
BiP Heterozigosity Aggravates Pathological Deterioration in Experimental Amyotrophic Lateral Sclerosis

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
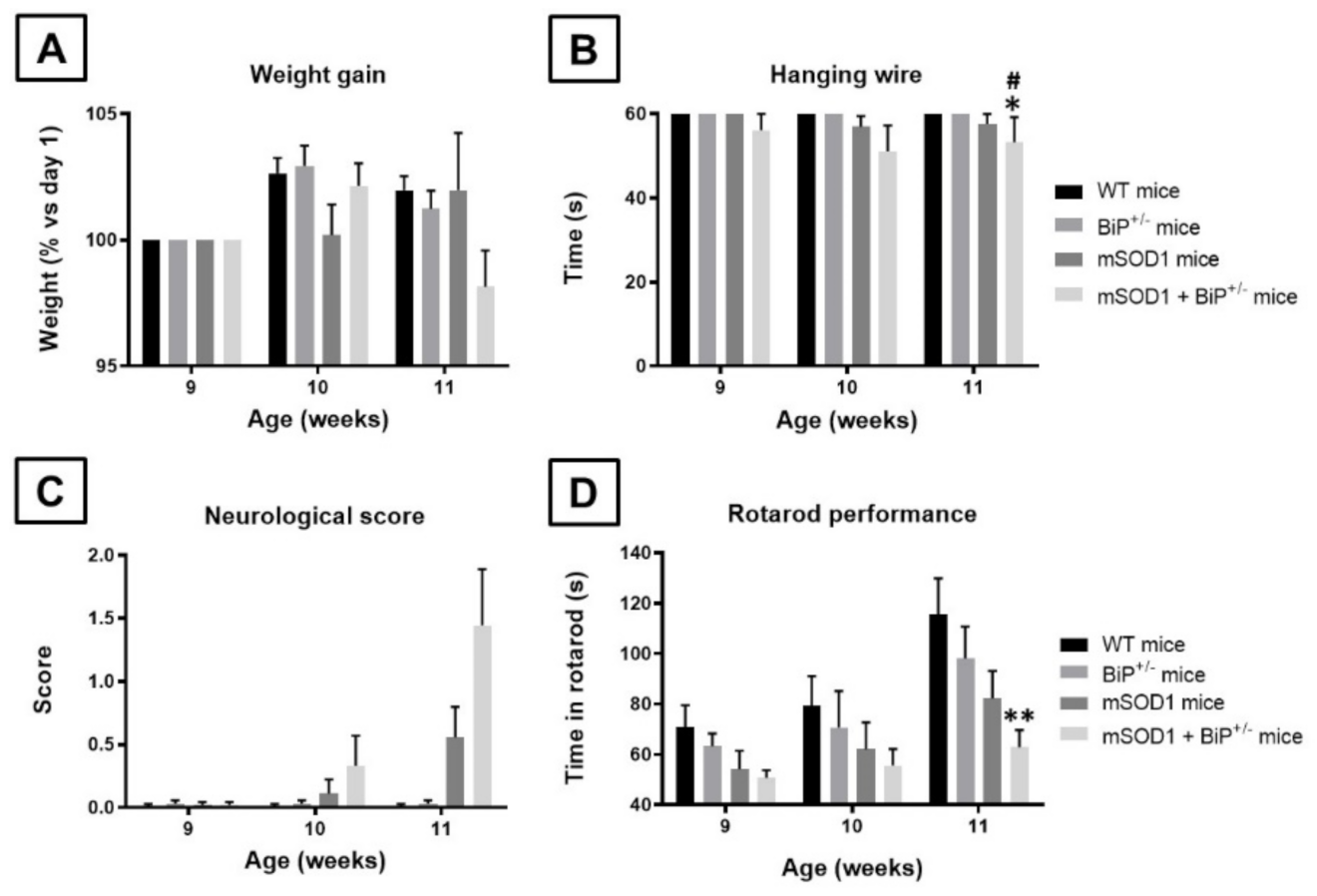
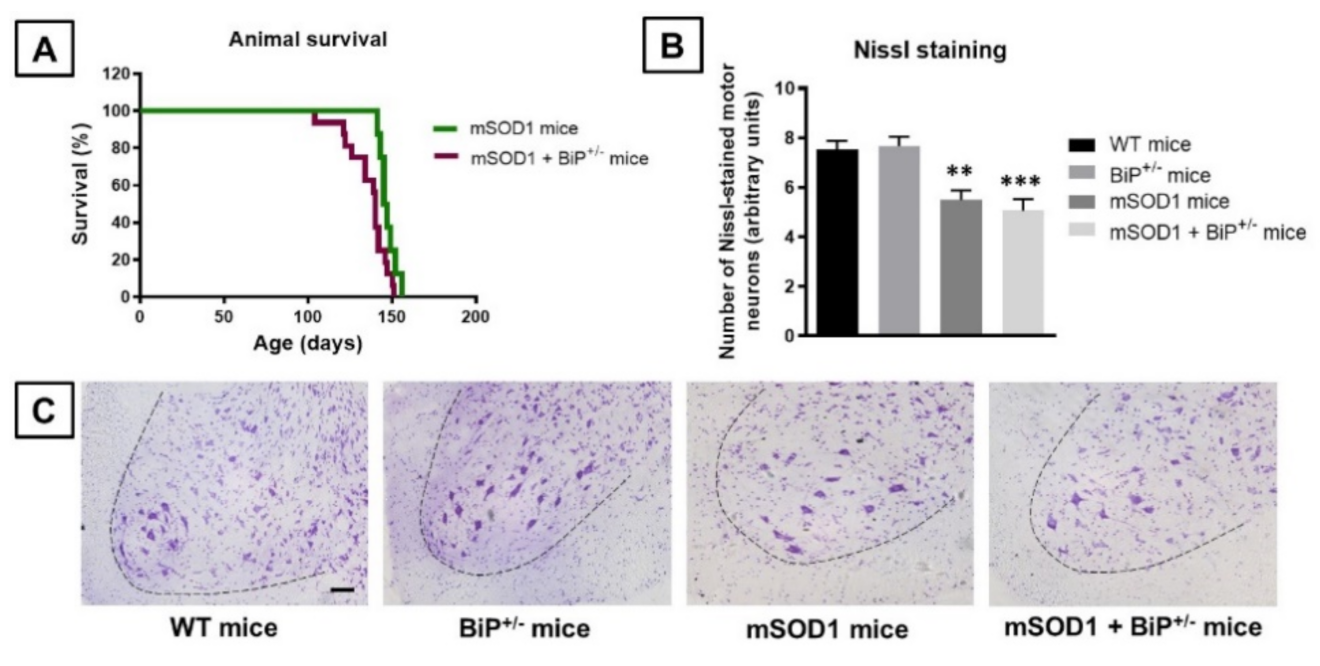
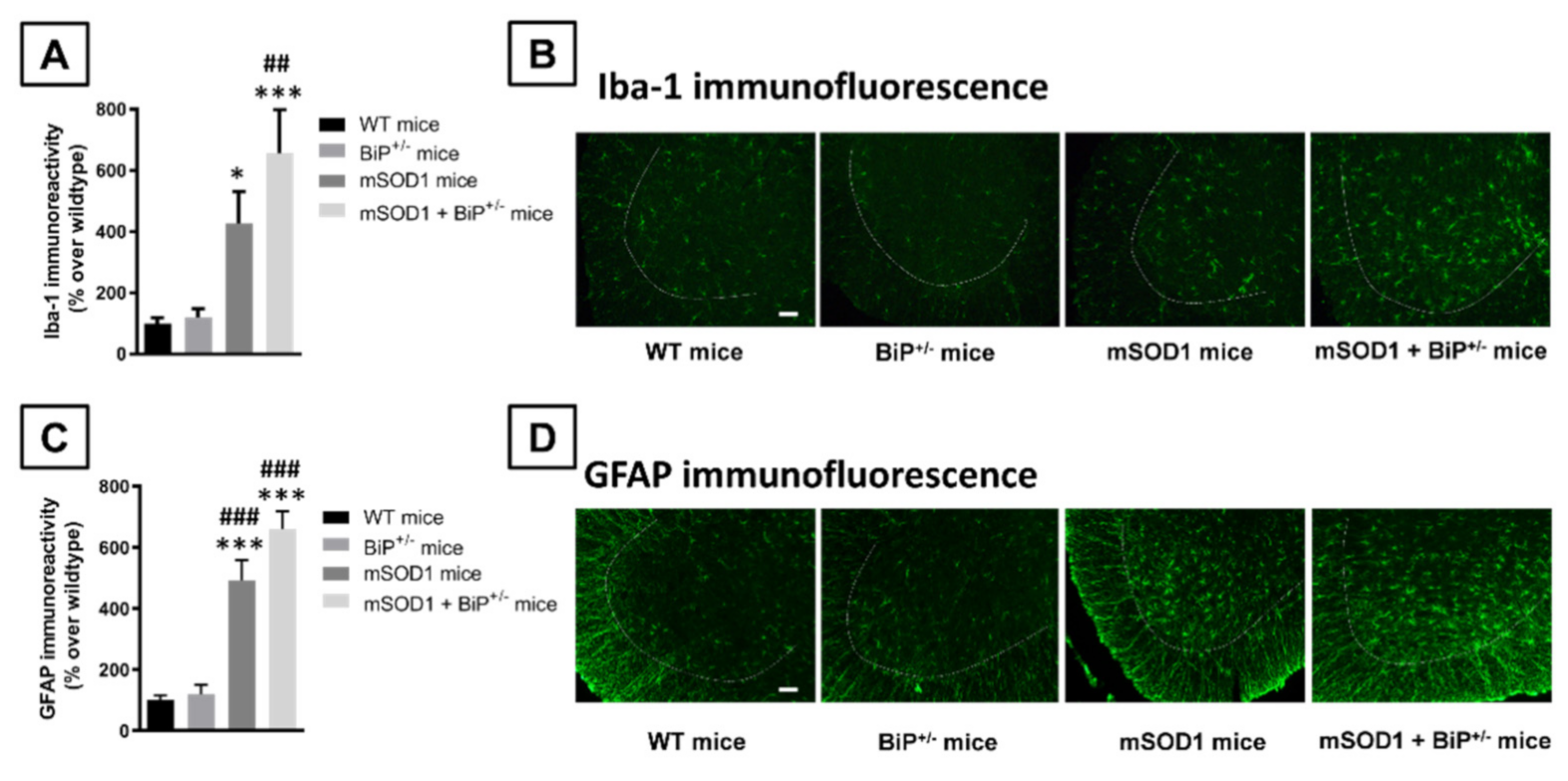
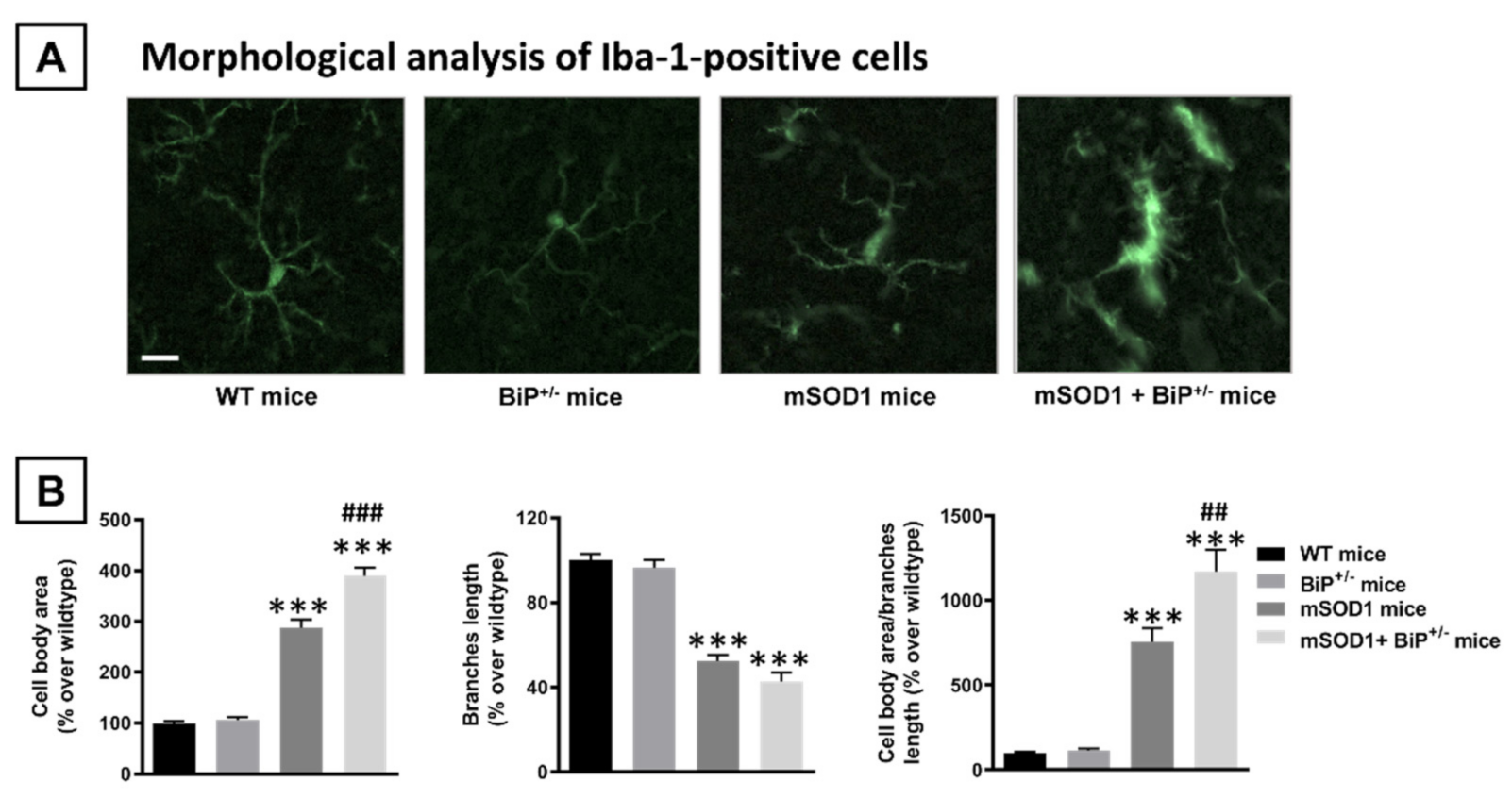
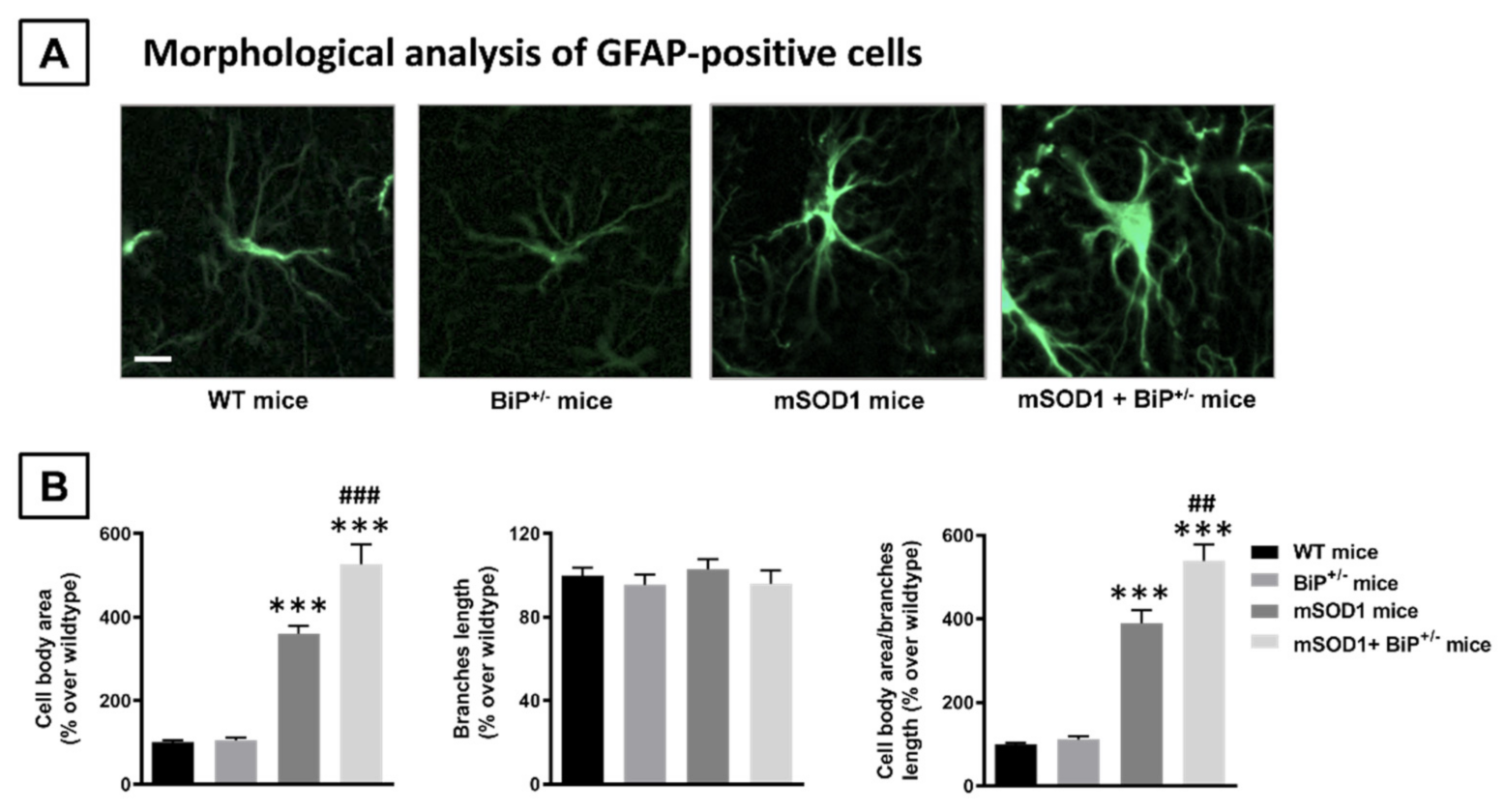
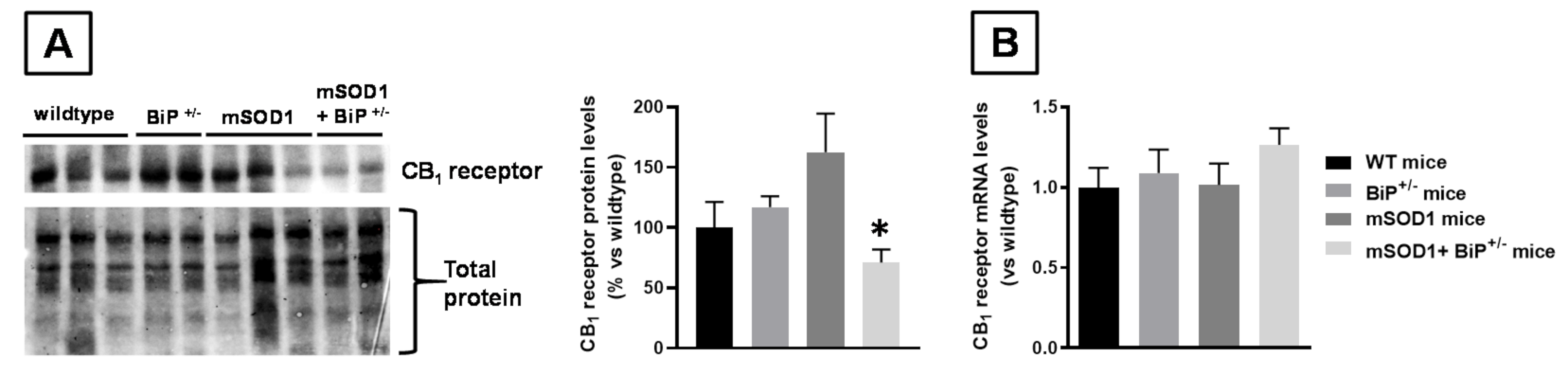
2.1. Studies in Experimental ALS: Generation of Double Mutants (mSOD1/BiP+/− Mice)
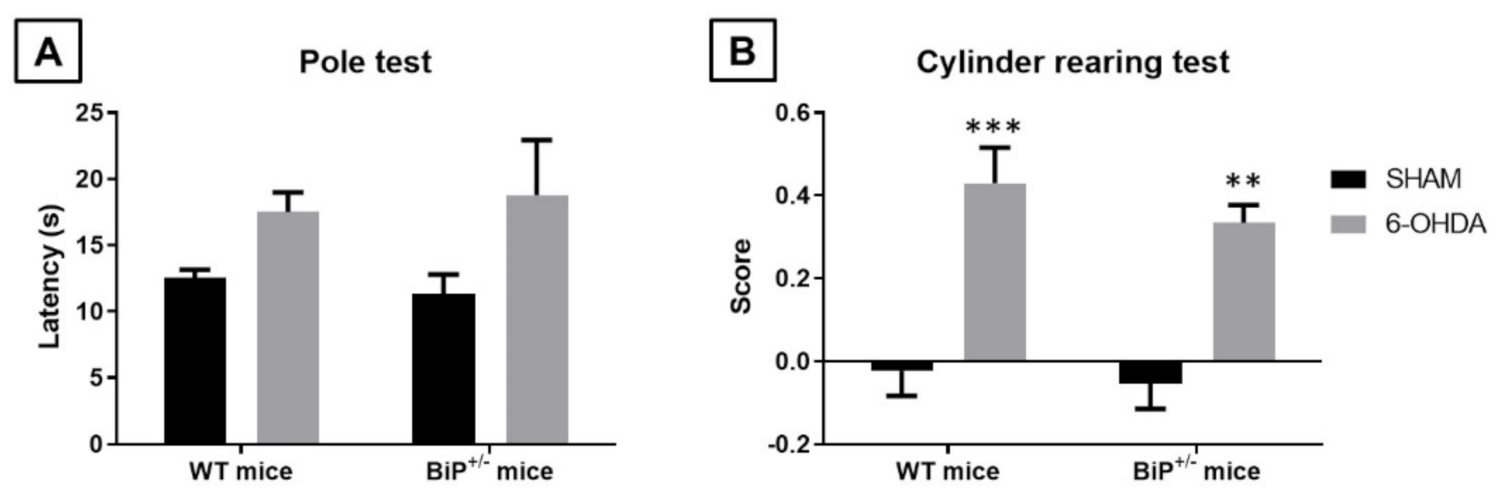
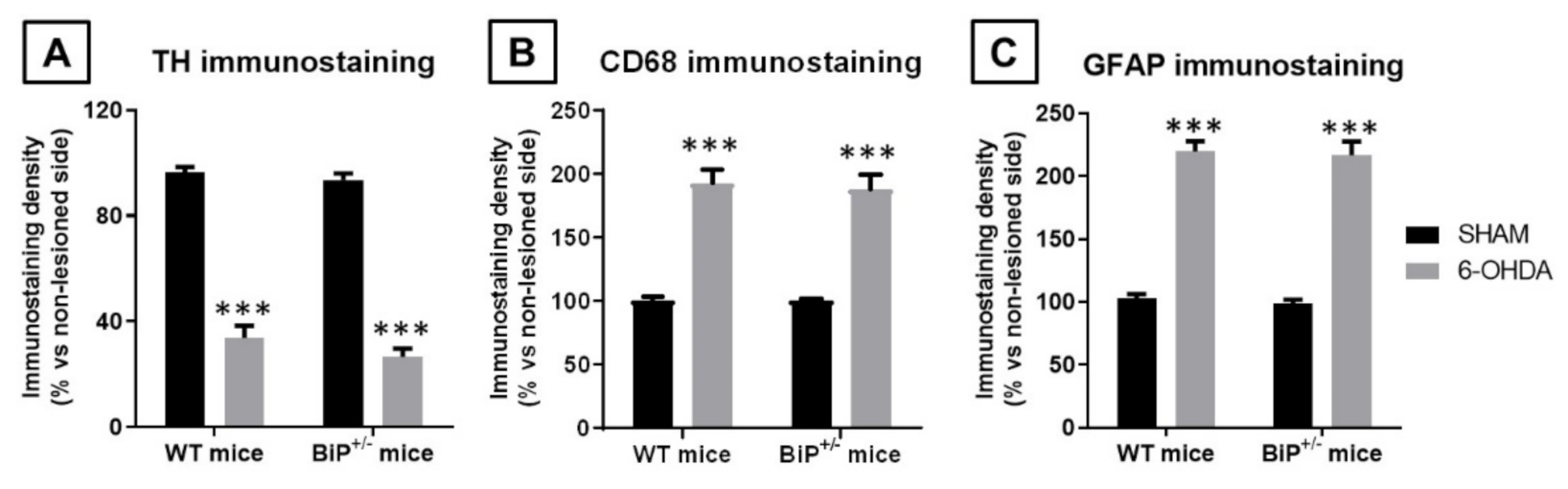
2.2. Studies in Experimental PD: Unilateral 6-OHDA Lesions in Wildtype and BiP+/− Mice
3. Discussion
4. Materials and Methods
4.1. Animals, Experiments and Sampling
4.2. Behavioral Recording
4.3. Histological Procedures
4.4. Real Time RT-qPCR Analysis
4.5. Western Blot Analysis
4.6. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fernández-Ruiz, J. The biomedical challenge of neurodegenerative disorders: An opportunity for cannabinoid-based ther-apies to improve on the poor current therapeutic outcomes. Br. J. Pharmacol. 2019, 176, 1370–1383. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ruiz, J.; Moro, M.A.; Martinez-Orgado, J. Cannabinoids in Neurodegenerative Disorders and Stroke/Brain Trauma: From Preclinical Models to Clinical Applications. Neurotherapeutics 2015, 12, 793–806. [Google Scholar] [CrossRef] [Green Version]
