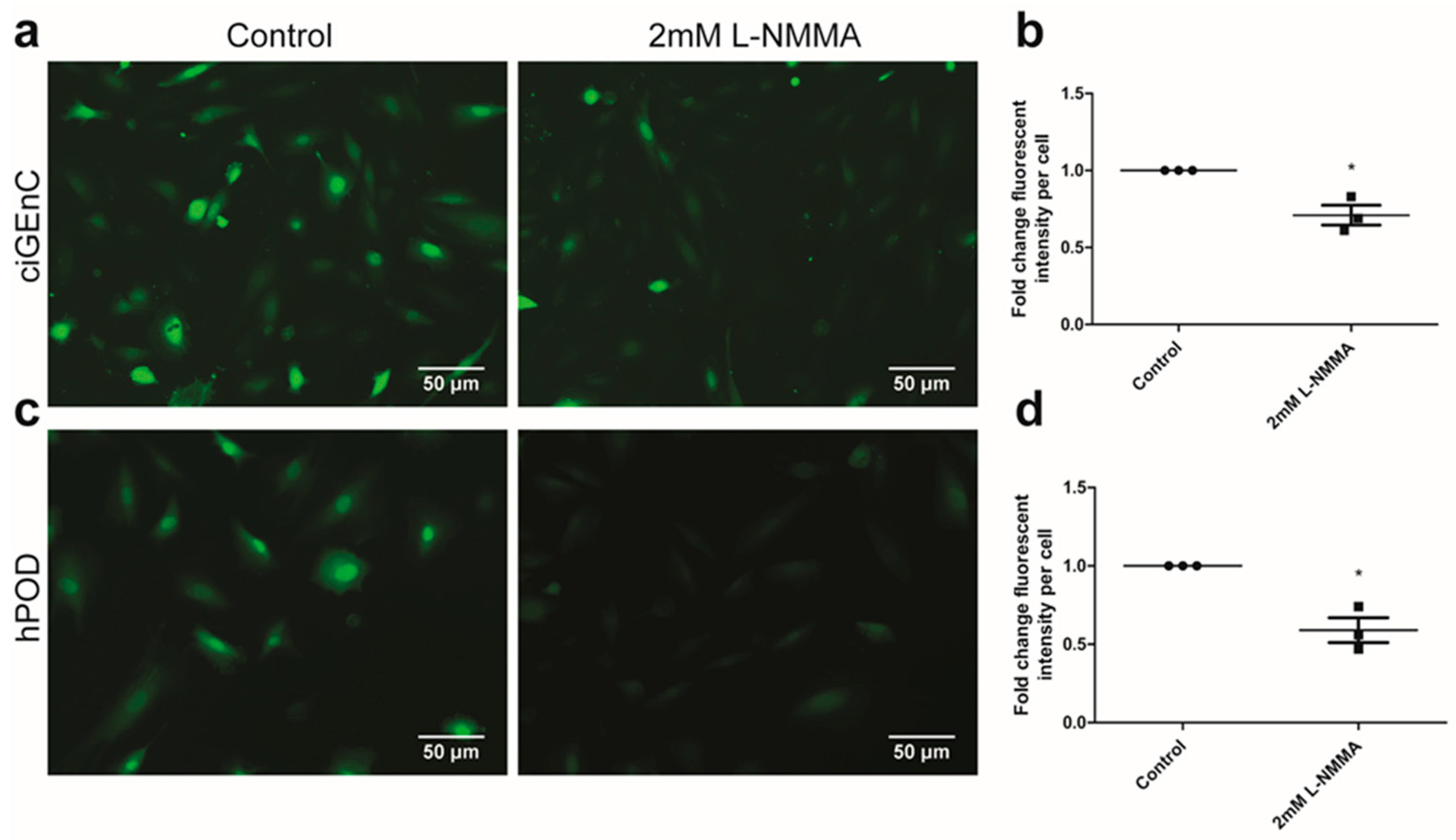
2.1. Glomerular Endothelial Cells and Podocytes Express eNOS and Produce NO
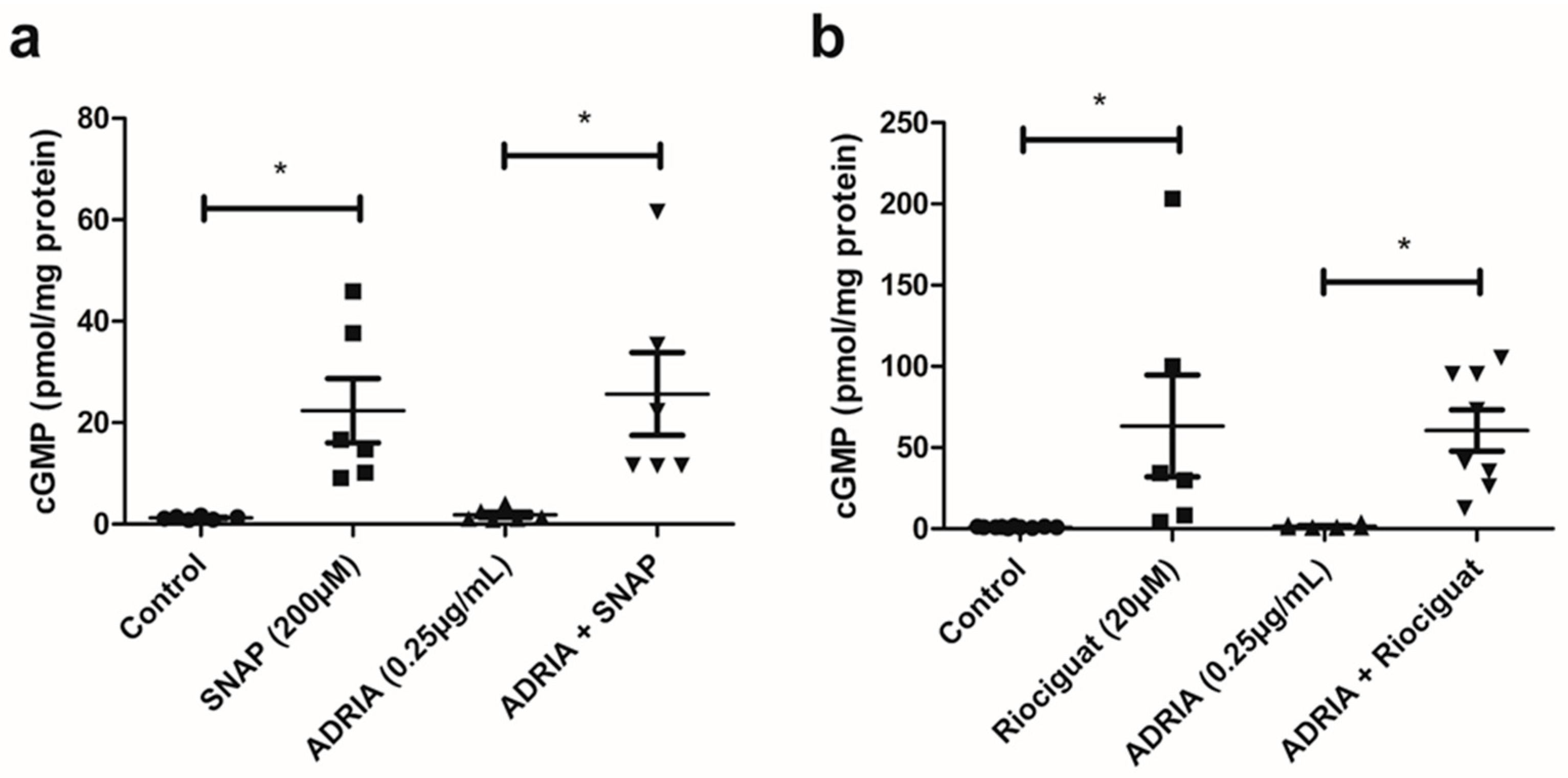
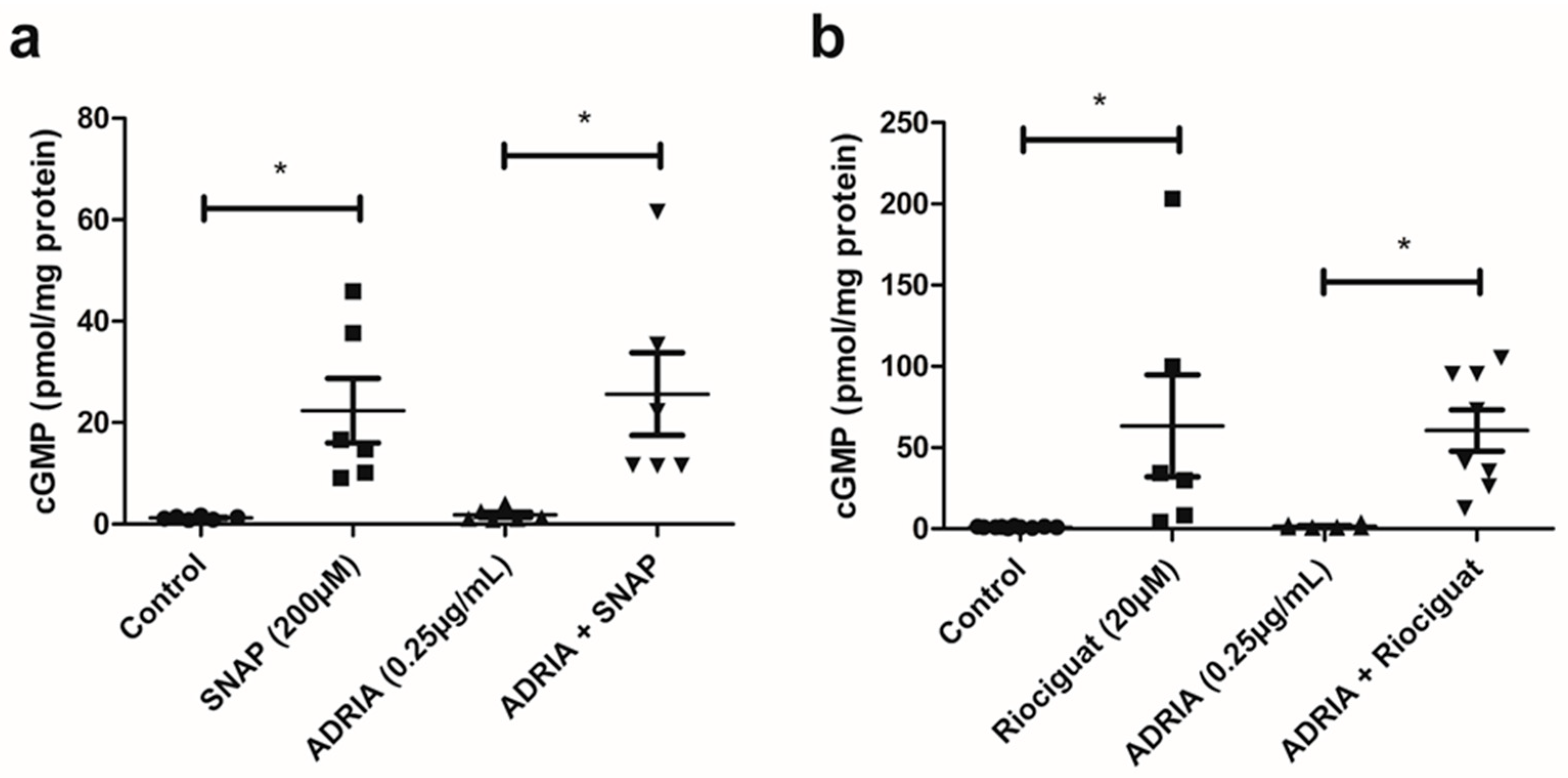
2.2. NO and Riociguat Increase cGMP Production and Downregulate TRPC6 Expression in hPOD
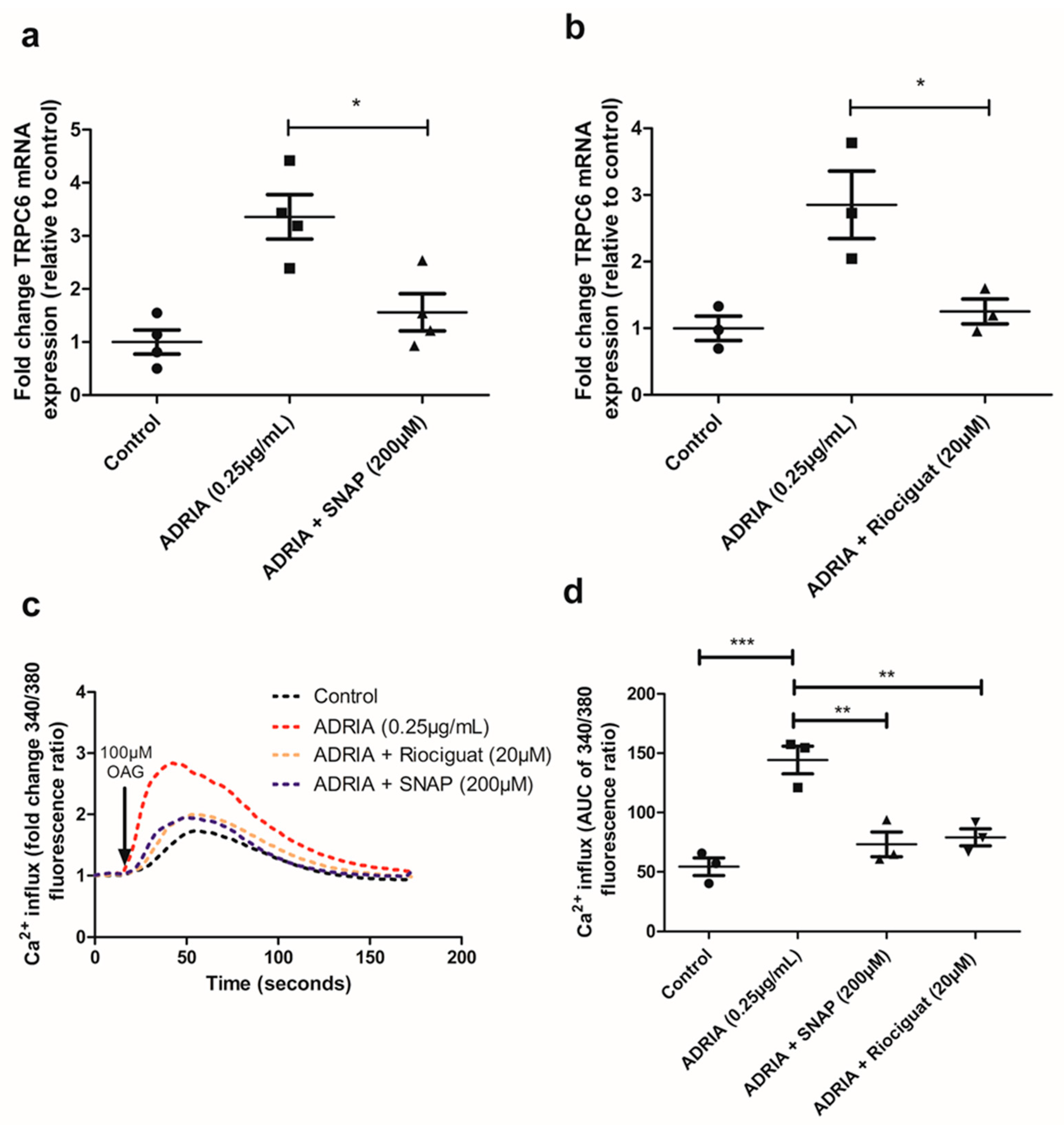
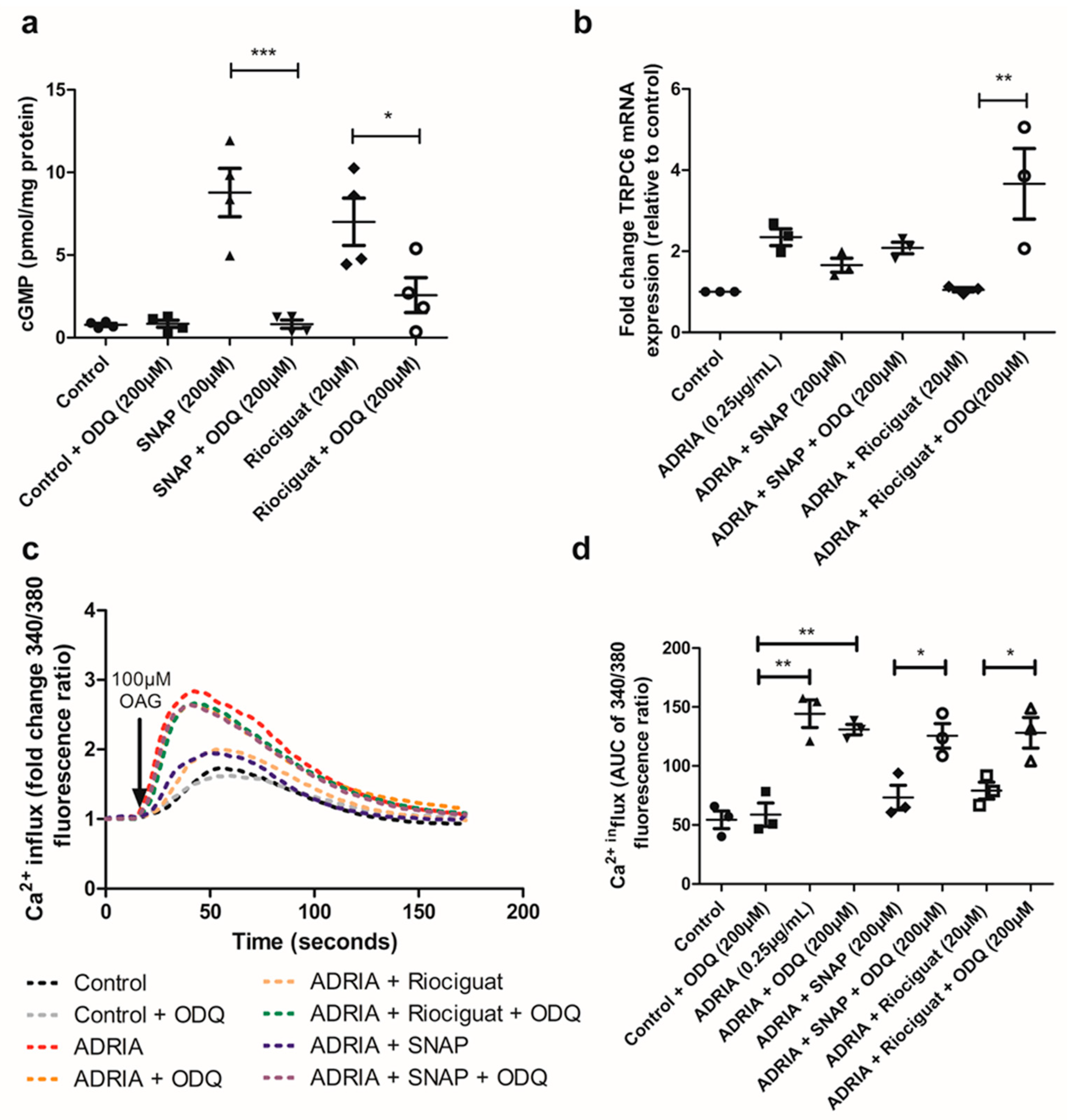
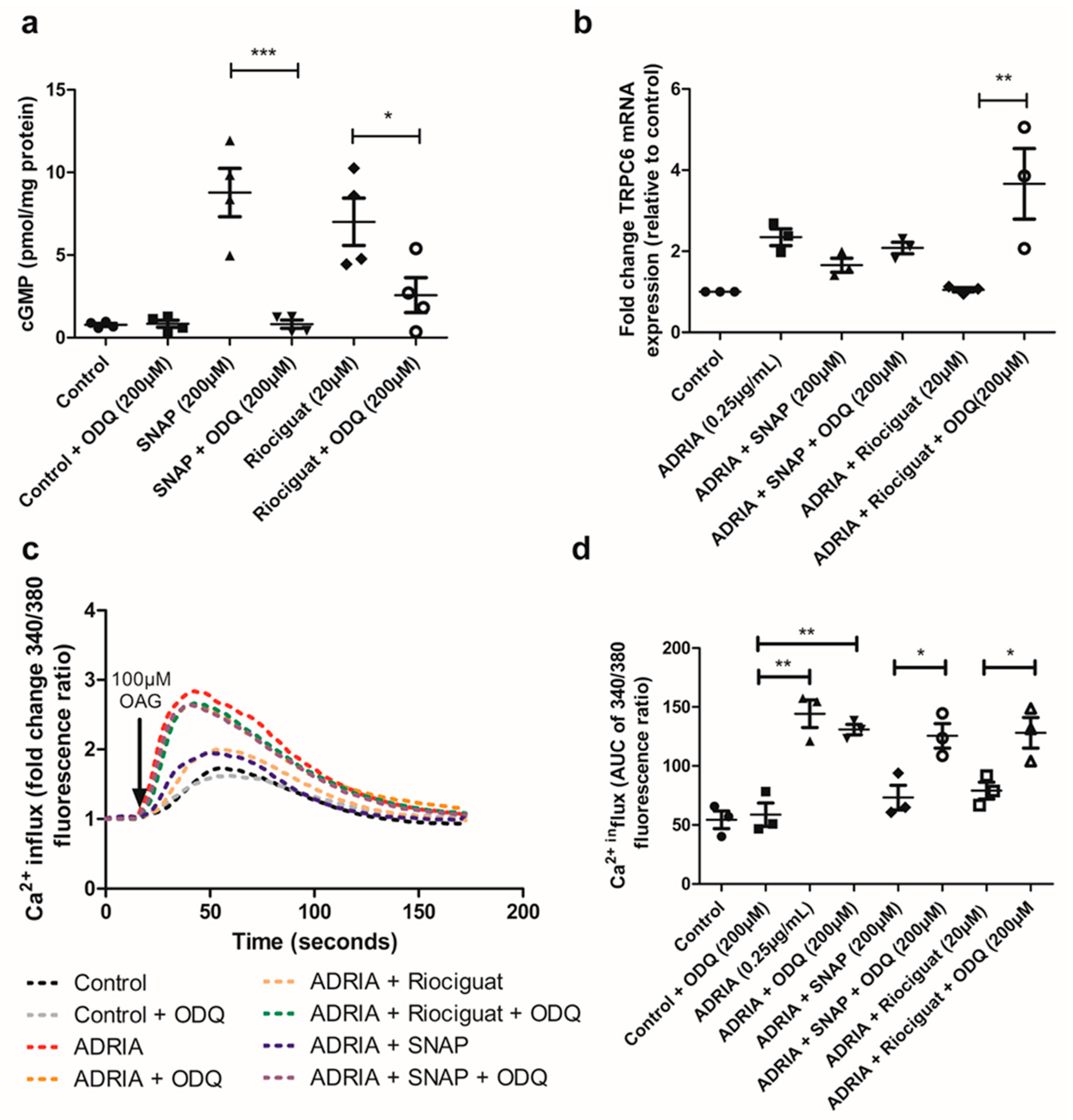
2.3. NO and Riociguat Downregulate Injury-Induced TRPC6 Expression via sGC Activation
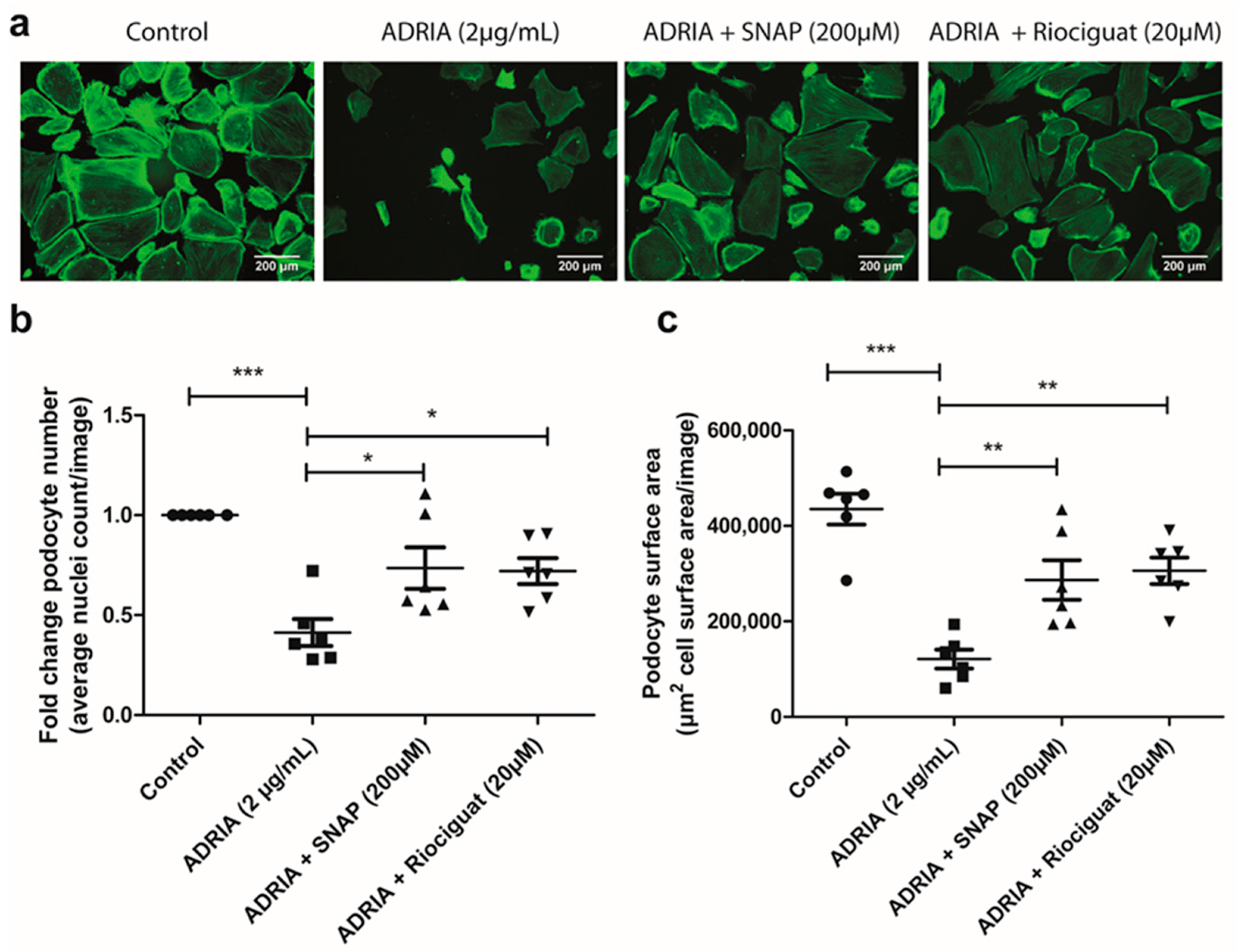
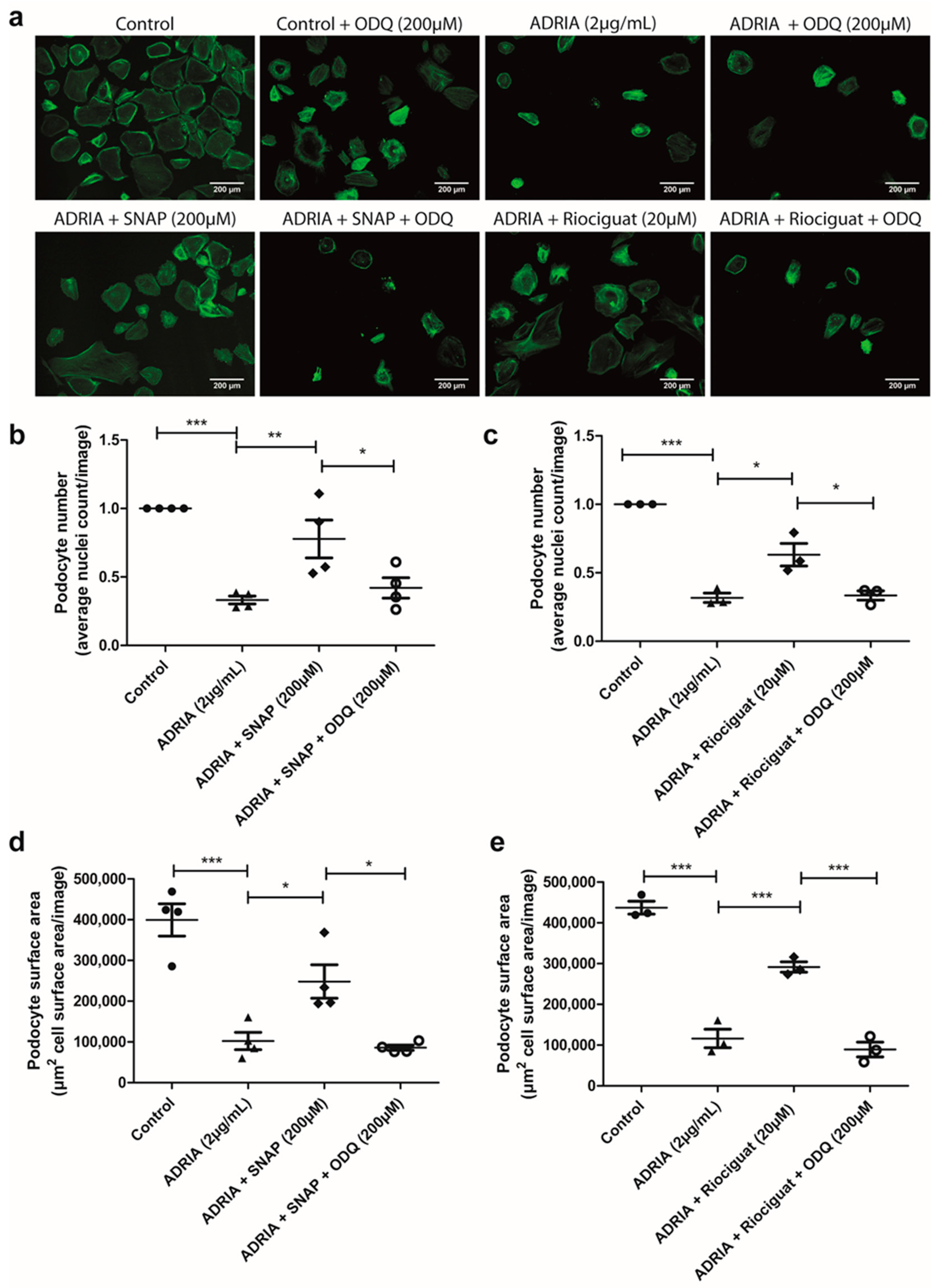
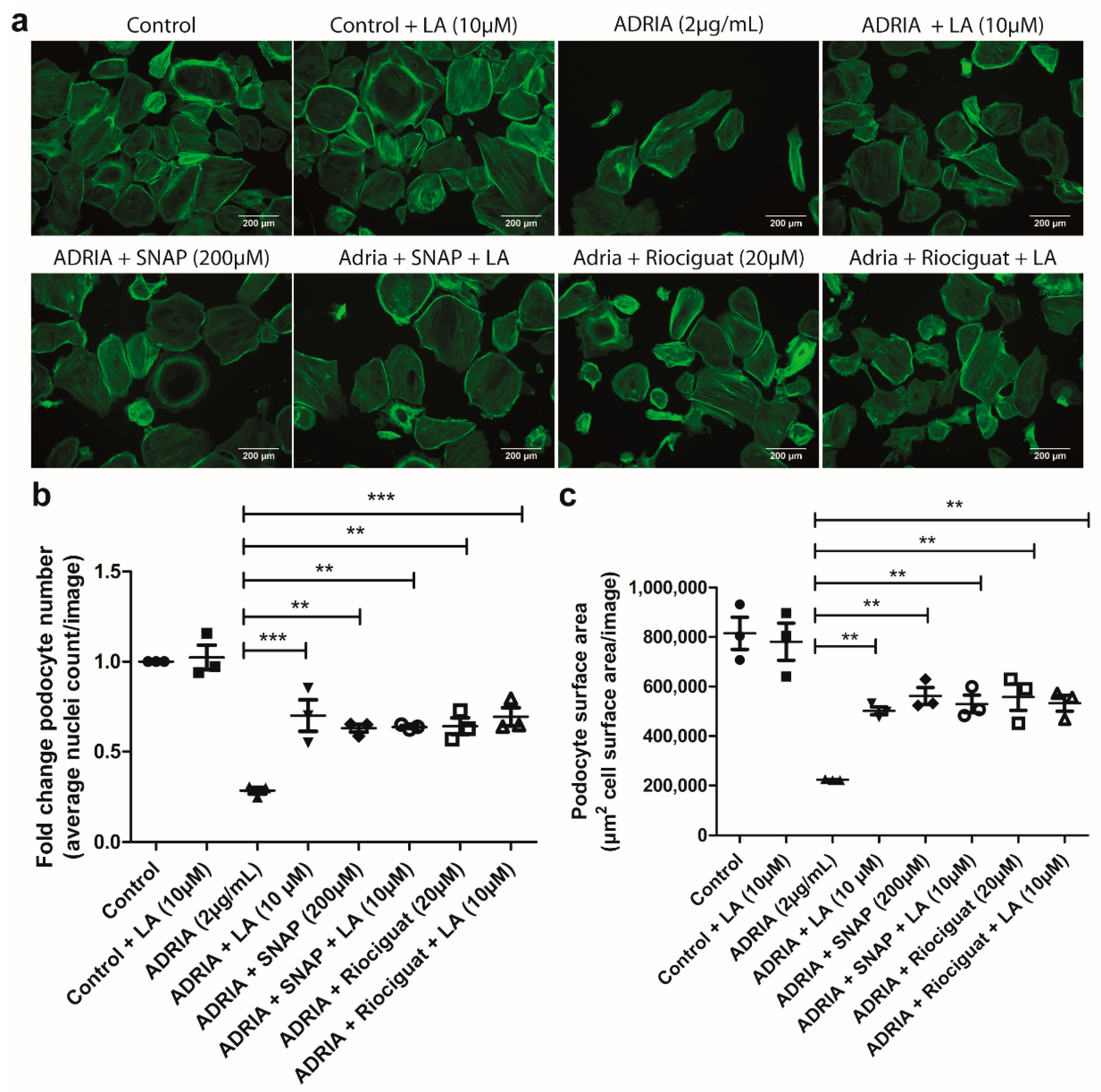
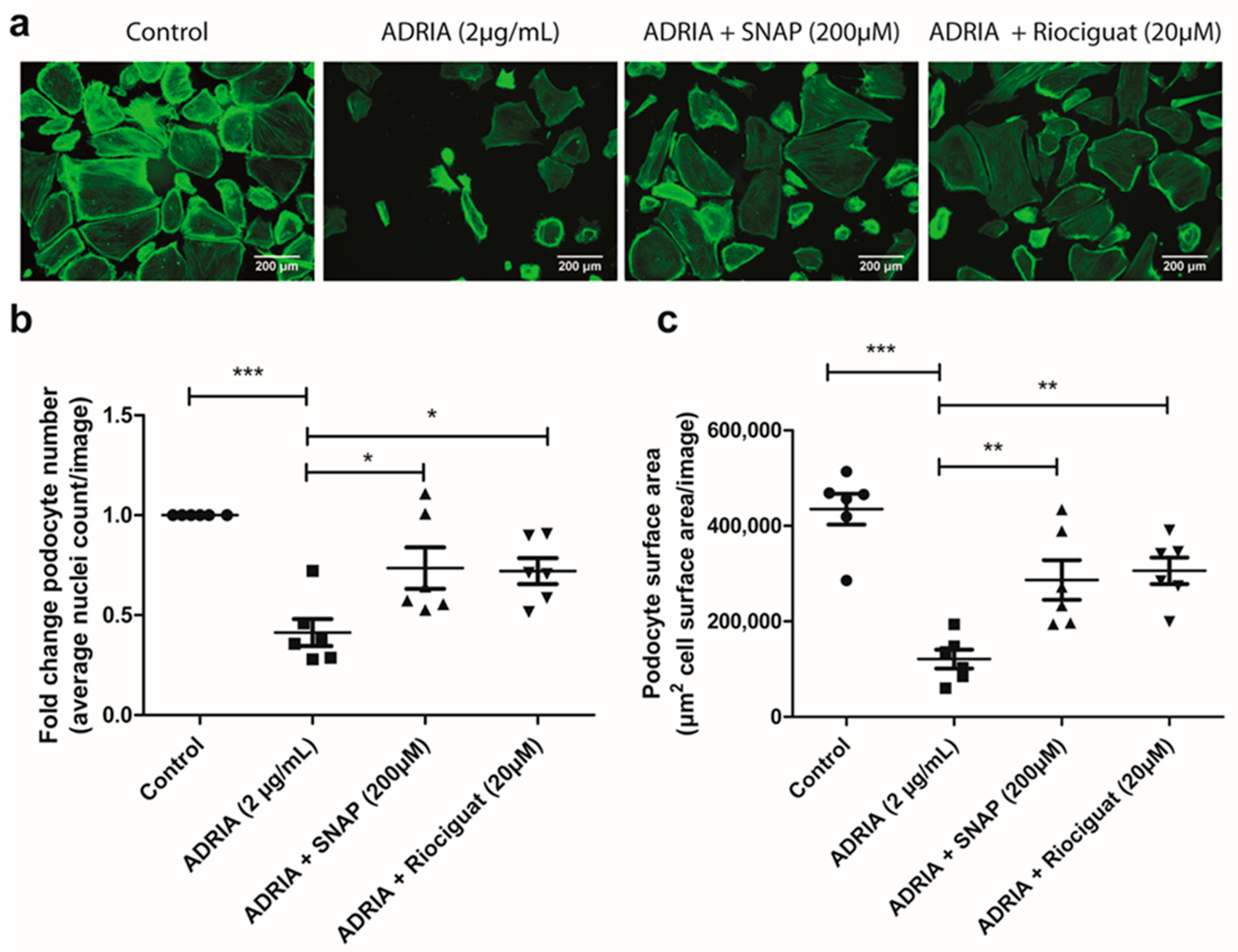
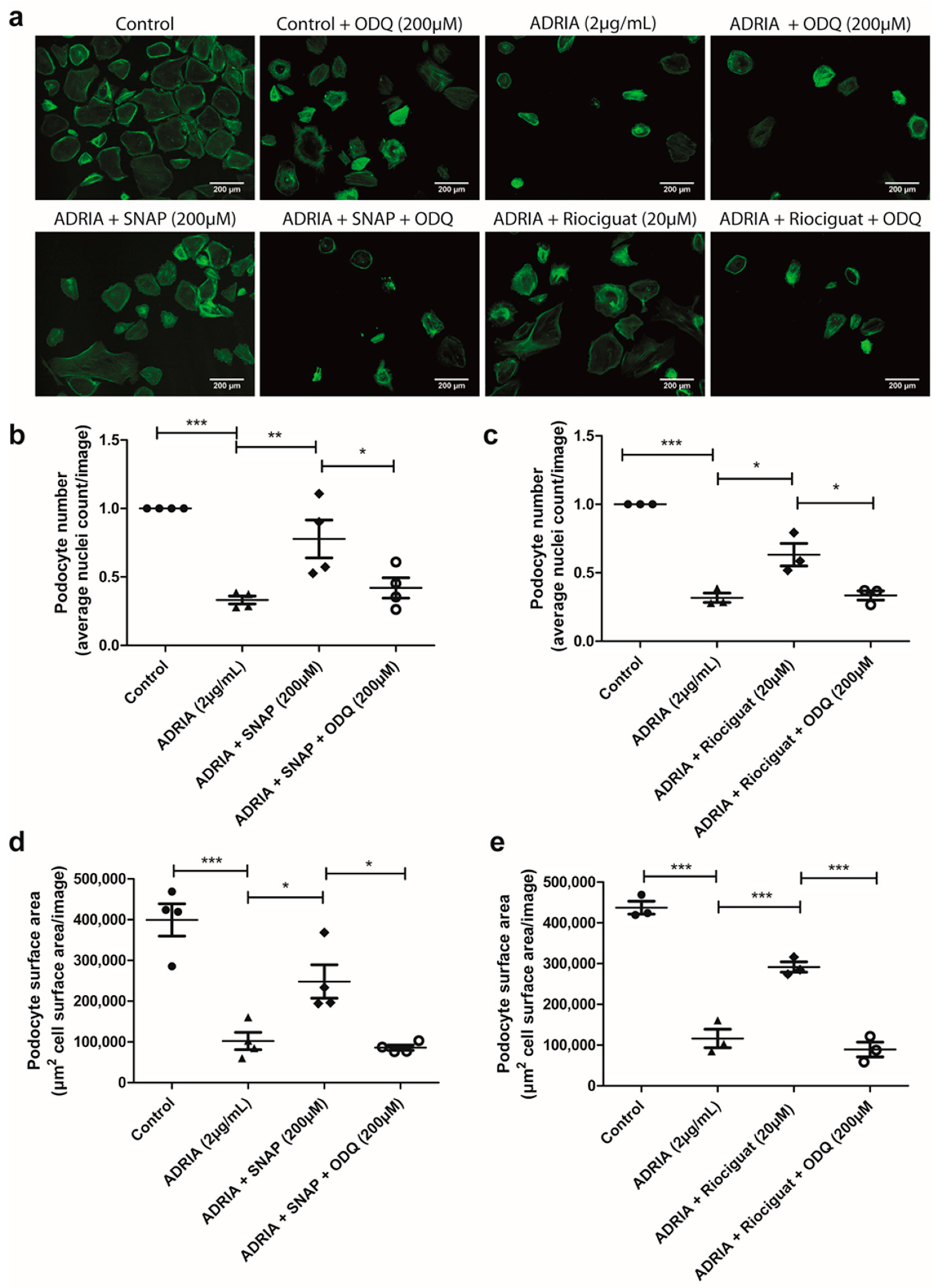
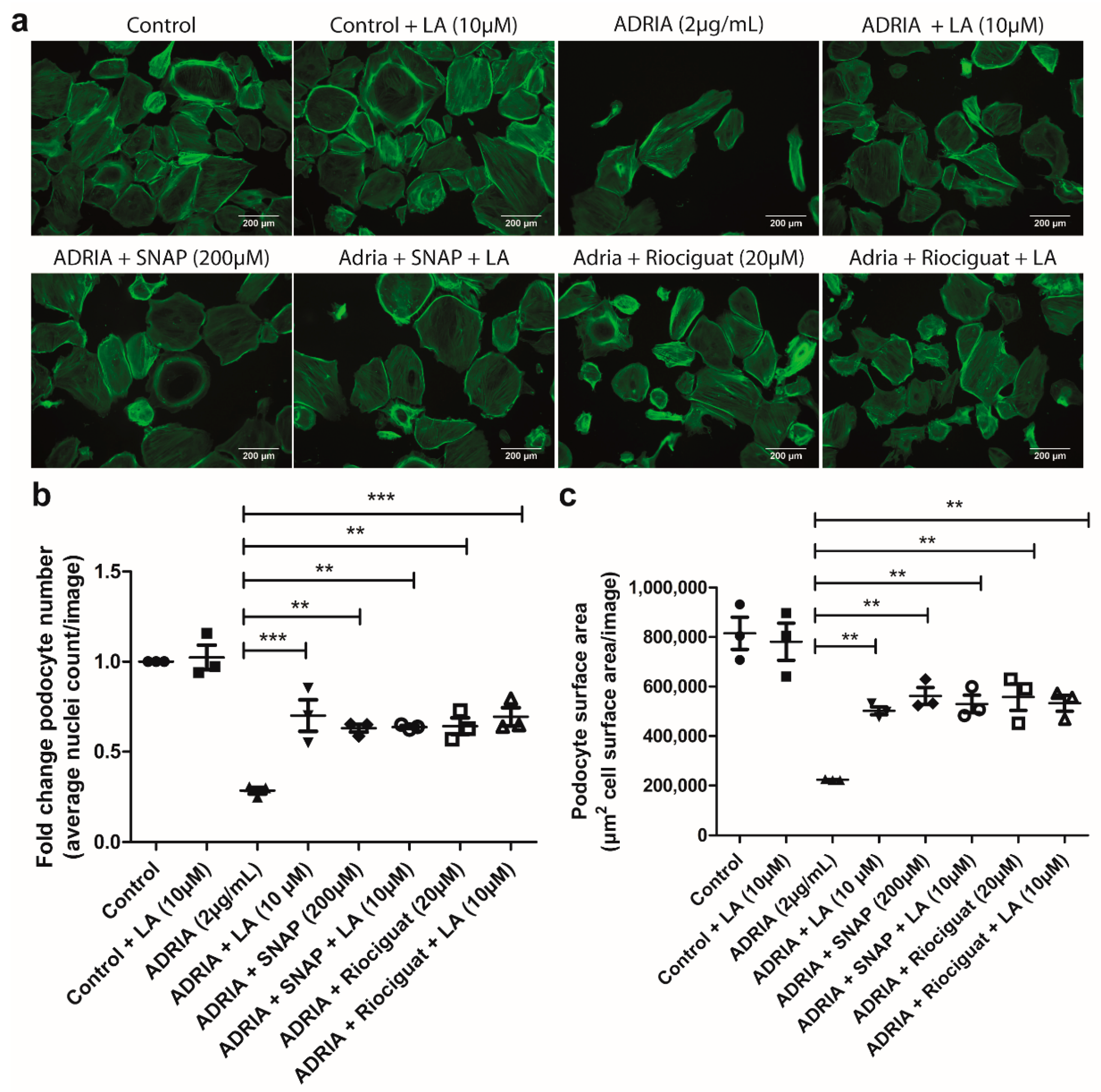
2.4. sGC Activation Prevents PODOCYTE Injury via TRPC6 Inhibition
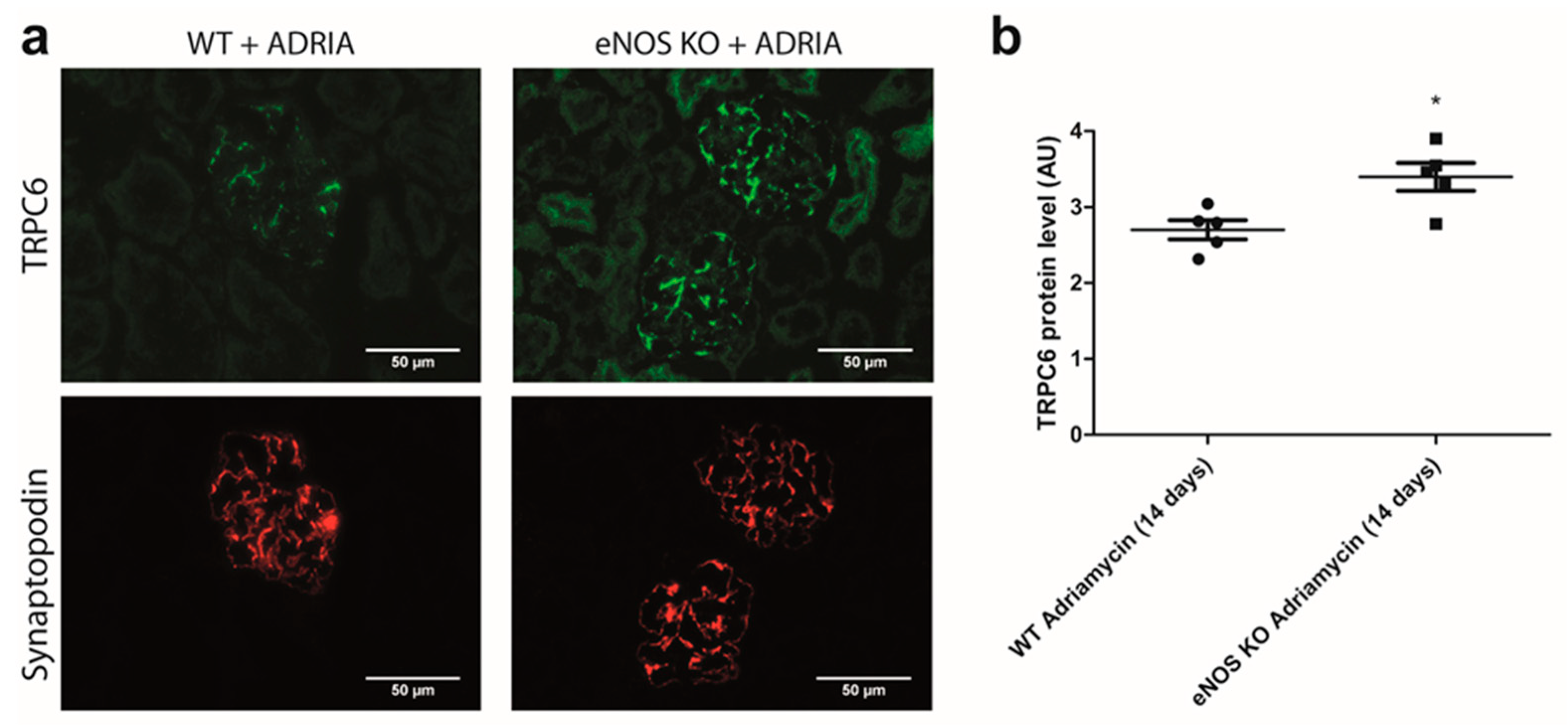
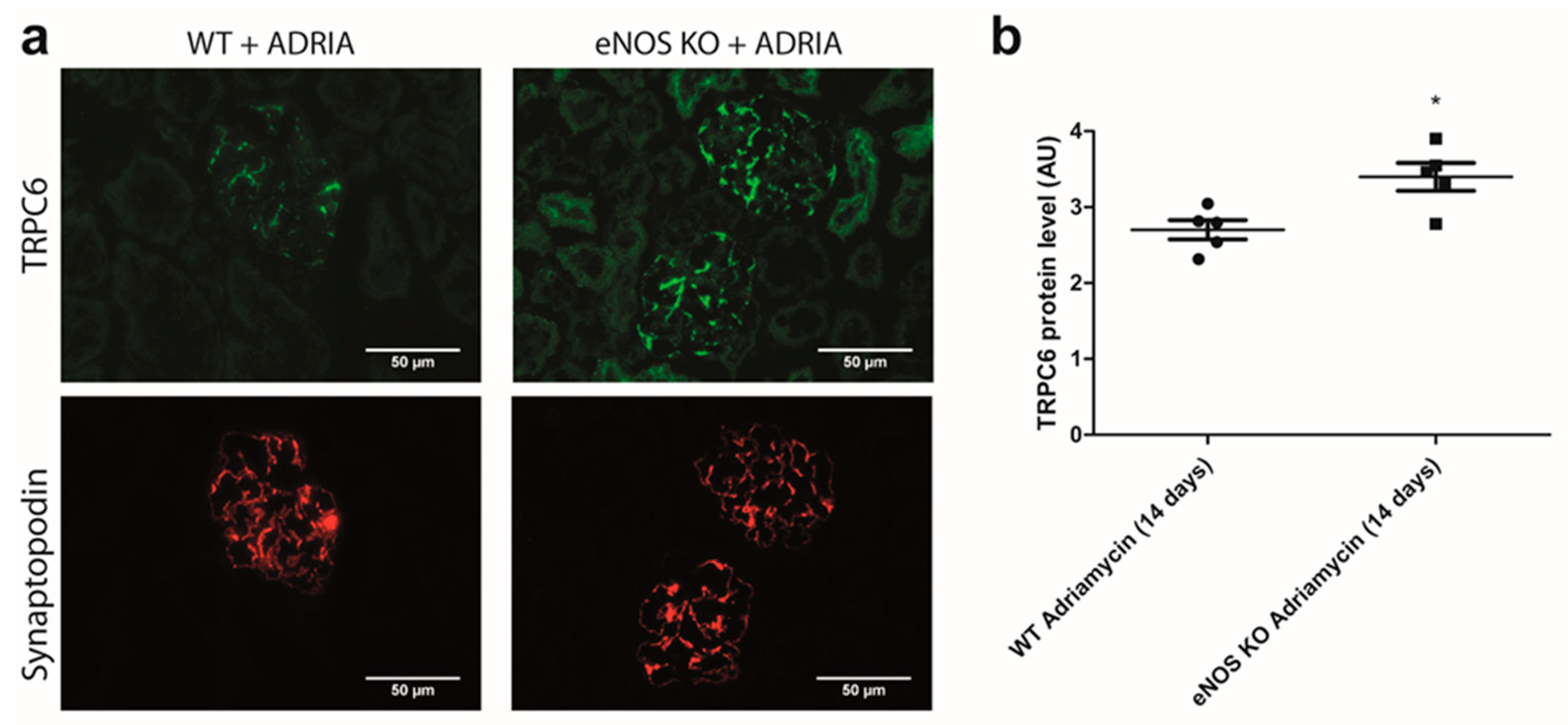
2.5. NO Reduces Glomerular TRPC6 Overexpression In Vivo
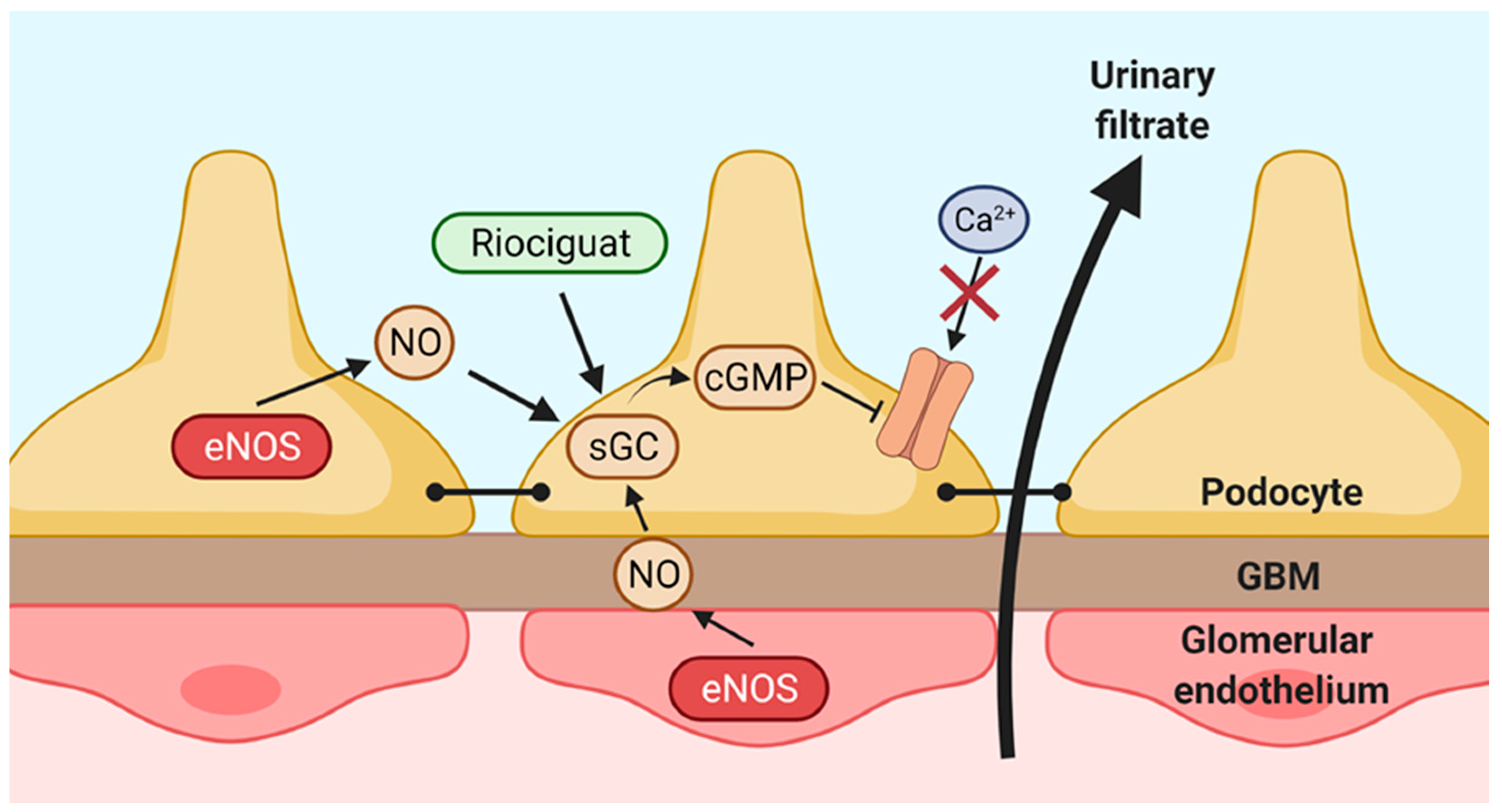
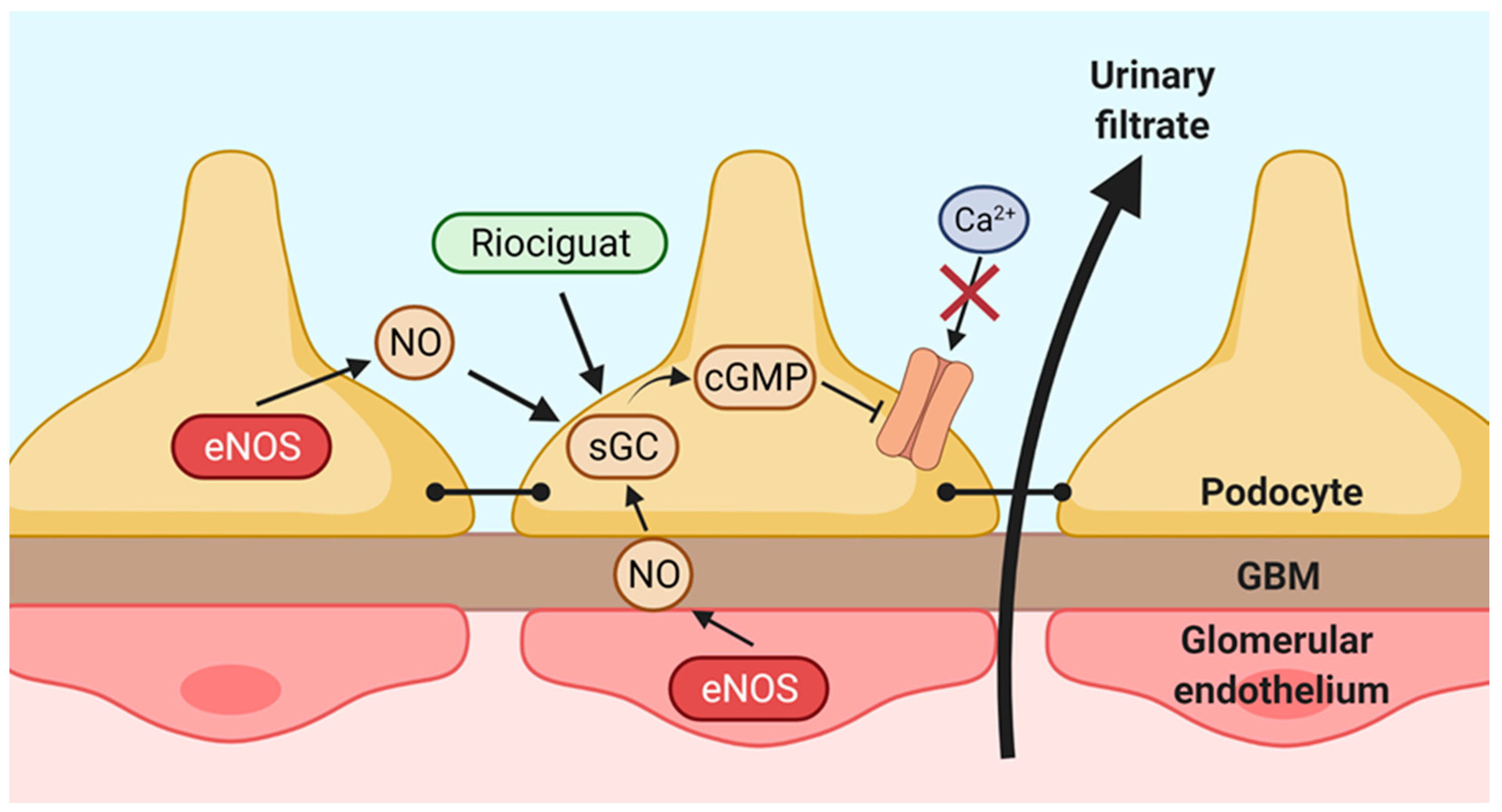
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Animal Studies
4.3. RNA Isolation and Quantitative PCR Analysis
4.4. Nitric Oxide Detection
4.5. cGMP ELISA
4.6. Immunohistochemistry
4.7. Intracellular Ca2+ Measurements by Fura-2 Ratiometry
4.8. Western Blot Analyses
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jarad, G.; Miner, J.H. Update on the glomerular filtration barrier. Curr. Opin. Nephrol. Hypertens. 2009, 18, 226–232. [Google Scholar] [CrossRef] [Green Version]
- Torban, E.; Braun, F.; Wanner, N.; Takano, T.; Goodyer, P.R.; Lennon, R.; Ronco, P.; Cybulsky, A.V.; Huber, T.B. From podocyte biology to novel cures for glomerular disease. Kidney Int. 2019, 96, 850–861. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Polu, K.R.; Möller, C.C.; Kenlan, P.; Altintas, M.M.; Wei, C.; Faul, C.; Herbert, S.; Villegas, I.; Avila-Casado, C.; et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat. Genet. 2005, 37, 739–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möller, C.C.; Wei, C.; Altintas, M.; Li, J.; Greka, A.; Ohse, T.; Pippin, J.W.; Rastaldi, M.P.; Wawersik, S.; Schiavi, S.; et al. Induction of TRPC6 Channel in Acquired Forms of Proteinuric Kidney Disease. J. Am. Soc. Nephrol. 