
The Somatic Mutation Paradigm in Congenital Malformations: Hirschsprung Disease as a Model

 , , and
, , and 
Abstract
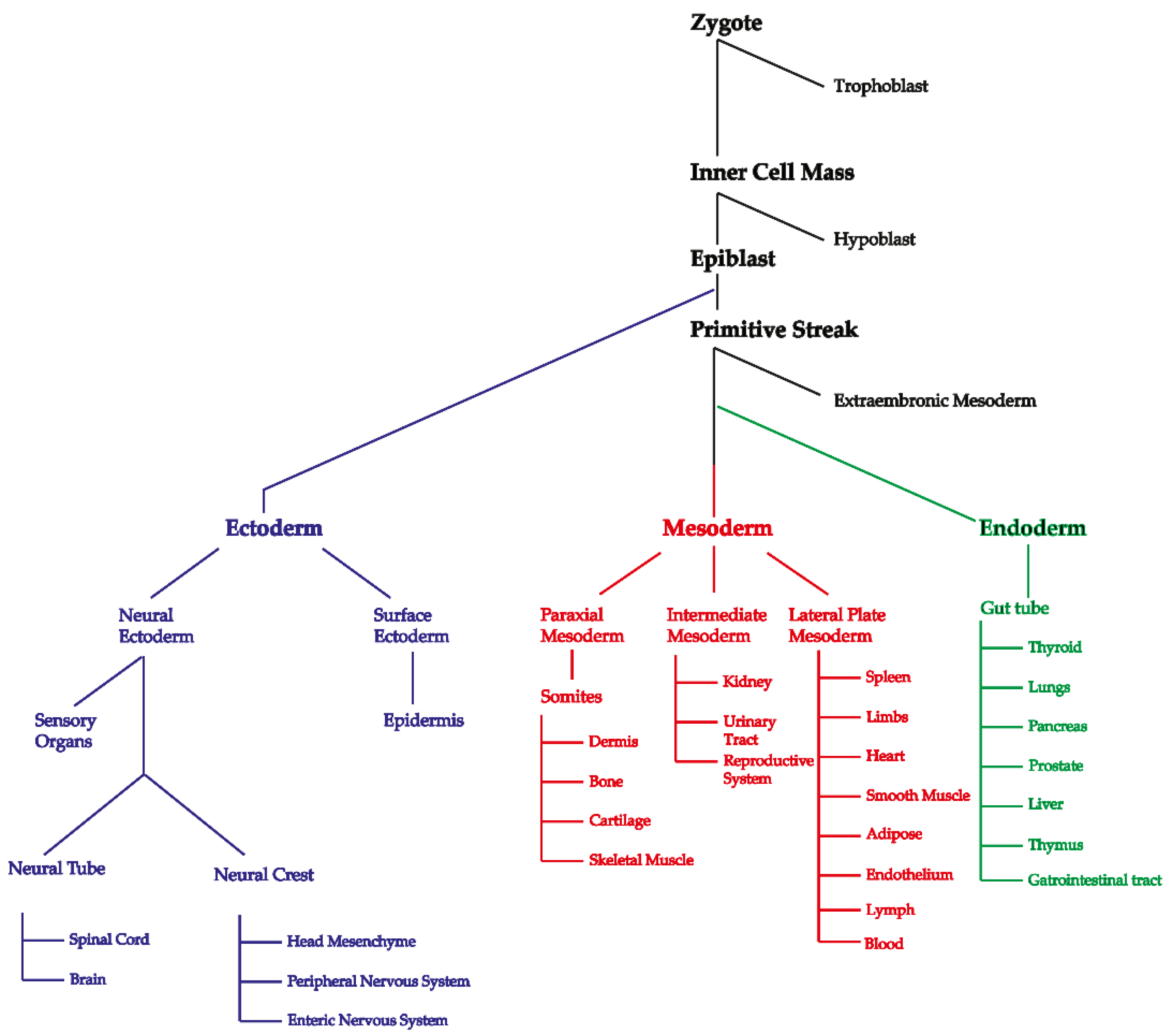
:1. Introduction
2. Results
2.1. Identification of Germline Mutations
2.2. Identification of Somatic Variants
2.3. Analysis of Somatic Copy Number Changes
3. Discussion
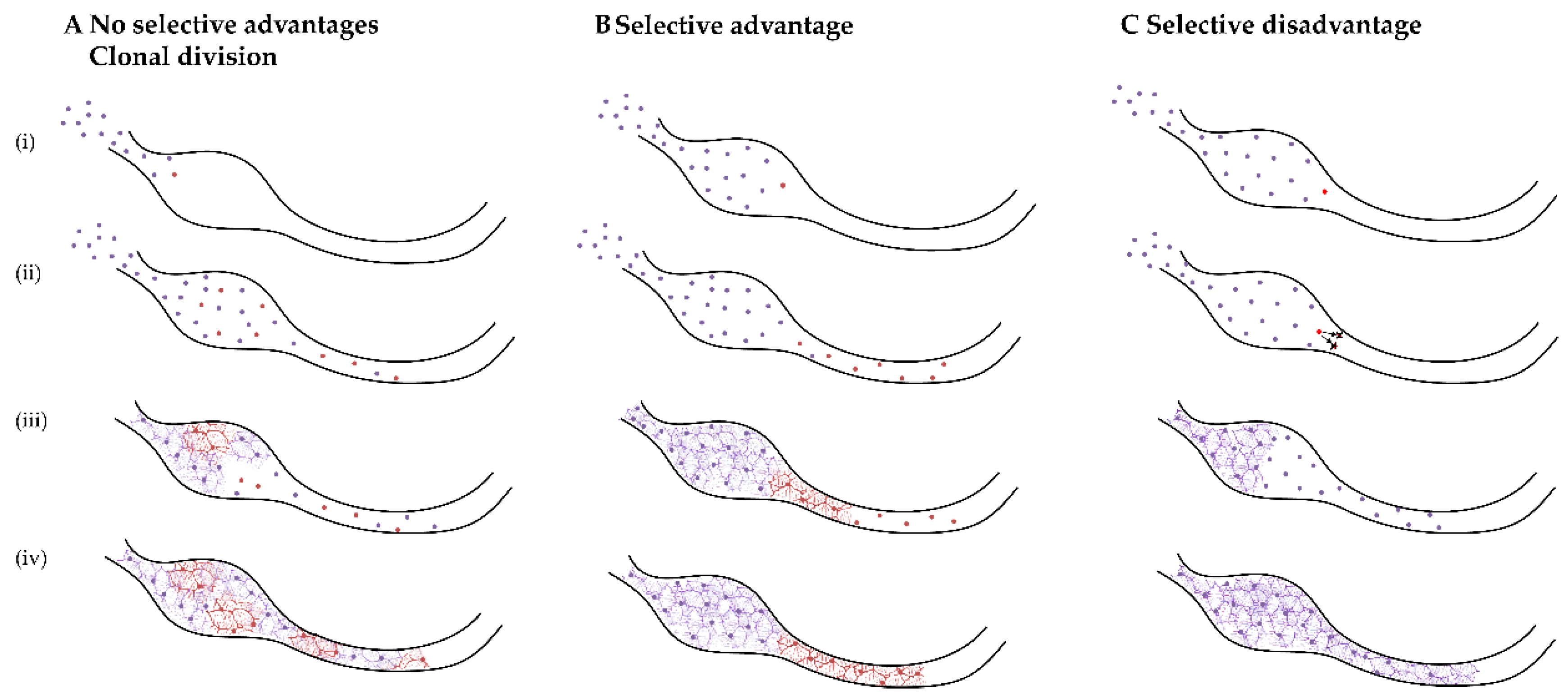
3.1. Detection of Somatic Mosaicism
3.2. ENS-Specific Somatic Changes in DNA Copy Number
3.3. Would ENCCs with a Somatic Variant Remain to Be Sampled?
4. Materials and Methods
4.1. Patients
4.2. Sample Collection
4.3. Cell Culture and Fluorescence-Activated Cell Sorting
4.4. Amplicon-Based WES
4.5. Data Analysis and Selection of Somatic Variants
4.6. Validation of Putative Mosaic Differences
4.7. Analysis of Somatic Copy Number Changes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dumitrescu, C.E.; Collins, M.T. Mccune-albright syndrome. Orphanet J. Rare Dis. 2008, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- O’Malley, M.P.; Haake, A.; Goldsmith, L.; Berg, D. Localized darier disease: Implications for genetic studies. Arch. Dermatol. 1997, 133, 1134–1138. [Google Scholar] [CrossRef] [PubMed]
- Vijg, J. Somatic mutations, genome mosaicism, cancer and aging. Curr. Opin. Genet. Dev. 2014, 26, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biesecker, L.G.; Spinner, N.B. A genomic view of mosaicism and human disease. Nat. Rev. Genet. 2013, 14, 307. [Google Scholar] [CrossRef] [PubMed]
- Erickson, R.P. Somatic gene mutation and human disease other than cancer: An update. Mutat. Res. Rev. Mutat. Res. 2010, 705, 96–106. [Google Scholar] [CrossRef]
- Youssoufian, H.; Pyeritz, R.E. Mechanisms and consequences of somatic mosaicism in humans. Nat. Rev. Genet. 2002, 3, 748. [Google Scholar] [CrossRef] [PubMed]
- Newgreen, D.F.; Zhang, D.; Cheeseman, B.L.; Binder, B.J.; Landman, K.A. Differential clonal expansion in an invading cell population: Clonal advantage or dumb luck? Cells Tissues Organs 2017, 203, 105–113. [Google Scholar] [CrossRef]
- Fernández, L.C.; Torres, M.; Real, F.X. Somatic mosaicism: On the road to cancer. Nat. Rev. Cancer 2016, 16, 43. [Google Scholar] [CrossRef]