- Aymerich, M.S.; Aso, E.; Abellanas, M.A.; Tolon, R.M.; Ramos, J.A.; Ferrer, I.; Romero, J.; Fernández-Ruiz, J. Cannabinoid pharma-cology/therapeutics in chronic degenerative disorders affecting the central nervous system. Biochem. Pharmacol. 2018, 157, 67–84. [Google Scholar] [CrossRef] [Green Version]
- Chiarlone, A.; Bellocchio, L.; Blázquez, C.; Resel, E.; Soria-Gómez, E.; Cannich, A.; Ferrero, J.J.; Sagredo, O.; Benito, C.; Romero, J.; et al. A restricted population of CB1 cannabinoid re-ceptors with neuroprotective activity. Proc. Natl. Acad. Sci. USA 2014, 111, 8257–8262. [Google Scholar] [CrossRef] [Green Version]
- Hiebel, C.; Behl, C. The complex modulation of lysosomal degradation pathways by cannabinoid receptors 1 and 2. Life Sci. 2015, 138, 3–7. [Google Scholar] [CrossRef]
- Aso, E.; Palomer, E.; Juvés, S.; Maldonado, R.; Muñoz, F.J.; Ferrer, I. CB1 Agonist ACEA Protects Neurons and Reduces the Cognitive Impairment of AβPP/PS1 Mice. J. Alzheimer’s Dis. 2012, 30, 439–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, G.; Borroto-Escuela, D.; Angelats, E.; Etayo, Í.; Reyes-Resina, I.; Pulido-Salgado, M.; Rodríguez-Pérez, A.I.; Canela, E.I.; Saura, J.; Lanciego, J.L.; et al. Receptor-heteromer mediated regulation of endocan-nabinoid signaling in activated microglia. Role of CB1 and CB2 receptors and relevance for Alzheimer’s disease and levo-dopa-induced dyskinesia. Brain Behav. Immun. 2018, 67, 139–151. [Google Scholar] [CrossRef]
