
Dissimilar Ligands Bind in a Similar Fashion: A Guide to Ligand Binding-Mode Prediction with Application to CELPP Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Comparison of the Ligand Binding Modes
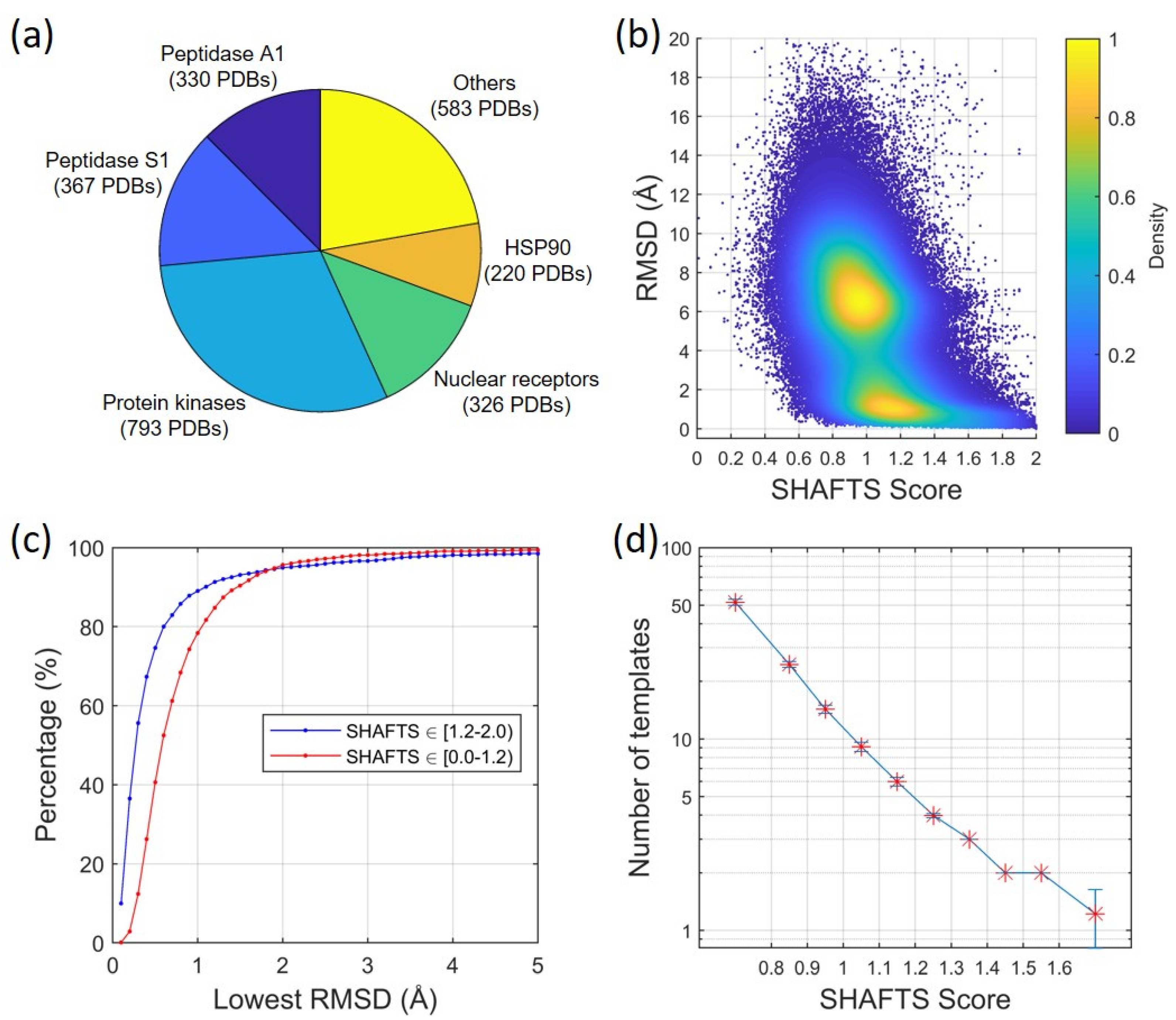
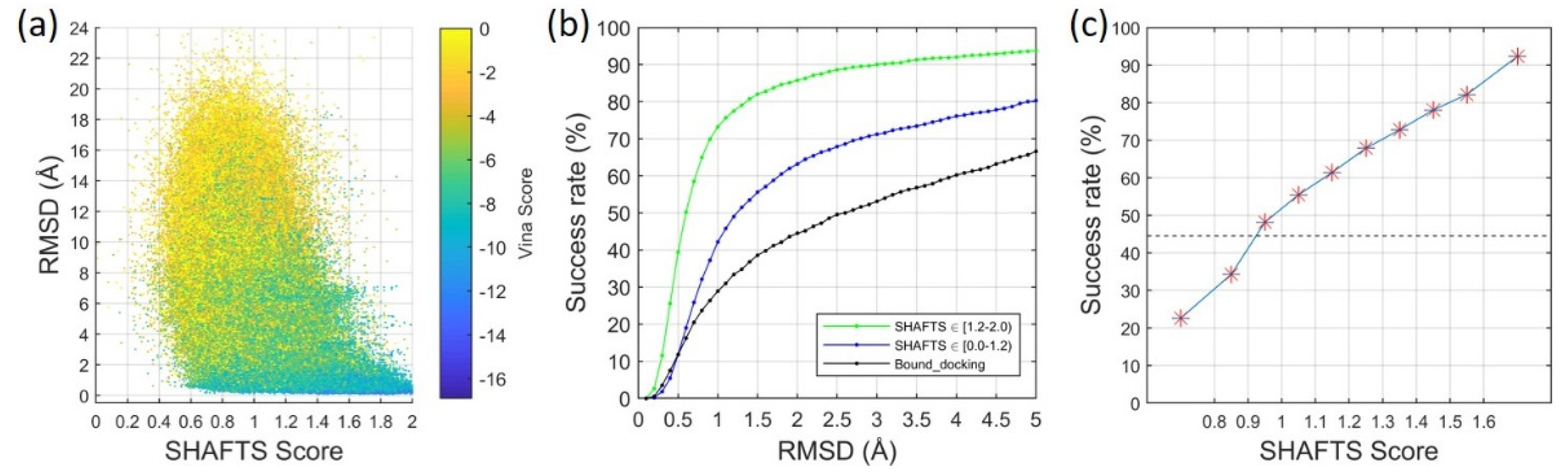