2006, 18, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.P.; Conlon, P.J.; Lynn, K.L.; Farrington, M.K.; Creazzo, T.; Hawkins, A.F.; Daskalakis, N.; Kwan, S.Y.; Ebersviller, S.; Burchette, J.L.; et al. A Mutation in the TRPC6 Cation Channel Causes Familial Focal Segmental Glomerulosclerosis. Science 2005, 308, 1801–1804. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.Y.; Shotorbani, P.Y.; Dryer, S.E. Trpc6 inactivation confers protection in a model of severe nephrosis in rats. J. Mol. Med. 2018, 96, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Spires, D.; Ilatovskaya, D.V.; Levchenko, V.; North, P.E.; Geurts, A.M.; Palygin, O.; Staruschenko, A. Protective role of Trpc6 knockout in the progression of diabetic kidney disease. Am. J. Physiol. Physiol. 2018, 315, F1091–F1097. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Ding, Y.; Smedley, C.; Wang, Y.; Chaudhari, S.; Birnbaumer, L.; Ma, R. Increased glomerular filtration rate and impaired contractile function of mesangial cells in TRPC6 knockout mice. Sci. Rep. 2017, 7, 4145. [Google Scholar] [CrossRef]
- Nijenhuis, T.; Sloan, A.J.; Hoenderop, J.G.; Flesche, J.; van Goor, H.; Kistler, A.D.; Bakker, M.; Bindels, R.J.; de Boer, R.A.; Möller, C.C.; et al. Angiotensin II Contributes to Podocyte Injury by Increasing TRPC6 Expression via an NFAT-Mediated Positive Feedback Signaling Pathway. Am. J. Pathol. 2011, 179, 1719–1732. [Google Scholar] [CrossRef]
- Verheijden, K.A.; Sonneveld, R.; Bebber, M.B.-V.; Wetzels, J.F.; van der Vlag, J.; Nijenhuis, T. The Calcium-Dependent Protease Calpain-1 Links TRPC6 Activity to Podocyte Injury. J. Am. Soc. Nephrol. 2018, 29, 2099–2109. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Jarad, G.; Tripathi, P.; Pan, M.; Cunningham, J.; Martin, D.R.; Liapis, H.; Miner, J.H.; Chen, F. Activation of NFAT Signaling in Podocytes Causes Glomerulosclerosis. J. Am. Soc. Nephrol. 2010, 21, 1657–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonneveld, R.; Hoenderop, J.G.; Isidori, A.; Henique, C.; Dijkman, H.B.; Berden, J.H.; Tharaux, P.-L.; Van Der Vlag, J.; Nijenhuis, T. Sildenafil Prevents Podocyte Injury via PPAR-γ–Mediated TRPC6 Inhibition. J. Am. Soc. Nephrol. 2016, 28, 1491–1505. [Google Scholar] [CrossRef] [Green Version]