- Crunfli, F.; Vrechi, T.A.; Costa, A.P.; Torrão, A.S. Cannabinoid Receptor Type 1 Agonist ACEA Improves Cognitive Deficit on STZ-Induced Neurotoxicity through Apoptosis pathway and NO Modulation. Neurotox. Res. 2019, 35, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, C.; Chiarlone, A.; Sagredo, O.; Aguado, T.; Pazos, M.R.; Resel, E.; Palazuelos, J.; Julien, B.; Salazar, M.; Börner, C.; et al. Loss of striatal type 1 cannabinoid receptors is a key pathogenic factor in Huntington’s disease. Brain 2011, 134, 119–136. [Google Scholar] [CrossRef] [Green Version]
- Blazquez, C.; Chiarlone, A.; Bellocchio, L.; Resel, E.; Pruunsild, P.; García-Rincón, D.; Sendtner, M.; Timmusk, T.; Lutz, B.; Galve-Roperh, I.; et al. The CB1 cannabinoid receptor signals striatal neuroprotection via a PI3K/Akt/mTORC1/BDNF pathway. Cell Death Differ. 2015, 22, 1618–1629. [Google Scholar] [CrossRef]
- Maya-López, M.; Colín-González, A.L.; Aguilera, G.; De Lima, M.E.; Colpo-Ceolin, A.; Rangel-Lopez, E.; Villeda-Hernández, J.; Rembao-Bojórquez, D.; Túnez, I.; Luna-López, A.; et al. Neuroprotective effect of WIN55,212-2 against 3-nitropropionic acid-induced toxicity in the rat brain: Involvement of CB1 and NMDA receptors. Am. J. Transl. Res. 2017, 9, 261–274. [Google Scholar]
- Ruiz-Calvo, A.; Maroto, I.B.; Bajo-Grañeras, R.; Chiarlone, A.; Gaudioso, A.; Ferrero, J.J.; Resel, E.; Sánchez-Prieto, J.; Rodríguez-Navarro, A.J.; Marsicano, G.; et al. Pathway-Specific Control of Striatal Neuron Vulnerability by Corticostriatal Cannabinoid CB1 Receptors. Cereb. Cortex 2018, 28, 307–322. [Google Scholar] [CrossRef]
