
Τhe Greek Variant in APP Gene: The Phenotypic Spectrum of APP Mutations

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Case Report
1.2. Genetic Analysis
2. Methods
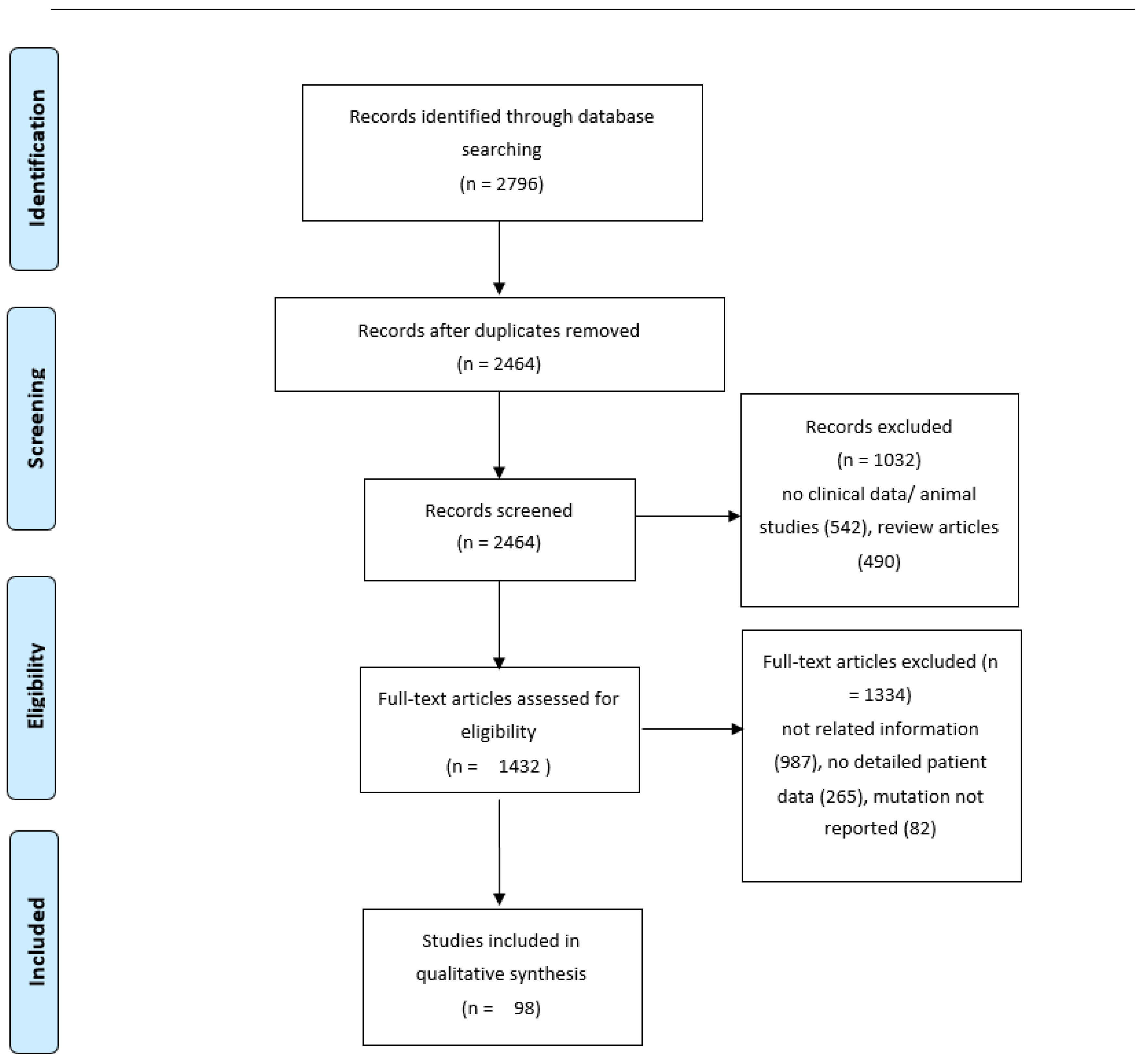
2.1. Review of the Literature
2.2. Statistical Analysis
3. Results
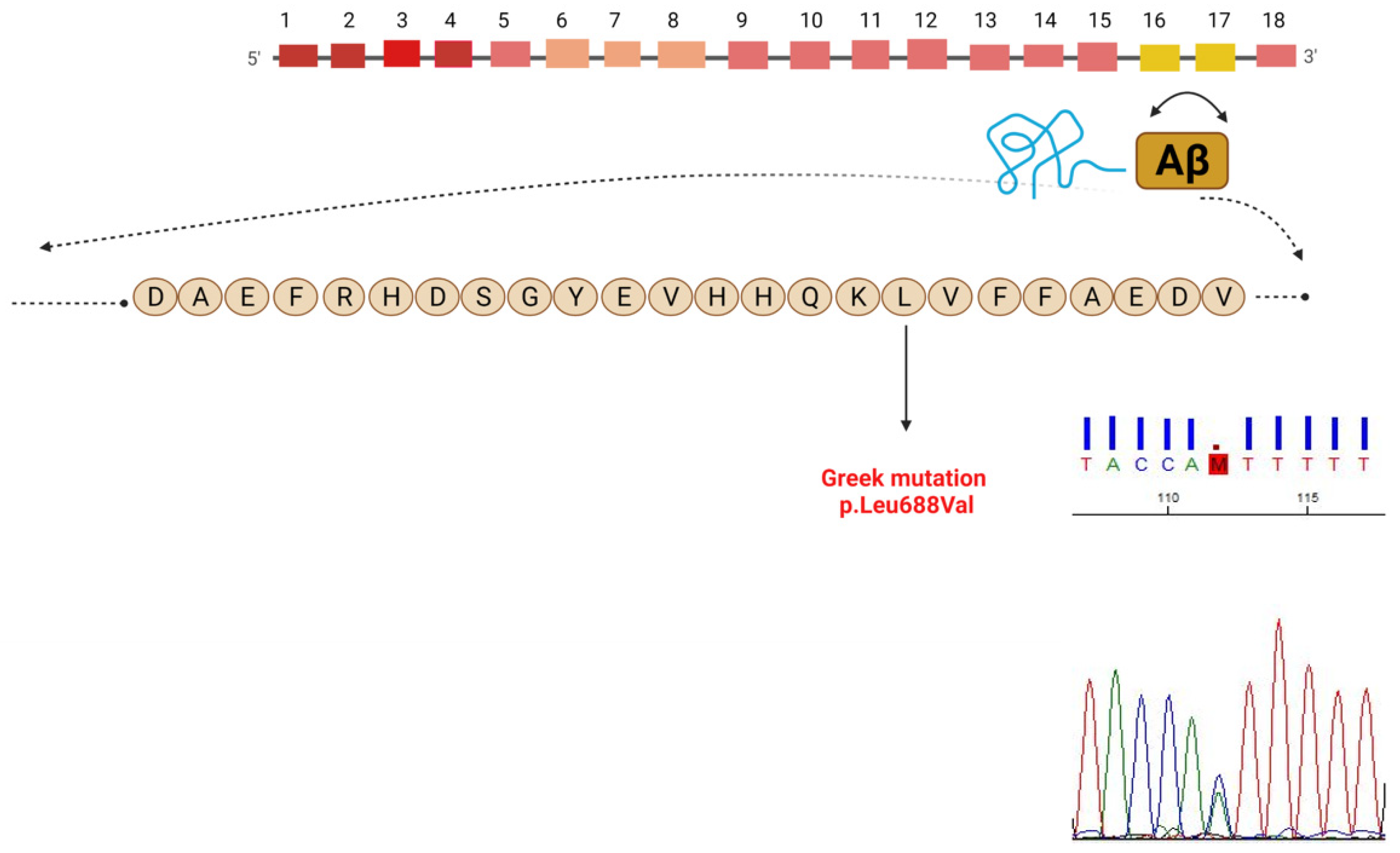
3.1. Genetic Analysis
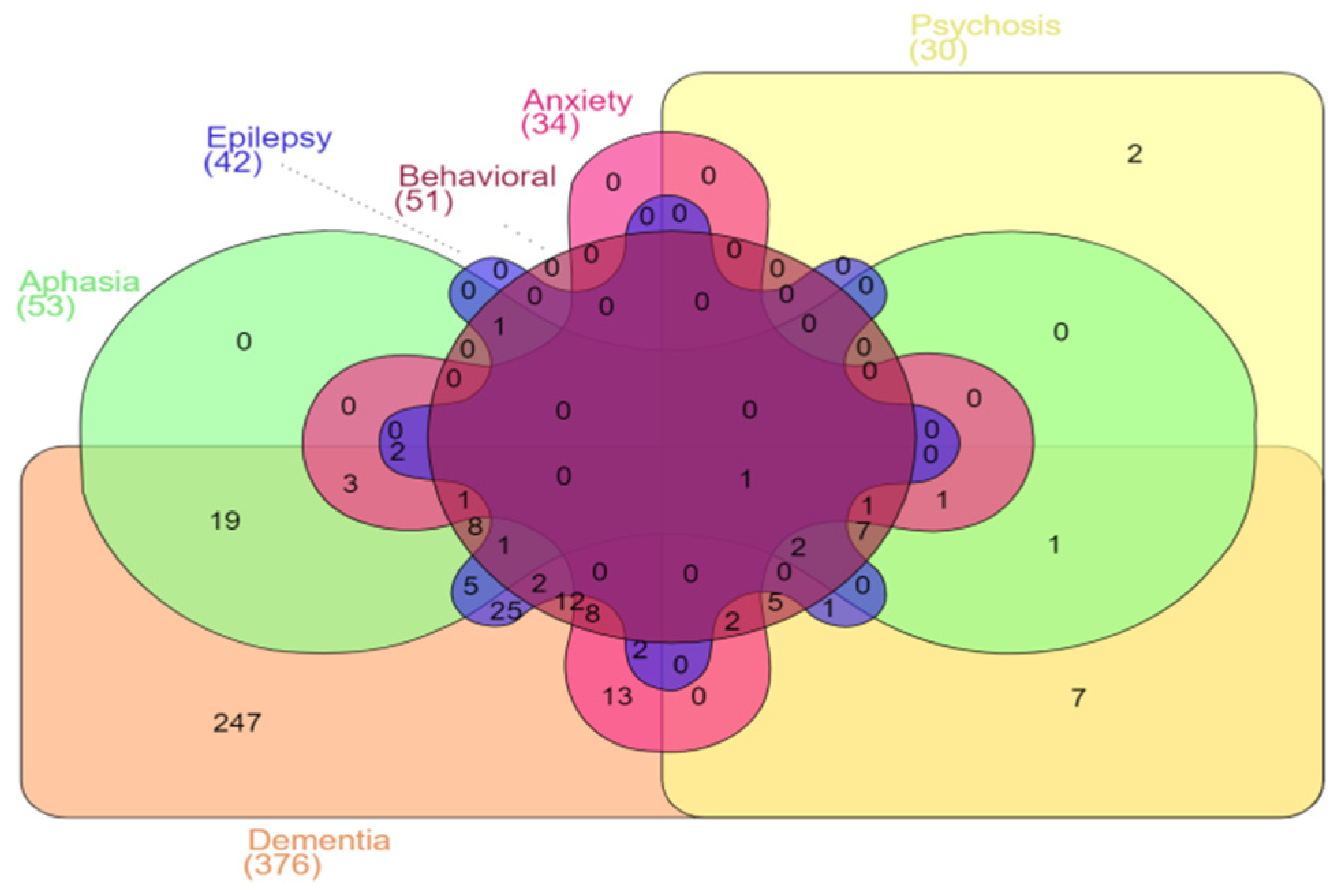
3.2. Demographics and Clinical Phenotype
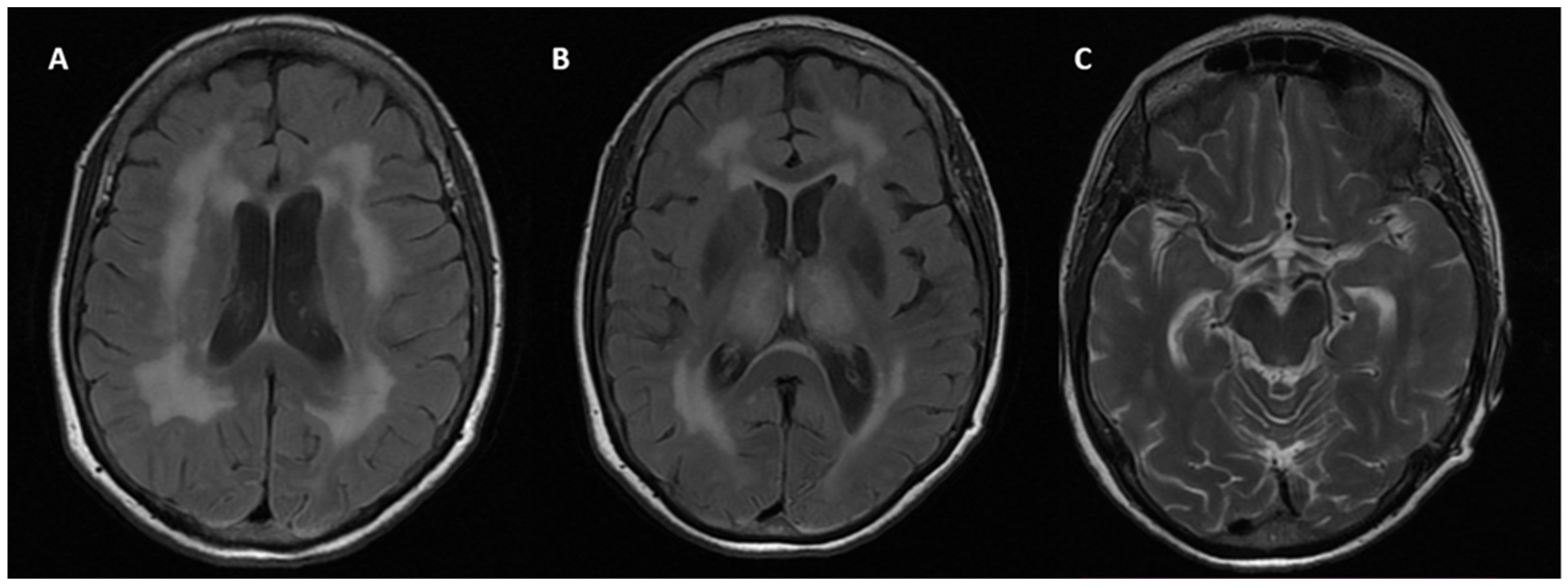
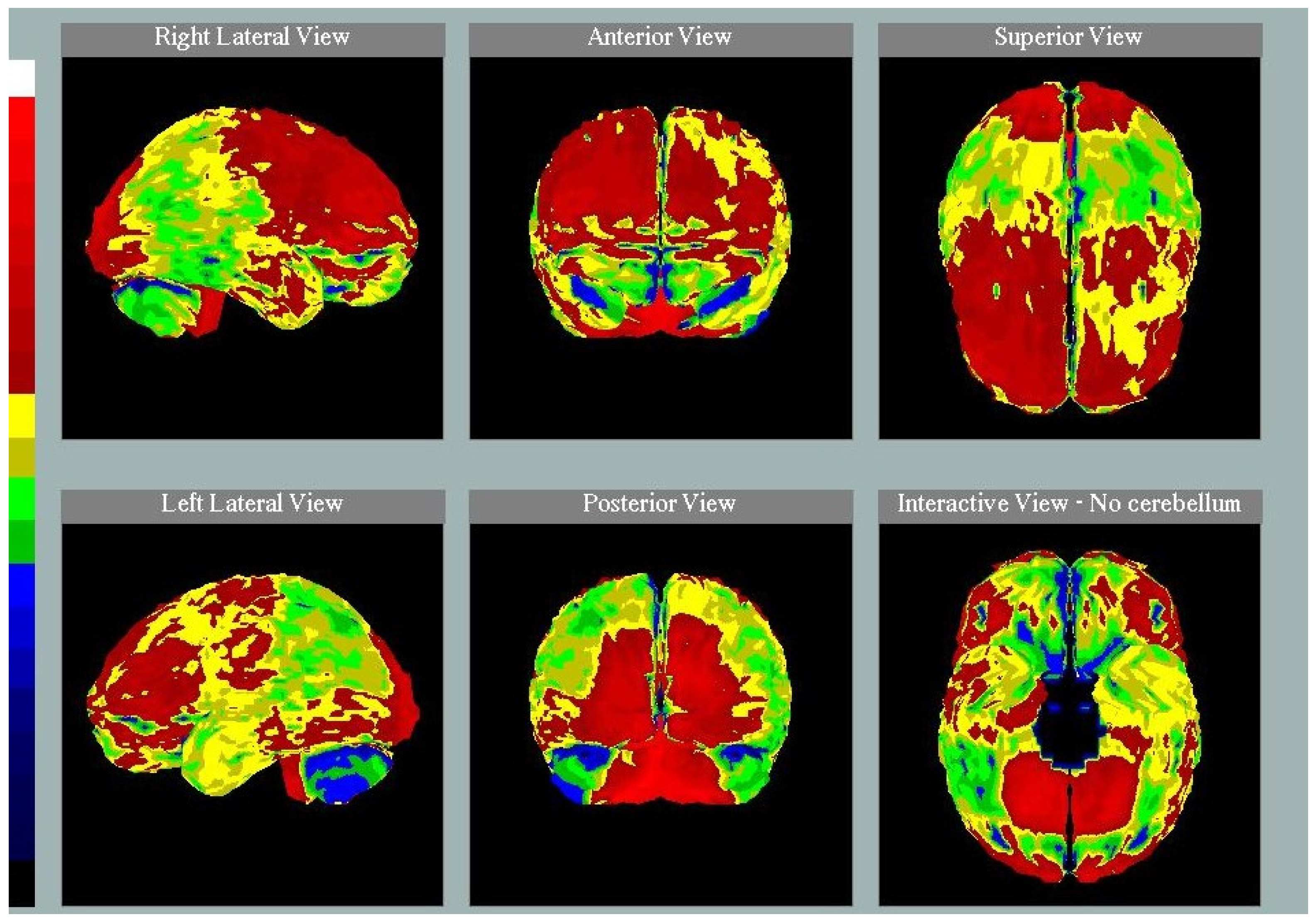
3.3. Neuroimaging
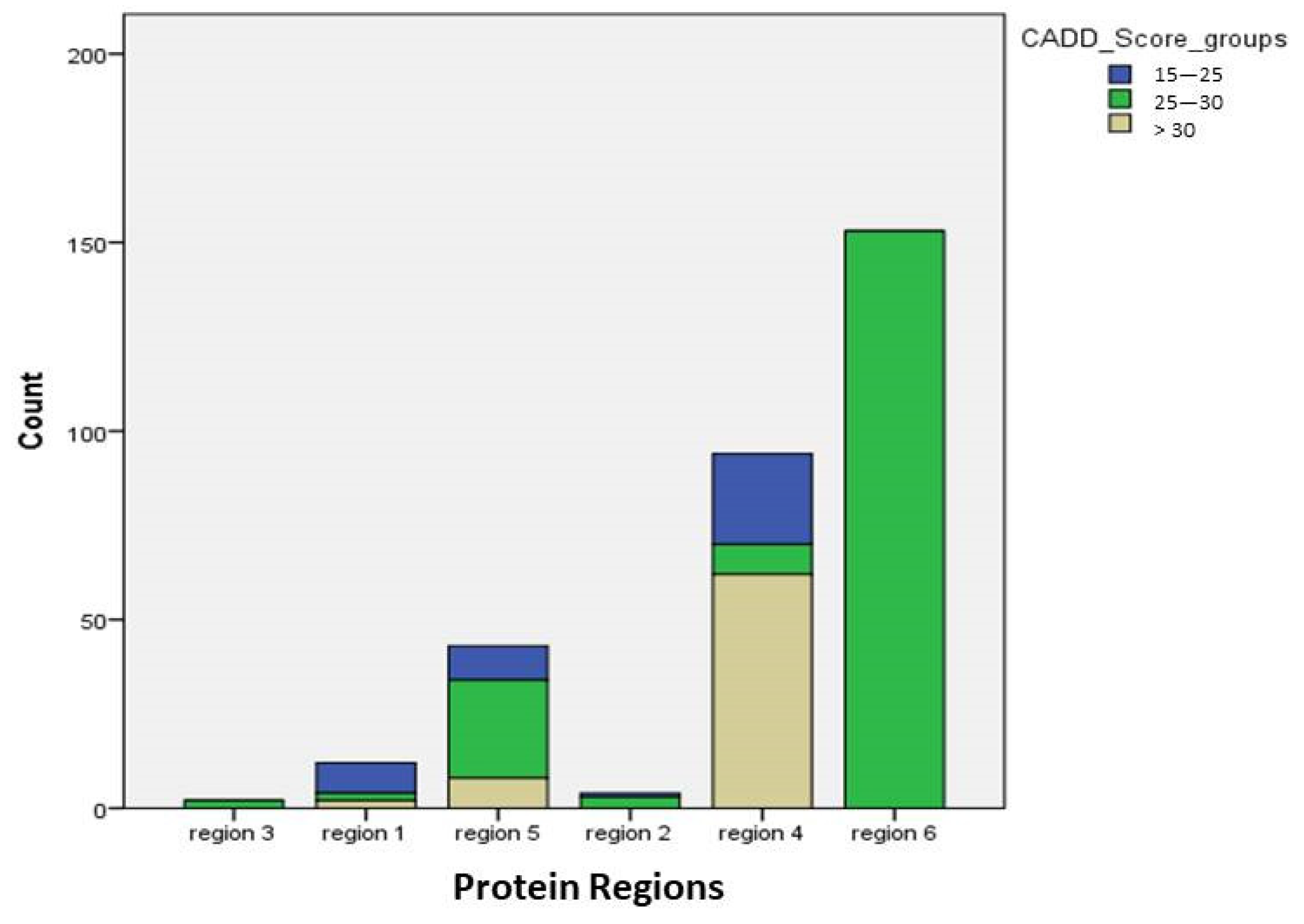
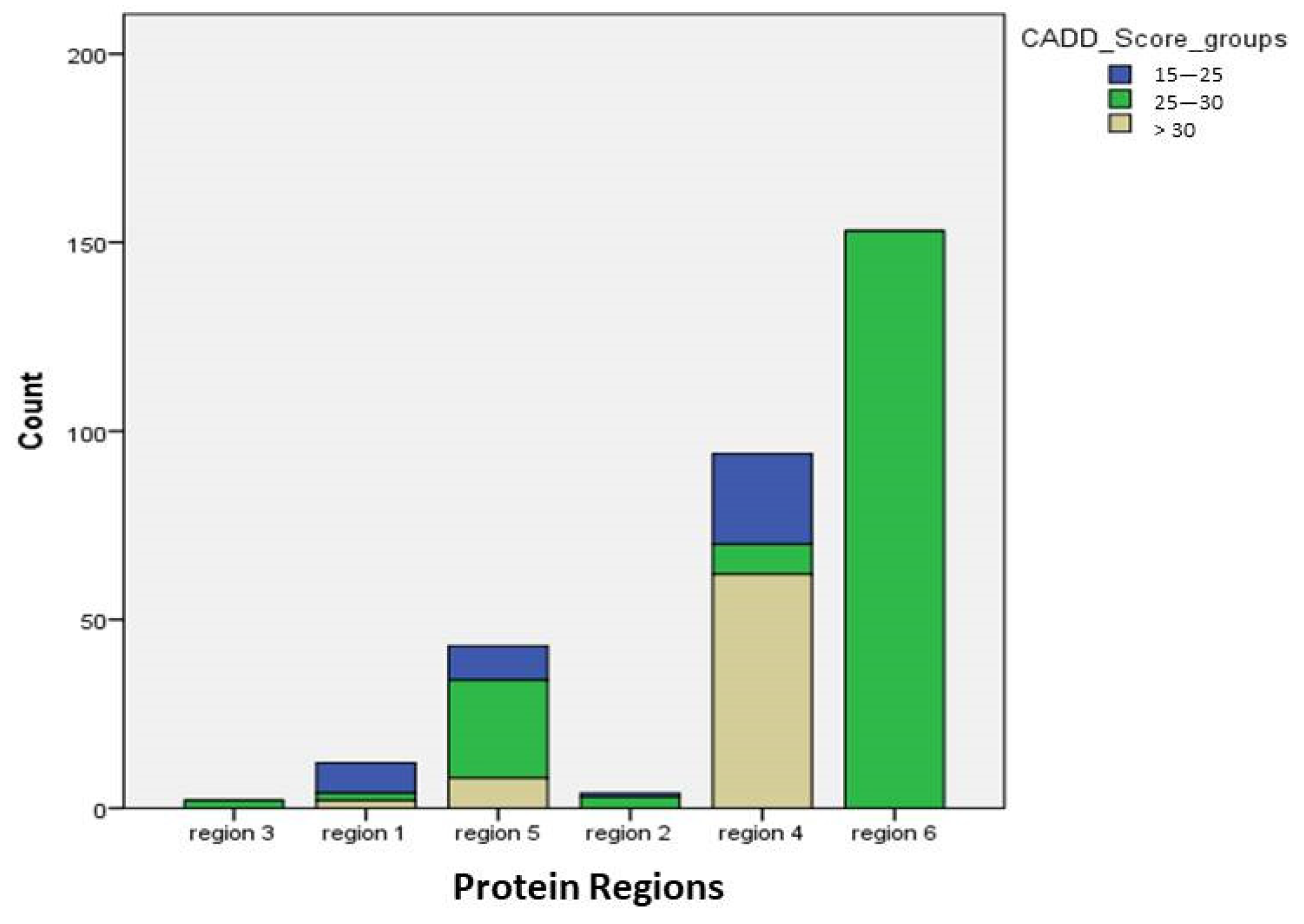
3.4. Mutations
4. Discussion
4.1. Clinical Spectrum of APP Mutations: Age of Onset and Symptoms
4.2. Mutations in Amyloid Precursor Protein (APP) Gene: Location and Pathogenicity
4.3. Phenotypic Variability of APP Mutations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. 2015, 1, 15056. [Google Scholar] [CrossRef]
- Cacace, R.; Sleegers, K.; Van, B.C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimer Dement. J. Alzheimer Assoc. 2016, 12, 733–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shea, Y.F.; Chu, L.W.; Chan, A.O.; Ha, J.; Li, Y.; Song, Y.Q. A systematic review of familial Alzheimer’s disease: Differences in presentation of clinical features among three mutated genes and potential ethnic differences. J. Formos. Med. Assoc. Taiwan Yi Zhi 2016, 115, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Rosa-Neto, P.; Hsiung, G.Y.; Sadovnick, A.D.; Masellis, M.; Black, S.E.; Jia, J. Gauthier SEarly-onset familial Alzheimer’s disease (EOFAD). Can. J. Neurol. Sci. 2012, 39, 436–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilotto, A.; Padovani, A.; Borroni, B. Clinical, biological, and imaging features of monogenic Alzheimer’s Disease. Biomed Res. Int. 2013, 2013, 689591. [Google Scholar] [CrossRef] [Green Version]