- Amiel, J.; Sproat-Emison, E.; Garcia-Barcelo, M.; Lantieri, F.; Burzynski, G.; Borrego, S.; Pelet, A.; Arnold, S.; Miao, X.; Griseri, P.; et al. Hirschsprung disease, associated syndromes and genetics: A review. J. Med. Genet. 2008, 45, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Gui, H.; Schriemer, D.; Cheng, W.W.; Chauhan, R.K.; Antiňolo, G.; Berrios, C.; Bleda, M.; Brooks, A.S.; Brouwer, R.W.; Burns, A.J.; et al. Whole exome sequencing coupled with unbiased functional analysis reveals new hirschsprung disease genes. Genome Biol. 2017, 18, 48. [Google Scholar] [CrossRef] [Green Version]
- Emison, E.S.; Garcia-Barcelo, M.; Grice, E.A.; Lantieri, F.; Amiel, J.; Burzynski, G.; Fernandez, R.M.; Hao, L.; Kashuk, C.; West, K.; et al. Differential contributions of rare and common, coding and noncoding ret mutations to multifactorial hirschsprung disease liability. Am. J. Hum. Genet. 2010, 87, 60–74. [Google Scholar] [CrossRef] [Green Version]
- Hofstra, R.M.; Wu, Y.; Stulp, R.P.; Elfferich, P.; Osinga, J.; Maas, S.M.; Siderius, L.; Brooks, A.S.; vd Ende, J.J.; Heydendael, V.M.; et al. Ret and gdnf gene scanning in hirschsprung patients using two dual denaturing gel systems. Hum. Mutat. 2000, 15, 418–429. [Google Scholar] [CrossRef]
- Tang, C.S.; Gui, H.; Kapoor, A.; Kim, J.H.; Luzon-Toro, B.; Pelet, A.; Burzynski, G.; Lantieri, F.; So, M.T.; Berrios, C.; et al. Trans-ethnic meta-analysis of genome-wide association studies for hirschsprung disease. Hum. Mol. Genet. 2016, 25, 5265–5275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Kapoor, A.; Akiyama, J.A.; Auer, D.R.; Lee, D.; Gabriel, S.; Berrios, C.; Pennacchio, L.A.; Chakravarti, A. Enhancer variants synergistically drive dysfunction of a gene regulatory network in hirschsprung disease. Cell 2016, 167, 355–368.e10. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Jiang, Q.; Chatterjee, S.; Chakraborty, P.; Sosa, M.X.; Berrios, C.; Chakravarti, A. Population variation in total genetic risk of hirschsprung disease from common ret, sema3 and nrg1 susceptibility polymorphisms. Hum. Mol. Genet. 2015, 24, 2997–3003. [Google Scholar] [CrossRef] [Green Version]
- Alves, M.M.; Sribudiani, Y.; Brouwer, R.W.W.; Amiel, J.; Antiñolo, G.; Borrego, S.; Ceccherini, I.; Chakravarti, A.; Fernández, R.M.; Garcia-Barcelo, M.-M.; et al. Contribution of rare and common variants determine complex diseases—Hirschsprung disease as a model. Dev. Biol. 2013, 382, 320–329. [Google Scholar] [CrossRef]