- Rossi, S.; Furlan, R.; De Chiara, V.; Muzio, L.; Musella, A.; Motta, C.; Studer, V.; Cavasinni, F.; Bernardi, G.; Martino, G.; et al. Cannabinoid CB1 receptors regulate neuronal TNF-α effects in experimental auto-immune encephalomyelitis. Brain Behav. Immun. 2011, 25, 1242–1248. [Google Scholar] [CrossRef]
- Moreno-Martet, M.; Feliú, A.; Espejo-Porras, F.; Mecha, M.; Carrillo-Salinas, F.J.; Fernández-Ruiz, J.; Guaza, C.; de Lago, E. The disease-modifying effects of a Sativex-like combination of phytocannabinoids in mice with experimental autoimmune en-cephalomyelitis are preferentially due to Δ9-tetrahydrocannabinol acting through CB1 receptors. Mult. Scler. Relat. Disord. 2015, 4, 505–511. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.C.; Bok, E.; Huh, S.H.; Park, J.Y.; Yoon, S.H.; Kim, S.R.; Kim, Y.S.; Maeng, S.; Park, S.H.; Jin, B.K. Cannabinoid receptor type 1 protects nigrostriatal dopaminergic neurons against MPTP neurotoxicity by inhibiting microglial activation. J. Immunol. 2011, 187, 6508–6517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Rial, S.; García-Gutiérrez, M.S.; Molina, J.A.; Pérez-Nievas, B.G.; Ledent, C.; Leiva, C.; Leza, J.C.; Manzanares, J. Increased vulnerability to 6-hydroxydopamine lesion and reduced development of dyskinesias in mice lacking CB1 cannabinoid receptors. Neurobiol. Aging 2011, 32, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Pinilla, E.; Aguinaga, D.; Navarro, G.; Rico, A.J.; Oyarzábal, J.; Sánchez-Arias, J.A.; Lanciego, J.L.; Franco, R. Targeting CB1 and GPR55 Endocannabinoid Receptors as a Potential Neuroprotective Approach for Parkinson’s Disease. Mol. Neurobiol. 2019, 56, 5900–5910. [Google Scholar] [CrossRef]