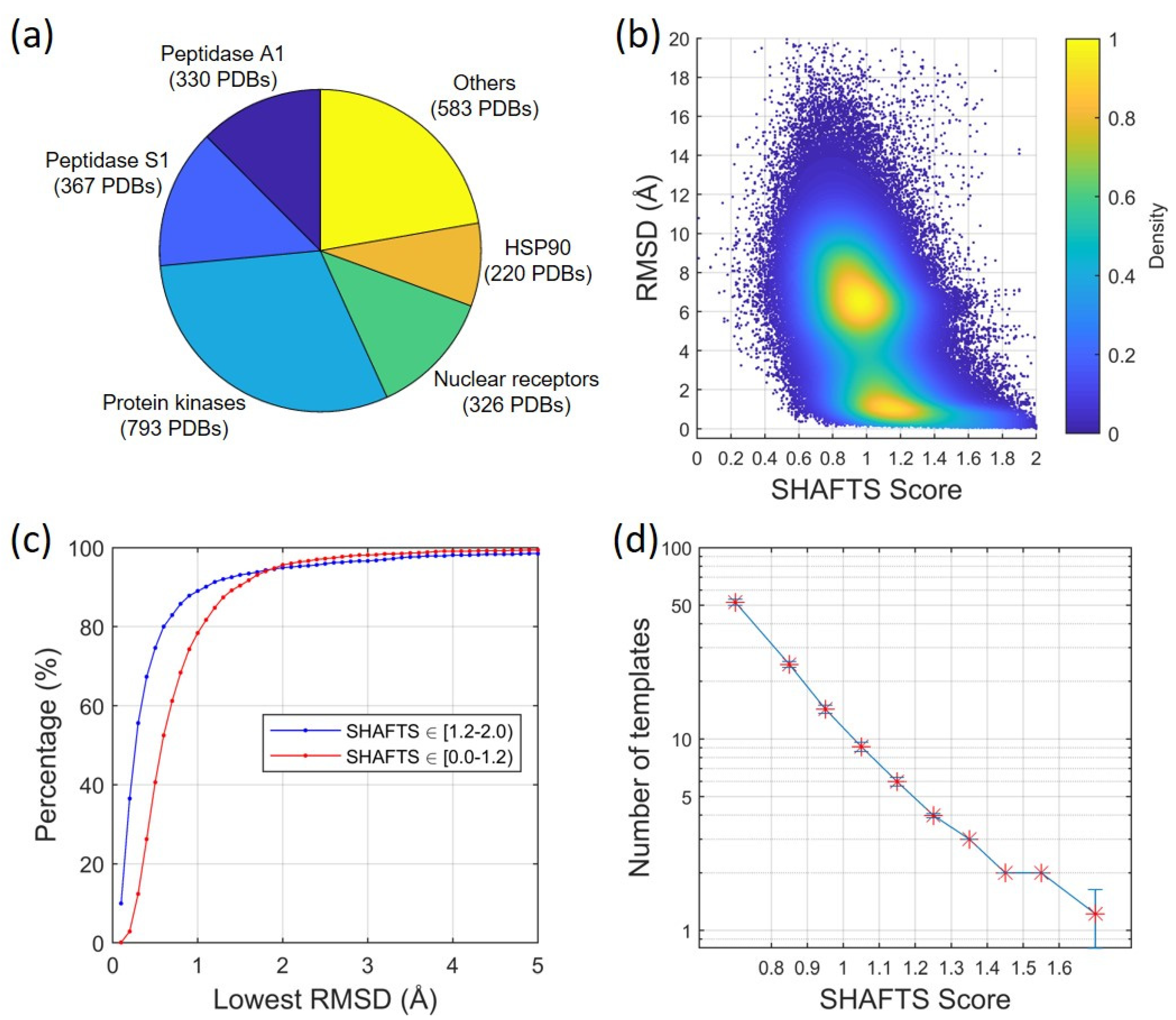
2.1.1. Ligand Root-Mean-Square Deviation (RMSD) vs. Molecular Similarity