- Sribudiani, Y.; Chauhan, R.K.; Alves, M.M.; Petrova, L.; Brosens, E.; Harrison, C.; Wabbersen, T.; de Graaf, B.M.; Rugenbrink, T.; Burzynski, G.; et al. Identification of variants in ret and ihh pathway members in a large family with history of hirschsprung disease. Gastroenterology 2018, 155, 118–129.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, S.W.; Zaahl, M.G. Tissue specific somatic mutations and aganglionosis in hirschsprung’s disease. J. Pediatr. Surg. 2014, 49, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jiang, Q.; Li, Q.; Cheng, W.; Qiao, G.; Xiao, P.; Gan, L.; Su, L.; Miao, C.; Li, L. Genotyping analysis of 3 ret polymorphisms demonstrates low somatic mutation rate in chinese hirschsprung disease patients. Int. J. Clin. Exp. Pathol. 2015, 8, 5528. [Google Scholar]
- Jiang, Q.; Liu, F.; Miao, C.; Li, Q.; Zhang, Z.; Xiao, P.; Su, L.; Yu, K.; Chen, X.; Zhang, F.; et al. Ret somatic mutations are underrecognized in hirschsprung disease. Genet. Med. 2018, 20, 770–777. [Google Scholar] [CrossRef]
- Brosens, E.; MacKenzie, K.C.; Alves, M.M.; Hofstra, R.M.W. Do ret somatic mutations play a role in hirschsprung disease? Genet. Med. 2018, 20, 1477–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theda, C.; Hwang, S.H.; Czajko, A.; Loke, Y.J.; Leong, P.; Craig, J.M. Quantitation of the cellular content of saliva and buccal swab samples. Sci. Rep. 2018, 8, 6944. [Google Scholar] [CrossRef]
- Schriemer, D.; Sribudiani, Y.; Ijpma, A.; Natarajan, D.; MacKenzie, K.C.; Metzger, M.; Binder, E.; Burns, A.J.; Thapar, N.; Hofstra, R.M.W. Regulators of gene expression in enteric neural crest cells are putative hirschsprung disease genes. Dev. Biol. 2016, 416, 255–265. [Google Scholar] [CrossRef]
- Conlin, L.K.; Thiel, B.D.; Bonnemann, C.G.; Medne, L.; Ernst, L.M.; Zackai, E.H.; Deardorff, M.A.; Krantz, I.D.; Hakonarson, H.; Spinner, N.B. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum. Mol. Genet. 2010, 19, 1263–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, B.; Beitel, L.K.; Alvarado, C.; Trifiro, M.A. Selection and mutation in the “new” genetics: An emerging hypothesis. Hum. Genet. 2010, 127, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.M.; Yuan, B.; Robberecht, C.; Pfundt, R.; Szafranski, P.; McEntagart, M.E.; Nagamani, S.C.; Erez, A.; Bartnik, M.; Wisniowiecka-Kowalnik, B.; et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am. J. Hum. Genet. 2014, 95, 173–182. [Google Scholar] [CrossRef] [Green Version]