- Ringman, J.M.; Goate, A.; Masters, C.L.; Cairns, N.J.; Danek, A.; Graff-Radford, N.; Ghetti, B.; Morris, J.C. Genetic heterogeneity in Alzheimer disease and implications for treatment strategies. Curr. Neurol. Neurosci. 2014, 14, 499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tcw, J.; Goate, A.M. Genetics of β-Amyloid Precursor Protein in Alzheimer’s Disease. Csh. Perspect. Med. 2017, 7, a024539. [Google Scholar] [CrossRef] [PubMed]
- Giri, M.; Zhang, M.; Lü, Y. Genes associated with Alzheimer’s disease: An overview and current status. Clin. Interv. Aging 2016, 11, 665–681. [Google Scholar] [CrossRef] [Green Version]
- Carmona, S.; Hardy, J.; Guerreiro, R. The genetic landscape of Alzheimer disease. Handb. Clin. Neurol. 2018, 148, 395–408. [Google Scholar]
- Hardy, J. The discovery of Alzheimer-causing mutations in the APP gene and the formulation of the “amyloid cascade hypothesis”. FEBS J. 2017, 284, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Talbot, P.R.; Lloyd, J.J.; Snowden, J.S.; Neary, D.; Testa, H.J. Choice of reference region in the quantification of single-photon emission tomography in primary degenerative dementia. Eur. J. Nucl. Med. 1994, 21, 503–508. [Google Scholar] [CrossRef]
- Valotassiou, V.; Papatriantafyllou, J.; Sifakis, N.; Tzavara, C.; Tsougos, I.; Psimadas, D.; Fezoulidis, I.; Kapsalaki, E.; Hadjigeorgiou, G.; Georgoulias, P. Clinical Evaluation of Brain Perfusion SPECT with Brodmann Areas Mapping in Early Diagnosis of Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 47, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Witten, D.; Cooper, G.J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Psychogios, Κ.; Xiromerisiou, G.; Kargiotis, O.; Safouris, A.; Fiolaki, A.; Bonakis, A.; Paraskevas, G.; Giannopoulos, S.; Tsivgoulis, G. Hereditary cerebral amyloid angiopathy mimicking CADASIL syndrome. Eur. J. Neurol. 2021, 28, 3866–3869. [Google Scholar] [CrossRef] [PubMed]