- Abood, M.E.; Rizvi, G.; Sallapudi, N.; McAllister, S.D. Activation of the CB1 cannabinoid receptor protects cultured mouse spinal neurons against excitotoxicity. Neurosci. Lett. 2001, 309, 197–201. [Google Scholar] [CrossRef]
- Zhao, P.; Ignacio, S.; Beattie, E.C.; Abood, M.E. Altered presymptomatic AMPA and cannabinoid receptor trafficking in motor neurons of ALS model mice: Implications for excitotoxicity. Eur. J. Neurosci. 2008, 27, 572–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, S.; De Chiara, V.; Musella, A.; Cozzolino, M.; Bernardi, G.; Maccarrone, M.; Mercuri, N.B.; Carrì, M.T.; Centonze, D. Abnormal sensitivity of cannabinoid CB1 receptors in the striatum of mice with experimental amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2019, 11, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Oliver, E.E.; Hughes, E.K.; Puckett, M.K.; Chen, R.; Lowther, W.T.; Howlett, A.C. Cannabinoid Receptor Interacting Protein 1a (CRIP1a) in Health and Disease. Biomolecules 2020, 10, 1609. [Google Scholar] [CrossRef]
- Costas-Insua, C.; Moreno, E.; Maroto, I.B.; Ruiz-Calvo, A.; Bajo-Grañeras, R.; Martín-Gutiérrez, D.; Diez-Alarcia, R.; Vilaró, M.T.; Cortés, R.; García-Font, N.; et al. Identification of BiP as a CB1 Receptor-Interacting Protein That Fine-Tunes Cannabinoid Signaling in the Mouse Brain. J. Neurosci. 2021, 41, 7924–7941. [Google Scholar] [CrossRef]