- McConnell, M.J.; Lindberg, M.R.; Brennand, K.J.; Piper, J.C.; Voet, T.; Cowing-Zitron, C.; Shumilina, S.; Lasken, R.S.; Vermeesch, J.R.; Hall, I.M.; et al. Mosaic copy number variation in human neurons. Science 2013, 342, 632–637. [Google Scholar] [CrossRef] [Green Version]
- Villela, D.; Suemoto, C.K.; Leite, R.; Pasqualucci, C.A.; Grinberg, L.T.; Pearson, P.; Rosenberg, C. Increased DNA copy number variation mosaicism in elderly human brain. Neural Plast. 2018, 2018, 2406170. [Google Scholar] [CrossRef] [Green Version]
- Veenma, D.; Beurskens, N.; Douben, H.; Eussen, B.; Noomen, P.; Govaerts, L.; Grijseels, E.; Lequin, M.; de Krijger, R.; Tibboel, D.; et al. Comparable low-level mosaicism in affected and non affected tissue of a complex cdh patient. PLoS ONE 2010, 5, e15348. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.B.; Newgreen, D.F.; Young, H.M. Neural crest and the development of the enteric nervous system. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar] [CrossRef]
- Cheeseman, B.L.; Zhang, D.; Binder, B.J.; Newgreen, D.F.; Landman, K.A. Cell lineage tracing in the developing enteric nervous system: Superstars revealed by experiment and simulation. J. R. Soc. Interface 2014, 11, 20130815. [Google Scholar] [CrossRef]
- Nishiyama, C.; Uesaka, T.; Manabe, T.; Yonekura, Y.; Nagasawa, T.; Newgreen, D.F.; Young, H.M.; Enomoto, H. Trans-mesenteric neural crest cells are the principal source of the colonic enteric nervous system. Nat. Neurosci. 2012, 15, 1211. [Google Scholar] [CrossRef]
- Lindhurst, M.J.; Parker, V.E.R.; Payne, F.; Sapp, J.C.; Rudge, S.; Harris, J.; Witkowski, A.M.; Zhang, Q.; Groeneveld, M.P.; Scott, C.E.; et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in pik3ca. Nat. Genet. 2012, 44, 928. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.J.; Champeval, D.; Le Douarin, N.M. Sacral neural crest cells colonise aganglionic hindgut in vivo but fail to compensate for lack of enteric ganglia. Dev. Biol. 2000, 219, 30–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzger, M.; Caldwell, C.; Barlow, A.J.; Burns, A.J.; Thapar, N. Enteric nervous system stem cells derived from human gut mucosa for the treatment of aganglionic gut disorders. Gastroenterology 2009, 136, 2214–2225.e3. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, R.W.W.; van den Hout, M.; Kockx, C.E.M.; Brosens, E.; Eussen, B.; de Klein, A.; Sleutels, F.; van, I.W.F.J. Nimbus: A design-driven analyses suite for amplicon based ngs data. Bioinformatics 2018, 34, 2732–2739. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Jian, X.; Liu, X. In silico prediction of deleteriousness for nonsynonymous and splice-altering single nucleotide variants in the human genome. Methods Mol. Biol. 2017, 1498, 191–197. [Google Scholar]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Carter, H.; Douville, C.; Stenson, P.D.; Cooper, D.N.; Karchin, R. Identifying mendelian disease genes with the variant effect scoring tool. BMC Genom. 2013, 14 (Suppl. S3), S3. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. Mutationtaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Quang, D.; Chen, Y.; Xie, X. Dann: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous snvs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shihab, H.A.; Rogers, M.F.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.; Gaunt, T.R.; Campbell, C. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 2015, 31, 1536–1543. [Google Scholar] [CrossRef] [Green Version]
- Douville, C.; Masica, D.L.; Stenson, P.D.; Cooper, D.N.; Gygax, D.M.; Kim, R.; Ryan, M.; Karchin, R. Assessing the pathogenicity of insertion and deletion variants with the variant effect scoring tool (vest-indel). Hum. Mutat. 2016, 37, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. Dbnsfp v3.0: A one-stop database of functional predictions and annotations for human nonsynonymous and splice-site snvs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Memic, F.; Knoflach, V.; Morarach, K.; Sadler, R.; Laranjeira, C.; Hjerling-Leffler, J.; Sundström, E.; Pachnis, V.; Marklund, U. Transcription and signaling regulators in developing neuronal subtypes of mouse and human enteric nervous system. Gastroenterology 2018, 154, 624–636. [Google Scholar] [CrossRef] [Green Version]
- McCann, C.J.; Alves, M.M.; Brosens, E.; Natarajan, D.; Perin, S.; Chapman, C.; Hofstra, R.M.; Burns, A.J.; Thapar, N. Neuronal development and onset of electrical activity in the human enteric nervous system. Gastroenterology 2019, 156, 1483–1495.e6. [Google Scholar] [CrossRef] [Green Version]