- Ryman, D.C.; Acosta-Baena, N.; Aisen, P.S.; Bird, T.; Danek, A.; Fox, N.C.; Goate, A.; Frommelt, P.; Ghetti, B.; Langbaum, J.B.S.; et al. Symptom onset in autosomal dominant Alzheimer disease: A systematic review and meta-analysis. Neurology 2014, 83, 253–260. [Google Scholar] [CrossRef]
- Ryan, N.S.; Rossor, M.N. Correlating familial Alzheimer’s disease gene mutations with clinical phenotype. Biomark. Med. 2010, 4, 99–112. [Google Scholar] [CrossRef] [Green Version]
- Thajeb, P.; Wang, P.; Chien, C.L.; Harrigan, R. Novel polymorphisms of the amyloid precursor protein (APP) gene in Chinese/Taiwanese patients with Alzheimer’s disease. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2009, 16, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Talarico, G.; Piscopo, P.; Gasparini, M.; Salati, E.; Pignatelli, M.; Pietracupa, S.; Malvezzi-Campeggi, L.; Crestini, A.; Boschi, S.; Lenzi, G.L.; et al. The London APP mutation (Val717Ile) associated with early shifting abilities and behavioral changes in two Italian families with early-onset Alzheimer’s disease. Dement. Geriatr. Cogn. 2010, 29, 484–490. [Google Scholar] [CrossRef]
- Rosenberg, C.K.; Pericak-Vance, M.A.; Saunders, A.M.; Gilbert, J.R.; Gaskell, P.C.; Hulette, C.M. Lewy body and Alzheimer pathology in a family with the amyloid-beta precursor protein APP717 gene mutation. Acta Neuropathol. 2000, 100, 145–152. [Google Scholar] [CrossRef]
- Zhang, G.; Xie, Y.; Wang, W.; Feng, X.; Jia, J. Clinical characterization of an APP mutation (V717I) in five Han Chinese families with early-onset Alzheimer’s disease. J. Neurol. Sci. 2017, 372, 379–386. [Google Scholar] [CrossRef]