- Hall, G.; Rowell, J.; Farinelli, F.; Gbadegesin, R.A.; Lavin, P.; Wu, G.; Homstad, A.; Malone, A.; Lindsey, T.; Jiang, R.; et al. Phosphodiesterase 5 inhibition ameliorates angiontensin II-induced podocyte dysmotility via the protein kinase G-mediated downregulation of TRPC6 activity. Am. J. Physiol. Physiol. 2014, 306, F1442–F1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, S.; Lin, H.; Geshi, N.; Mori, Y.; Kawarabayashi, Y.; Takami, N.; Mori, M.X.; Honda, A.; Inoue, R. Nitric oxide-cGMP-protein kinase G pathway negatively regulates vascular transient receptor potential channel TRPC6. J. Physiol. 2008, 586, 4209–4223. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Berka, V.; Tsai, A.; Murad, F. Soluble Guanylyl Cyclase: The Nitric Oxide Receptor. Methods Enzymol. 2005, 396, 478–492. [Google Scholar] [CrossRef]
- Raij, L.; Baylis, C. Glomerular actions of nitric oxide. Kidney Int. 1995, 48, 20–32. [Google Scholar] [CrossRef] [Green Version]
- Delles, C.; Klingbeil, A.U.; Schneider, M.P.; Handrock, R.; Schäufele, T.; Schmieder, R.E. The role of nitric oxide in the regulation of glomerular haemodynamics in humans. Nephrol. Dial. Transplant. 2004, 19, 1392–1397. [Google Scholar] [CrossRef]
- Heeringa, P.; van Goor, H.; Itoh-Lindstrom, Y.; Maeda, N.; Falk, R.J.; Assmann, K.J.; Kallenberg, C.G.; Jennette, J.C. Lack of Endothelial Nitric Oxide Synthase Aggravates Murine Accelerated Anti-Glomerular Basement Membrane Glomerulonephritis. Am. J. Pathol. 2000, 156, 879–888. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.J.; Wang, S.; Cheng, H.; Zhang, M.-Z.; Takahashi, T.; Fogo, A.B.; Breyer, M.D.; Harris, R.C. Endothelial Nitric Oxide Synthase Deficiency Produces Accelerated Nephropathy in Diabetic Mice. J. Am. Soc. Nephrol. 2006, 17, 2664–2669. [Google Scholar] [CrossRef]
- Garsen, M.; Rops, A.L.; Li, J.; Van Beneden, K.; Branden, C.V.D.; Berden, J.H.; Rabelink, T.; Van Der Vlag, J. Endothelial Nitric Oxide Synthase Prevents Heparanase Induction and the Development of Proteinuria. PLoS ONE 2016, 11, e0160894. [Google Scholar] [CrossRef] [Green Version]