- Gorbatyuk, M.; Gorbatyuk, O. Review: Retinal degeneration: Focus on the unfolded protein response. Mol. Vis. 2013, 19, 1985. [Google Scholar] [PubMed]
- Casas, C. GRP78 at the Centre of the Stage in Cancer and Neuroprotection. Front. Neurosci. 2017, 11, 177. [Google Scholar] [CrossRef]
- Jin, H.; Komita, M.; Aoe, T. The Role of BiP Retrieval by the KDEL Receptor in the Early Secretory Pathway and its Effect on Protein Quality Control and Neurodegeneration. Front. Mol. Neurosci. 2017, 10, 222. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Mao, C.; Lee, B.; Lee, A.S. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol. Cell Biol. 2006, 26, 5688–5697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbatyuk, M.; Shabashvili, A.; Chen, W.; Meyers, C.; Sullivan, L.F.; Salganik, M.; Lin, J.H.; Lewin, A.; Muzyczka, N.; Gorbatyuk, O.S. Glucose Regulated Protein 78 Diminishes α-Synuclein Neurotoxicity in a Rat Model of Parkinson Disease. Mol. Ther. 2012, 20, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Jeon, G.S.; Shim, Y.-M.; Lee, D.-Y.; Kim, J.-S.; Kang, M.; Ahn, S.H.; Shin, J.-Y.; Geum, D.; Hong, Y.-H.; Sung, J.-J. Pathological Modification of TDP-43 in Amyotrophic Lateral Sclerosis with SOD1 Mutations. Mol. Neurobiol. 2019, 56, 2007–2021. [Google Scholar] [CrossRef] [Green Version]
- Cueto, C.R.; Santos-García, I.; García-Toscano, L.; Espejo-Porras, F.; Bellido, M.; Fernández-Ruiz, J.; Munoz, E.; de Lago, E. Neuroprotective effects of the cannabigerol quinone derivative VCE-003.2 in SOD1G93A transgenic mice, an experimental model of amyotrophic lateral sclerosis. Biochem. Pharmacol. 2018, 157, 217–226. [Google Scholar] [CrossRef]