- Guerreiro, R.J.; Baquero, M.; Blesa, R.; Boada, M.; Brás, J.M.; Bullido, M.J.; Calado, A.; Crook, R.; Ferreira, C.; Frank, A.; et al. Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol. Aging 2010, 31, 725–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suárez-Calvet, M.; Belbin, O.; Pera, M.; Badiola, N.; Magrané, J.; Guardia-Laguarta, C.; Muñoz, L.; Colom-Cadena, M.; Clarimón, J.; Lleó, A. Autosomal-dominant Alzheimer’s disease mutations at the same codon of amyloid precursor protein differentially alter Aβ production. J. Neurochem. 2014, 128, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Guardia-Laguarta, C.; Pera, M.; Clarimón, J.; Molinuevo, J.L.; Sánchez-Valle, R.; Lladó, A.; Coma, M.; Gómez-Isla, T.; Blesa, R.; Ferrer, I.; et al. Clinical, neuropathologic, and biochemical profile of the amyloid precursor protein I716F mutation. J. Neuropath. Exp. Neur. 2010, 69, 53–59. [Google Scholar] [CrossRef]
- Warrington, E.K.; Agnew, S.K.; Kennedy, A.M.; Rossor, M.N. Neuropsychological profiles of familial Alzheimer’s disease associated with mutations in the presenilin 1 and amyloid precursor protein genes. J. Neurol. 2001, 248, 45–50. [Google Scholar] [CrossRef]
- Qin, Q.; Yin, Y.; Wang, Y.; Lu, Y.; Tang, Y.; Jia, J. Gene mutations associated with early onset familial Alzheimer’s disease in China: An overview and current status. Mol. Genet. Genom. Ed. 2020, 8, e1443. [Google Scholar] [CrossRef]
- Minkeviciene, R.; Rheims, S.; Dobszay, M.B.; Zilberter, M.; Hartikainen, J.; Fülöp, L.; Penke, B.; Zilberter, Y.; Harkany, T.; Pitkänen, A.; et al. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 3453–3462. [Google Scholar] [CrossRef] [PubMed]
- McNaughton, D.; Knight, W.; Guerreiro, R.; Ryan, N.; Lowe, J.; Poulter, M.; Nicholl, D.J.; Hardy, J.; Revesz, T.; Lowe, J.; et al. Duplication of amyloid precursor protein (APP), but not prion protein (PRNP) gene is a significant cause of early onset dementia in a large UK series. Neurobiol. Aging 2012, 33, e413–e421. [Google Scholar] [CrossRef] [Green Version]