- Bevan, H.S.; Slater, S.C.; Clarke, H.; Cahill, P.A.; Mathieson, P.W.; Welsh, G.I.; Satchell, S.C. Acute laminar shear stress reversibly increases human glomerular endothelial cell permeability via activation of endothelial nitric oxide synthase. Am. J. Physiol. Physiol. 2011, 301, F733–F742. [Google Scholar] [CrossRef] [PubMed]
- Feliers, D.; Lee, D.-Y.; Gorin, Y.; Kasinath, B.S. Symmetric dimethylarginine alters endothelial nitric oxide activity in glomerular endothelial cells. Cell. Signal. 2015, 27, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Furusu, A.; Miyazaki, M.; Abe, K.; Tsukasaki, S.; Shioshita, K.; Sasaki, O.; Miyazaki, K.; Ozono, Y.; Koji, T.; Harada, T.; et al. Expression of endothelial and inducible nitric oxide synthase in human glomerulonephritis. Kidney Int. 1998, 53, 1760–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghofrani, H.-A.; Humbert, M.; Langleben, D.; Schermuly, R.; Stasch, J.-P.; Wilkins, M.R.; Klinger, J.R. Riociguat: Mode of Action and Clinical Development in Pulmonary Hypertension. Chest 2017, 151, 468–480. [Google Scholar] [CrossRef] [Green Version]
- Ghofrani, H.-A.; D’Armini, A.M.; Grimminger, F.; Hoeper, M.; Jansa, P.; Kim, N.H.; Mayer, E.; Simonneau, G.; Wilkins, M.R.; Fritsch, A.; et al. Riociguat for the Treatment of Chronic Thromboembolic Pulmonary Hypertension. N. Engl. J. Med. 2013, 369, 319–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghofrani, H.-A.; Galie, N.; Grimminger, F.; Grünig, E.; Humbert, M.; Jing, Z.-C.; Keogh, A.M.; Langleben, D.; Kilama, M.O.; Fritsch, A.; et al. Riociguat for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2013, 369, 330–340. [Google Scholar] [CrossRef] [Green Version]
- Aires, V.; Hichami, A.; Boulay, G.; Khan, N.A. Activation of TRPC6 calcium channels by diacylglycerol (DAG)-containing arachidonic acid: A comparative study with DAG-containing docosahexaenoic acid. Biochimie 2007, 89, 926–937. [Google Scholar] [CrossRef]
- Urban, N.; Wang, L.; Kwiek, S.; Rademann, J.; Kuebler, W.M.; Schaefer, M. Identification and Validation of Larixyl Acetate as a Potent TRPC6 Inhibitor. Mol. Pharmacol. 2015, 89, 197–213. [Google Scholar] [CrossRef] [Green Version]