- Kuil, L.E.; MacKenzie, K.C.; Tang, C.S.; Windster, J.D.; Le, T.L.; Karim, A.; de Graaf, B.M.; van der Helm, R.; van Bever, Y.; Sloots, C.E.J.; et al. Size matters: Large copy number losses in hirschsprung disease patients reveal genes involved in enteric nervous system development. PLoS Genet. 2021, 17, e1009698. [Google Scholar] [CrossRef]
- Brosens, E.; Marsch, F.; de Jong, E.M.; Zaveri, H.P.; Hilger, A.C.; Choinitzki, V.G.; Holscher, A.; Hoffmann, P.; Herms, S.; Boemers, T.M.; et al. Copy number variations in 375 patients with oesophageal atresia and/or tracheoesophageal fistula. Eur. J. Hum. Genet. 2016, 24, 1715–1723. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, E.; Biezuner, T.; Linnarsson, S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 2013, 14, 618. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Patient | ENCC Only | Blood Only | PPAE | PPAB | VAPE | Validated @ |
|---|---|---|---|---|---|---|
| 1 | 50 | 43 | 8 | 11 | 5 | 0 |
| 2 | 16 | 28 | 2 | 4 | 1 | 0 |
| 3 | 25 | 33 | 0 | 1 | 0 | 0 |
| 4 # | 96 | 178 | 17 | 11 | 15 | 0 $$ |
| 5 | 29 | 35 | 2 | 1 | 2 | 0 |
| Patient | Gene | cDNA | Type | dbSNP | Class | GnomADe | GnomADg | MisZ | pLI | FE | ME |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | FMN2 | c.162delC | FD | VUS | 0.000000 | 0.000000 | 1.42 | 0.99 | no | yes | |
| 1 | YWHAE | c.G142A | M | VUS | 0.000000 | 0.000000 | 3.25 | 0.96 | yes | na | |
| 1 | YWHAE | c.T116C | M | VUS | 0.000000 | 0.000000 | 3.25 | 0.96 | yes | na | |
| 1 | PHAX | c.C379T | PS | VUS | 0.000000 | 0.000000 | –0.51 | 0.00 | yes | na | |
| 1 | POR | c.T1231C | M | VUS | 0.000000 | 0.000000 | −0.54 | 0.00 | yes | na | |
| 2 | DEPDC1 | c.T1459A | M | VUS | 0.000000 | 0.000000 | −0.32 | 0.00 | yes | na | |
| 4 | F5 | c.A1867G | M | VUS | 0.000000 | 0.000000 | −1.30 | 0.00 | no | na | |
| 4 | PHRF1 | c.G1075A | M | rs551874512 | VUS | 0.000033 | 0.000032 | −1.36 | 0.95 | yes | na |
| 4 | MYBPC3 | c.C482A | M | VUS | 0.000000 | 0.000000 | 0.69 | 0.00 | no | na | |
| 4 | PACS1 | c.G1069A | M | rs750459659 | VUS | 0.000041 | 0.000032 | 4.32 | 1.00 | yes | na |
| 4 | OAS3 | c.C1390T | M | rs750291946 | VUS | 0.000012 | 0.000000 | −0.60 | 0.00 | yes | na |
| 4 | MAN2A2 | c.G478A | M | rs374688808 | VUS | 0.000012 | 0.000032 | 1.28 | 0.00 | yes | yes |
| 4 | SNF8 | c.G578A | M | rs775611332 | VUS | 0.000025 | 0.000000 | 0.97 | 0.29 | yes | yes |
| 4 | MED15 | c.C730A | M | VUS | 0.000000 | 0.000000 | 2.50 | 0.96 | yes | na | |
| 4 | IQCF5 | c.C283T | M | rs772101978 | VUS | 0.000100 | 0.000000 | −1.59 | 0.43 | no | na |