- Kasuga, K.; Shimohata, T.; Nishimura, A.; Shiga, A.; Mizuguchi, T.; Tokunaga, J.; Ohno, T.; Miyashita, A.; Kuwano, R.; Matsumoto, N.; et al. Identification of independent APP locus duplication in Japanese patients with early-onset Alzheimer disease. J. Neurol. Neurosur. Psychiatry 2009, 80, 1050–1052. [Google Scholar] [CrossRef] [PubMed]
- Guyant-Marechal, I.; Berger, E.; Laquerrière, A.; Rovelet-Lecrux, A.; Viennet, G.; Frebourg, T.; Rumbach, L.; Campion, D.; Hannequin, D. Intrafamilial diversity of phenotype associated with app duplication. Neurology 2008, 71, 1925–1926. [Google Scholar] [CrossRef]
- Cabrejo, L.; Guyant-Maréchal, L.; Laquerrière, A.; Vercelletto, M.; De-la, F.F.; Thomas-Antérion, C.; Verny, C.; Letournel, F.; Pasquier, F.; Vital, A.; et al. Phenotype associated with APP duplication in five families. Brain A J. Neurol. 2006, 129, 2966–2976. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, T.J.; Cho, H.S.; Vonsattel, J.P.; Rebeck, G.W.; Greenberg, S.M. Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann. Neurol. 2001, 49, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Bagyinszky, E.; Kang, M.J.; Van, G.V.; Shim, K.; Pyun, J.M.; Suh, J.; An, S.S.; Kim, S.Y. Novel amyloid precursor protein mutation, Val669Leu (“Seoul APP”), in a Korean patient with early-onset Alzheimer’s disease. Neurobiol. Aging 2019, 84, e231–e236. [Google Scholar] [CrossRef] [PubMed]
- Basun, H.; Bogdanovic, N.; Ingelsson, M.; Almkvist, O.; Näslund, J.; Axelman, K.; Bird, T.D.; Nochlin, D.; Schellenberg, G.D.; Wahlund, L.-O.; et al. Clinical and neuropathological features of the arctic APP gene mutation causing early-onset Alzheimer disease. Arch. Neurol. 