- Garthwaite, J.; Southam, E.; Boulton, C.L.; Nielsen, E.B.; Schmidt, K.; Mayer, B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol. Pharmacol. 1995, 48, 184–188. [Google Scholar]
- Arif, E.; Solanki, A.K.; Nihalani, D. Adriamycin susceptibility among C57BL/6 substrains. Kidney Int. 2016, 89, 721–723. [Google Scholar] [CrossRef] [Green Version]
- Daehn, I.S. Glomerular Endothelial Cell Stress and Cross-Talk with Podocytes in Early Diabetic Kidney Disease. Front. Med. 2018, 5, 76. [Google Scholar] [CrossRef]
- Eremina, V.; Sood, M.; Haigh, J.; Nagy, A.; Lajoie, G.; Ferrara, N.; Gerber, H.-P.; Kikkawa, Y.; Miner, J.H.; Quaggin, S.E. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Investig. 2003, 111, 707–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogo, A.B. Talking back: The podocytes and endothelial cells duke it out. Kidney Int. 2016, 90, 1157–1159. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Lee, K.; Chuang, P.Y.; Liu, Z.-H.; He, J.C. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am. J. Physiol. Physiol. 2015, 308, F287–F297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhakar, S.; Starnes, J.; Shi, S.; Lonis, B.; Tran, R. Diabetic Nephropathy Is Associated with Oxidative Stress and Decreased Renal Nitric Oxide Production. J. Am. Soc. Nephrol. 2007, 18, 2945–2952. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, R.J.; Baylis, C. Total nitric oxide production is low in patients with chronic renal disease. Kidney Int. 2000, 58, 1261–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baylis, C. Nitric oxide deficiency in chronic kidney disease. Am. J. Physiol. Physiol. 2008, 294, F1–F9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, S.; Ozawa, S.; Mori, K.; Asanuma, K.; Yanagita, M.; Uchida, S.; Nakagawa, T. ENOS deficiency causes podocyte injury with mitochondrial abnormality. Free Radic. Biol. Med. 2015, 87, 181–192. [Google Scholar] [CrossRef]
- Yuen, D.A.; Stead, B.E.; Zhang, Y.; White, K.E.; Kabir, M.G.; Thai, K.; Advani, S.L.; Connelly, K.A.; Takano, T.; Zhu, L.; et al. eNOS Deficiency Predisposes Podocytes to Injury in Diabetes. J. Am. Soc. Nephrol. 2012, 23, 1810–1823. [Google Scholar] [CrossRef] [Green Version]