| 4 | TMEM165 | c.C782A | M | VUS | 0.000000 | 0.000000 | 1.83 | 0.94 | yes | na | |
| 4 | NOTCH4 | c.G1118A | M | rs745883985 | VUS | 0.000033 | 0.000032 | 2.45 | 0.00 | yes | na |
| 4 | DPPA5 | c.G214A | M | VUS | 0.000000 | 0.000000 | 1.64 | 0.00 | no | na | |
| 4 | SLC22A1 | c.C523T | M | rs768905186 | VUS | 0.000004 | 0.000000 | −0.28 | 0.00 | no | na |
| 4 | MGAM2 | c.G3015T | M | VUS | 0.000000 | 0.000000 | na | na | |||
| 4 | IKBKB | c.G809A | M | rs200841053 | VUS | 0.000024 | 0.000032 | 2.90 | 1.00 | yes | na |
| 5 | PCDH15 | c.G139A | M | VUS | 0.000000 | 0.000000 | −3.27 | 0.00 | no | yes | |
| 5 | ZNF592 | c.C3433A | M | VUS | 0.000000 | 0.000000 | 1.10 | 0.95 | yes | na |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MacKenzie, K.C.; Garritsen, R.; Chauhan, R.K.; Sribudiani, Y.; de Graaf, B.M.; Rugenbrink, T.; Brouwer, R.; van Ijcken, W.F.J.; de Blaauw, I.; Brooks, A.S.; et al. The Somatic Mutation Paradigm in Congenital Malformations: Hirschsprung Disease as a Model. Int. J. Mol. Sci. 2021, 22, 12354. https://doi.org/10.3390/ijms222212354
MacKenzie KC, Garritsen R, Chauhan RK, Sribudiani Y, de Graaf BM, Rugenbrink T, Brouwer R, van Ijcken WFJ, de Blaauw I, Brooks AS, et al. The Somatic Mutation Paradigm in Congenital Malformations: Hirschsprung Disease as a Model. International Journal of Molecular Sciences. 2021; 22(22):12354. https://doi.org/10.3390/ijms222212354
Chicago/Turabian StyleMacKenzie, Katherine C., Rhiana Garritsen, Rajendra K. Chauhan, Yunia Sribudiani, Bianca M. de Graaf, Tim Rugenbrink, Rutger Brouwer, Wilfred F. J. van Ijcken, Ivo de Blaauw, Alice S. Brooks, and et al. 2021. "The Somatic Mutation Paradigm in Congenital Malformations: Hirschsprung Disease as a Model" International Journal of Molecular Sciences 22, no. 22: 12354. https://doi.org/10.3390/ijms222212354
APA StyleMacKenzie, K. C., Garritsen, R., Chauhan, R. K., Sribudiani, Y., de Graaf, B. M., Rugenbrink, T., Brouwer, R., van Ijcken, W. F. J., de Blaauw, I., Brooks, A. S., Sloots, C. E. J., Meeuwsen, C. J. H. M., Wijnen, R. M., Newgreen, D. F., Burns, A. J., Hofstra, R. M. W., Alves, M. M., & Brosens, E. (2021). The Somatic Mutation Paradigm in Congenital Malformations: Hirschsprung Disease as a Model. International Journal of Molecular Sciences, 22(22), 12354. https://doi.org/10.3390/ijms222212354