2008, 65, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Brooks, W.S.; Kwok, J.B.; Halliday, G.M.; Godbolt, A.K.; Rossor, M.N.; Creasey, H.; Jones, A.O.; Schofield, P.R. Hemorrhage is uncommon in new Alzheimer family with Flemish amyloid precursor protein mutation. Neurology 2004, 63, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Giaccone, G.; Maletta, R.; Morbin, M.; Capobianco, R.; Mangieri, M.; Giovagnoli, A.R.; Bizzi, A.; Tomaino, C.; Perri, M.; et al. A family with Alzheimer disease and strokes associated with A713T mutation of the APP gene. Neurology 2004, 63, 910–912. [Google Scholar] [CrossRef] [PubMed]
- Wakutani, Y.; Watanabe, K.; Adachi, Y.; Wada-Isoe, K.; Urakami, K.; Ninomiya, H.; Saido, T.C.; Hashimoto, T.; Iwatsubo, T.; Nakashima, K.; et al. Novel amyloid precursor protein gene missense mutation (D678N) in probable familial Alzheimer’s disease. J. Neurol. Neurosur. Psychiatry 2004, 75, 1039–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, Y.; Kitamura, E.; Miyoshi, K.; Yamamoto, Y.; Furuyama, J.; Sugihara, T. Japanese siblings with missense mutation (717Val right arrow Ile) in amyloid precursor protein of early-onset Alzheimer’s disease. Neurology 1996, 46, 1721–1723. [Google Scholar] [CrossRef]
- Conidi, M.E.; Bernardi, L.; Puccio, G.; Smirne, N.; Muraca, M.G.; Curcio, S.A.; Colao, R.; Piscopo, P.; Gallo, M.; Anfossi, M.; et al. Homozygous carriers of APP A713T mutation in an autosomal dominant Alzheimer disease family. Neurology 2015, 84, 2266–2273. [Google Scholar] [CrossRef] [Green Version]
- Di, F.G.; Catania, M.; Morbin, M.; Rossi, G.; Suardi, S.; Mazzoleni, G.; Merlin, M.; Giovagnoli, A.R.; Prioni, S.; Erbetta, A.; et al. A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 2009, 323, 1473–1477. [Google Scholar]
- Tomiyama, T.; Nagata, T.; Shimada, H.; Teraoka, R.; Fukushima, A.; Kanemitsu, H.; Takuma, H.; Kuwano, R.; Imagawa, M.; Ataka, S.; et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann. Neurol. 2008, 63, 377–387. [Google Scholar] [CrossRef]