- Feliers, D.; Chen, X.; Akis, N.; Choudhury, G.G.; Madaio, M.; Kasinath, B.S. VEGF regulation of endothelial nitric oxide synthase in glomerular endothelial cells. Kidney Int. 2005, 68, 1648–1659. [Google Scholar] [CrossRef] [Green Version]
- Palygin, O.; Ilatovskaya, D.V.; Levchenko, V.; Endres, B.T.; Geurts, A.M.; Staruschenko, A. Nitric oxide production by glomerular podocytes. Nitric Oxide 2017, 72, 24–31. [Google Scholar] [CrossRef]
- Khayyat, N.H.; Kim, E.Y.; Dryer, S.E. TRPC6 inactivation does not protect against diabetic kidney disease in streptozotocin (STZ)-treated Sprague-Dawley rats. FASEB BioAdvances 2019, 1, 773–782. [Google Scholar] [CrossRef] [Green Version]
- Kistler, A.D.; Singh, G.; Altintas, M.; Yu, H.; Fernandez, I.C.; Gu, C.; Wilson, C.; Srivastava, S.K.; Dietrich, A.; Walz, K.; et al. Transient Receptor Potential Channel 6 (TRPC6) Protects Podocytes during Complement-mediated Glomerular Disease. J. Biol. Chem. 2013, 288, 36598–36609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iordache, A.M.; Docea, A.O.; Buga, A.M.; Zlatian, O.; Ciurea, M.E.; Rogoveanu, O.C.; Burada, F.; Sosoi, S.; Mitrut, R.; Mamoulakis, C.; et al. Sildenafil and tadalafil reduce the risk of contrast-induced nephropathy by modulating the oxidant/antioxidant balance in a murine model. Food Chem. Toxicol. 2019, 135, 111038. [Google Scholar] [CrossRef] [PubMed]
- Uijl, E.; Hart, D.C..; Roksnoer, L.C.; Groningen, M.C.C.-V.; van Veghel, R.; Garrelds, I.M.; de Vries, R.; van der Vlag, J.; Zietse, R.; Nijenhuis, T.; et al. Angiotensin–neprilysin inhibition confers renoprotection in rats with diabetes and hypertension by limiting podocyte injury. J. Hypertens. 2020, 38, 755–764. [Google Scholar] [CrossRef]
- Czirok, S.; Fang, L.; Radovits, T.; Szabó, G.; Szénási, G.; Rosivall, L.; Merkely, B.; Kökény, G. Cinaciguat ameliorates glomerular damage by reducing ERK1/2 activity and TGF-ß expression in type-1 diabetic rats. Sci. Rep. 2017, 7, 11218. [Google Scholar] [CrossRef] [PubMed]
- Harloff, M.; Prüschenk, S.; Seifert, R.; Schlossmann, J. Activation of soluble guanylyl cyclase signalling with cinaciguat improves impaired kidney function in diabetic mice. Br. J. Pharmacol. 2021. ahead of print. [Google Scholar] [CrossRef]