- García, C.; Palomo-Garo, C.; García-Arencibia, M.; Ramos, J.; Pertwee, R.; Fernández-Ruiz, J. Symptom-relieving and neuroprotective effects of the phytocannabinoid Δ9-THCV in animal models of Parkinson’s disease. Br. J. Pharmacol. 2011, 163, 1495–1506. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Fischer, D.; Henze, C.; Strenzke, C.; Westrich, J.; Ferger, B.; Höglinger, G.U.; Oertel, W.H.; Hartmann, A. Characterization of the striatal 6-OHDA model of Parkinson’s disease in wild type and alpha-synuclein-deleted mice. Exp. Neurol. 2008, 210, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.M.; Ekhator, O.R.; Ghisays, V. Assessment of sensorimotor function in mouse models of Parkinson’s disease. J. Vis. Exp. 2013, 76, 50303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, F.J.; Lafuente, H.; Rey-Santano, M.C.; Mielgo, E.V.; Gastiasoro, E.; Rueda, M.; Pertwee, R.G.; Castillo, I.A.; Romero, J.; Martínez-Orgado, J. Neuroprotective Effects of the Nonpsychoactive Cannabinoid Cannabidiol in Hypoxic-Ischemic Newborn Piglets. Pediatr. Res. 2008, 64, 653–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceprián, M.; Jiménez-Sánchez, L.; Vargas, C.; Barata, L.; Hind, W.; Martínez-Orgado, J. Cannabidiol reduces brain damage and improves functional recovery in a neonatal rat model of arterial ischemic stroke. Neuropharmacology 2017, 116, 151–159. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Almería, M.; Burgaz, S.; Costas-Insua, C.; Rodríguez-Cueto, C.; Santos-García, I.; Rodríguez-Crespo, I.; García, C.; Guzmán, M.; de Lago, E.; Fernández-Ruiz, J. BiP Heterozigosity Aggravates Pathological Deterioration in Experimental Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 12533. https://doi.org/10.3390/ijms222212533
Gómez-Almería M, Burgaz S, Costas-Insua C, Rodríguez-Cueto C, Santos-García I, Rodríguez-Crespo I, García C, Guzmán M, de Lago E, Fernández-Ruiz J. BiP Heterozigosity Aggravates Pathological Deterioration in Experimental Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2021; 22(22):12533. https://doi.org/10.3390/ijms222212533
Chicago/Turabian StyleGómez-Almería, Marta, Sonia Burgaz, Carlos Costas-Insua, Carmen Rodríguez-Cueto, Irene Santos-García, Ignacio Rodríguez-Crespo, Concepción García, Manuel Guzmán, Eva de Lago, and Javier Fernández-Ruiz. 2021. "BiP Heterozigosity Aggravates Pathological Deterioration in Experimental Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 22, no. 22: 12533. https://doi.org/10.3390/ijms222212533
APA StyleGómez-Almería, M., Burgaz, S., Costas-Insua, C., Rodríguez-Cueto, C., Santos-García, I., Rodríguez-Crespo, I., García, C., Guzmán, M., de Lago, E., & Fernández-Ruiz, J. (2021). BiP Heterozigosity Aggravates Pathological Deterioration in Experimental Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 22(22), 12533. https://doi.org/10.3390/ijms222212533