- Hooli, B.V.; Mohapatra, G.; Mattheisen, M.; Parrado, A.R.; Roehr, J.T.; Shen, Y.; Gusella, J.; Moir, R.; Saunders, A.J.; Lange, C.; et al. Role of common and rare APP DNA sequence variants in Alzheimer disease. Neurology 2012, 78, 1250–1257. [Google Scholar] [CrossRef] [Green Version]
- Dai, M.H.; Zheng, H.; Zeng, L.D.; Zhang, Y. The genes associated with early-onset Alzheimer’s disease. Oncotarget 2018, 9, 15132–15143. [Google Scholar] [CrossRef] [Green Version]
- Meraz-Ríos, M.A.; Franco-Bocanegra, D.; Toral, R.D.; Campos-Peña, V. Early onset Alzheimer’s disease and oxidative stress. Oxid. Med. Cell. Ongev. 2014, 2014, 375968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecimovic, S.; Wang, J.; Dolios, G.; Martinez, M.; Wang, R.; Goate, A.M. Mutations in APP have independent effects on Abeta and CTFgamma generation. Neurobiol. Dis. 2004, 17, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Carman, M.D.; Fernandez-Madrid, I.J.; Power, M.D.; Lieberburg, I.; Van, D.S.G.; Bots, G.T.; Luyendijk, W.; Frangione, B. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990, 248, 1124–1126. [Google Scholar] [CrossRef] [PubMed]
- Van, B.C.; Haan, J.; Bakker, E.; Hardy, J.A.; Van, H.W.; Wehnert, A.; Vegter-Van, D.V.M.; Roos, R.A. Amyloid beta protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch). Science 1990, 248, 1120–1122. [Google Scholar]
- Hendriks, L.; van, D.C.M.; Cras, P.; Cruts, M.; Van, H.W.; Van, H.F.; Warren, A.; McInnis, M.G.; Antonarakis, S.E.; Martin, J.J.; et al. Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of the beta-amyloid precursor protein gene. Nat. Genet. 1992, 1, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef]
- Cruchaga, C.; Haller, G.; Chakraverty, S.; Mayo, K.; Vallania, F.L.M.; Mitra, R.D.; Faber, K.; Williamson, J.; Bird, T.; Diaz-Arrastia, R.; et al. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PLoS ONE 2012, 7, e31039. [Google Scholar] [CrossRef]