- Tobin, J.V.; Zimmer, D.P.; Shea, C.; Germano, P.; Bernier, S.G.; Liu, G.; Long, K.; Miyashiro, J.; Ranganath, S.; Jacobson, S.; et al. Pharmacological Characterization of IW-1973, a Novel Soluble Guanylate Cyclase Stimulator with Extensive Tissue Distribution, Antihypertensive, Anti-Inflammatory, and Antifibrotic Effects in Preclinical Models of Disease. J. Pharmacol. Exp. Ther. 2018, 365, 664–675. [Google Scholar] [CrossRef]
- Sravani, S.; Saifi, M.A.; Godugu, C. Riociguat ameliorates kidney injury and fibrosis in an animal model. Biochem. Biophys. Res. Commun. 2020, 530, 706–712. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [Green Version]
- Frey, R.; Becker, C.; Unger, S.; Schmidt, A.; Wensing, G.; Mück, W. Assessment of the Effects of Renal Impairment and Smoking on the Pharmacokinetics of a Single Oral Dose of the Soluble Guanylate Cyclase Stimulator Riociguat (BAY 63-2521). Pulm. Circ. 2016, 6, S15–S26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, N.S.; Rahaghi, F.F.; Sood, N.; Frey, R.; Ghofrani, H.-A. Individual dose adjustment of riociguat in patients with pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Respir. Med. 2017, 129, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Saleh, S.; Becker, C.; Frey, R.; Mück, W. Population Pharmacokinetics of Single-Dose Riociguat in Patients with Renal or Hepatic Impairment. Pulm. Circ. 2016, 6, S75–S85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleem, M.A.; O’Hare, M.J.; Reiser, J.; Coward, R.J.; Inward, C.D.; Farren, T.; Xing, C.Y.; Ni, L.; Mathieson, P.W.; Mundel, P. A Conditionally Immortalized Human Podocyte Cell Line Demonstrating Nephrin and Podocin Expression. J. Am. Soc. Nephrol. 2002, 13, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Satchell, S.; Tasman, C.; Singh, A.; Ni, L.; Geelen, J.; von Ruhland, C.; O’Hare, M.; Saleem, M.; Heuvel, L.V.D.; Mathieson, P. Conditionally immortalized human glomerular endothelial cells expressing fenestrations in response to VEGF. Kidney Int. 2006, 69, 1633–1640. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.B.Y.; Qu, X.; Zhang, X.; Caruana, G.; Bertram, J.; Li, J. Glomerular Endothelial Cell Injury and Damage Precedes That of Podocytes in Adriamycin-Induced Nephropathy. PLoS ONE 2013, 8, e55027. [Google Scholar] [CrossRef] [Green Version]