- Mastromoro, G.; Gambardella, S.; Marchionni, E.; Campopiano, R.; Traversa, A.; Bonaventura, C.D.; Pizzuti, A. Unusual Segregation of APP Mutations in Monogenic Alzheimer Disease. Neurodegener. Dis. 2019, 19, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Theuns, J.; Brouwers, N.; Engelborghs, S.; Sleegers, K.; Bogaerts, V.; Corsmit, E.; De, P.T.; van, D.C.M.; De, D.P.P.; Van, B.C. Promoter mutations that increase amyloid precursor-protein expression are associated with Alzheimer disease. Am. J. Hum. Genet. 2006, 78, 936–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalampokini, S.; Georgouli, D.; Patrikiou, E.; Provatas, A.; Valotassiou, V.; Georgoulias, P.; Spanaki, C.; Hadjigeorgiou, G.M.; Xiromerisiou, G. Τhe Greek Variant in APP Gene: The Phenotypic Spectrum of APP Mutations. Int. J. Mol. Sci. 2021, 22, 12355. https://doi.org/10.3390/ijms222212355
Kalampokini S, Georgouli D, Patrikiou E, Provatas A, Valotassiou V, Georgoulias P, Spanaki C, Hadjigeorgiou GM, Xiromerisiou G. Τhe Greek Variant in APP Gene: The Phenotypic Spectrum of APP Mutations. International Journal of Molecular Sciences. 2021; 22(22):12355. https://doi.org/10.3390/ijms222212355
Chicago/Turabian StyleKalampokini, Stefania, Despoina Georgouli, Eleni Patrikiou, Antonios Provatas, Varvara Valotassiou, Panagiotis Georgoulias, Cleanthe Spanaki, Georgios M. Hadjigeorgiou, and Georgia Xiromerisiou. 2021. "Τhe Greek Variant in APP Gene: The Phenotypic Spectrum of APP Mutations" International Journal of Molecular Sciences 22, no. 22: 12355. https://doi.org/10.3390/ijms222212355
APA StyleKalampokini, S., Georgouli, D., Patrikiou, E., Provatas, A., Valotassiou, V., Georgoulias, P., Spanaki, C., Hadjigeorgiou, G. M., & Xiromerisiou, G. (2021). Τhe Greek Variant in APP Gene: The Phenotypic Spectrum of APP Mutations. International Journal of Molecular Sciences, 22(22), 12355. https://doi.org/10.3390/ijms222212355