- Satijn, D.P.; Gunster, M.J.; van der Vlag, J.; Hamer, K.M.; Schul, W.; Alkema, M.J.; Saurin, A.J.; Freemont, P.S.; van Driel, R.; Otte, A.P. RING1 is associated with the polycomb group protein complex and acts as a transcriptional repressor. Mol. Cell. Biol. 1997, 17, 4105–4113. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Gene Symbol | Forward Sequence | Reverse Sequence |
|---|---|---|---|
| GAPDH | GAPDH | AGATGGTGATGGGATTTC | TCCTCGCATGATTGTCACCC |
| TRPC6 | TRPC6 | AATGCTCCCAGAAATGGCAC | CCTCCACTGAACCCTGGAAA |
| nNOS | NOS1 | CTTCAAGAAGCTAGCAGAAGCTGT | ACAAGGACCAGAGTTTCATGTTC |
| iNOS | NOS2 | ACAACAAATTCAGGTACGCTGTG | TCTGATCAATGTCATGAGCAAAGG |
| eNOS | NOS3 | CCAGCTAGCCAAAGTCACCAT | GTCTCGGAGCCATACAGGATT |
| sGCα1 | GUCY1A1 | GCTCTTCTCAGACATCGTTGGG | ATAGGCATCGCCAATGGTCTCC |
| sGCα2 | GUCY1A2 | GCAGACTCTCAAGAGGACACTG | GTTGGAGTGGTCTGCATAGGAG |
| sGCβ1 | GUCY1B1 | GGAAATCCTCACTGACAGGCTAC | CAGCTCATTGGCAACAGACGGA |
| sGCβ2 | GUCY1B2 | TCTGCAACGCTTTTCCTTTCC | CGACTTTGCCATGCTACAGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
‘t Hart, D.; Li, J.; van der Vlag, J.; Nijenhuis, T. Repurposing Riociguat to Target a Novel Paracrine Nitric Oxide-TRPC6 Pathway to Prevent Podocyte Injury. Int. J. Mol. Sci. 2021, 22, 12485. https://doi.org/10.3390/ijms222212485
‘t Hart D, Li J, van der Vlag J, Nijenhuis T. Repurposing Riociguat to Target a Novel Paracrine Nitric Oxide-TRPC6 Pathway to Prevent Podocyte Injury. International Journal of Molecular Sciences. 2021; 22(22):12485. https://doi.org/10.3390/ijms222212485
Chicago/Turabian Style‘t Hart, Daan, Jinhua Li, Johan van der Vlag, and Tom Nijenhuis. 2021. "Repurposing Riociguat to Target a Novel Paracrine Nitric Oxide-TRPC6 Pathway to Prevent Podocyte Injury" International Journal of Molecular Sciences 22, no. 22: 12485. https://doi.org/10.3390/ijms222212485
APA Style‘t Hart, D., Li, J., van der Vlag, J., & Nijenhuis, T. (2021). Repurposing Riociguat to Target a Novel Paracrine Nitric Oxide-TRPC6 Pathway to Prevent Podocyte Injury. International Journal of Molecular Sciences, 22(22), 12485. https://doi.org/10.3390/ijms222212485