2.1.2. Ligand RMSD vs. Number/Quality of Templates
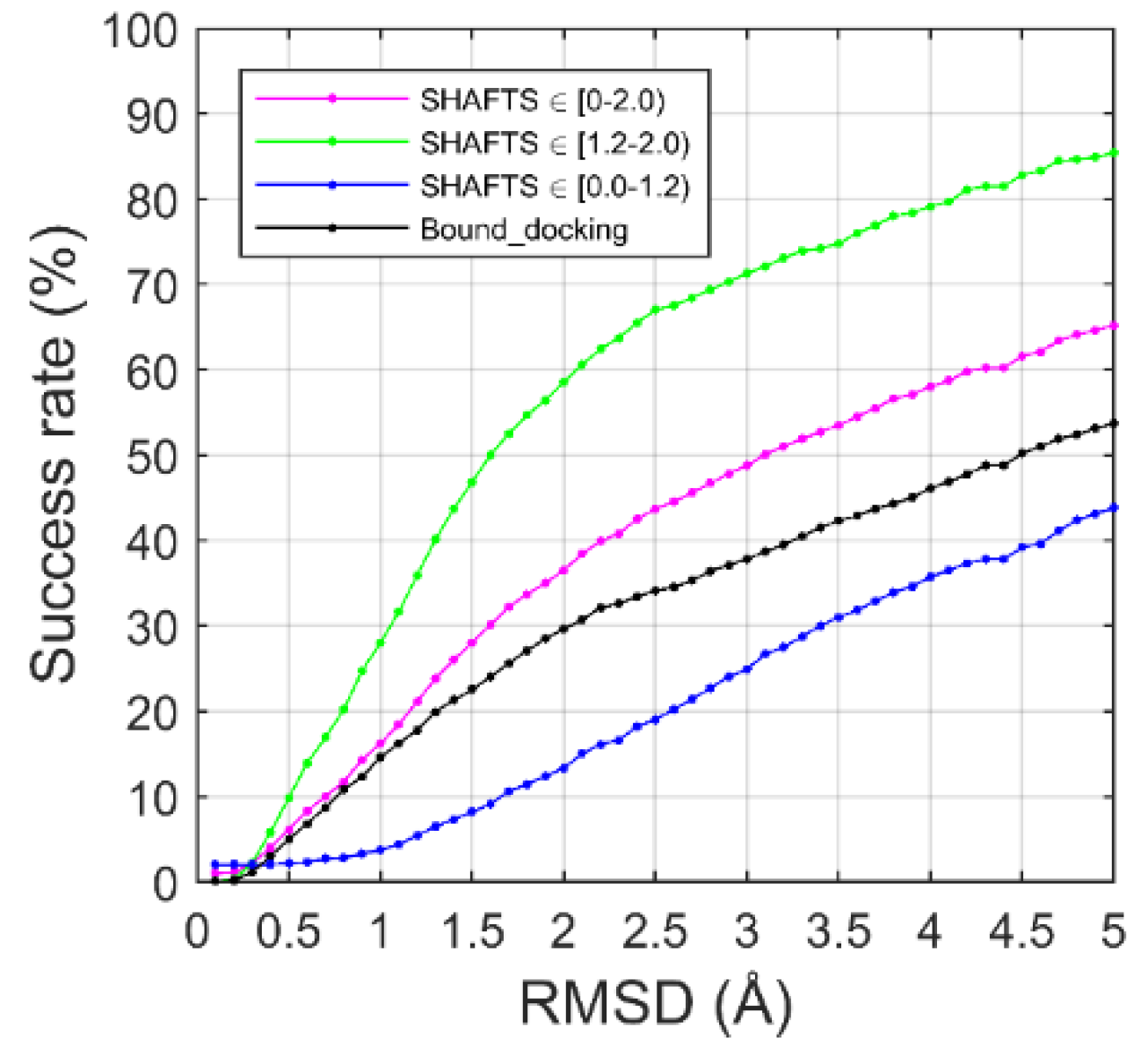
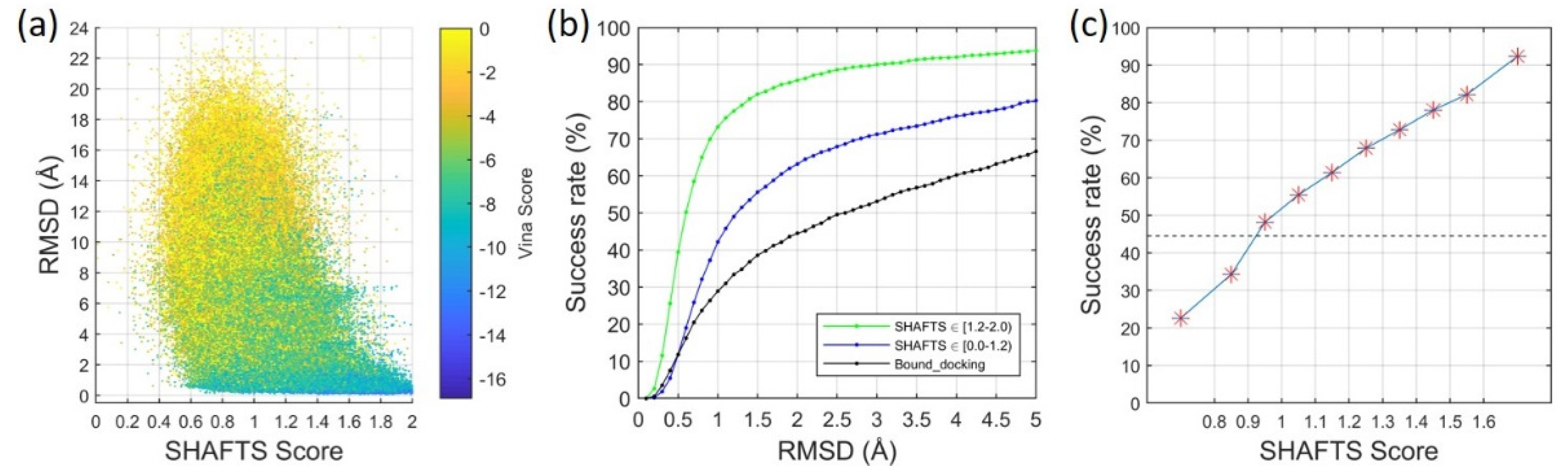
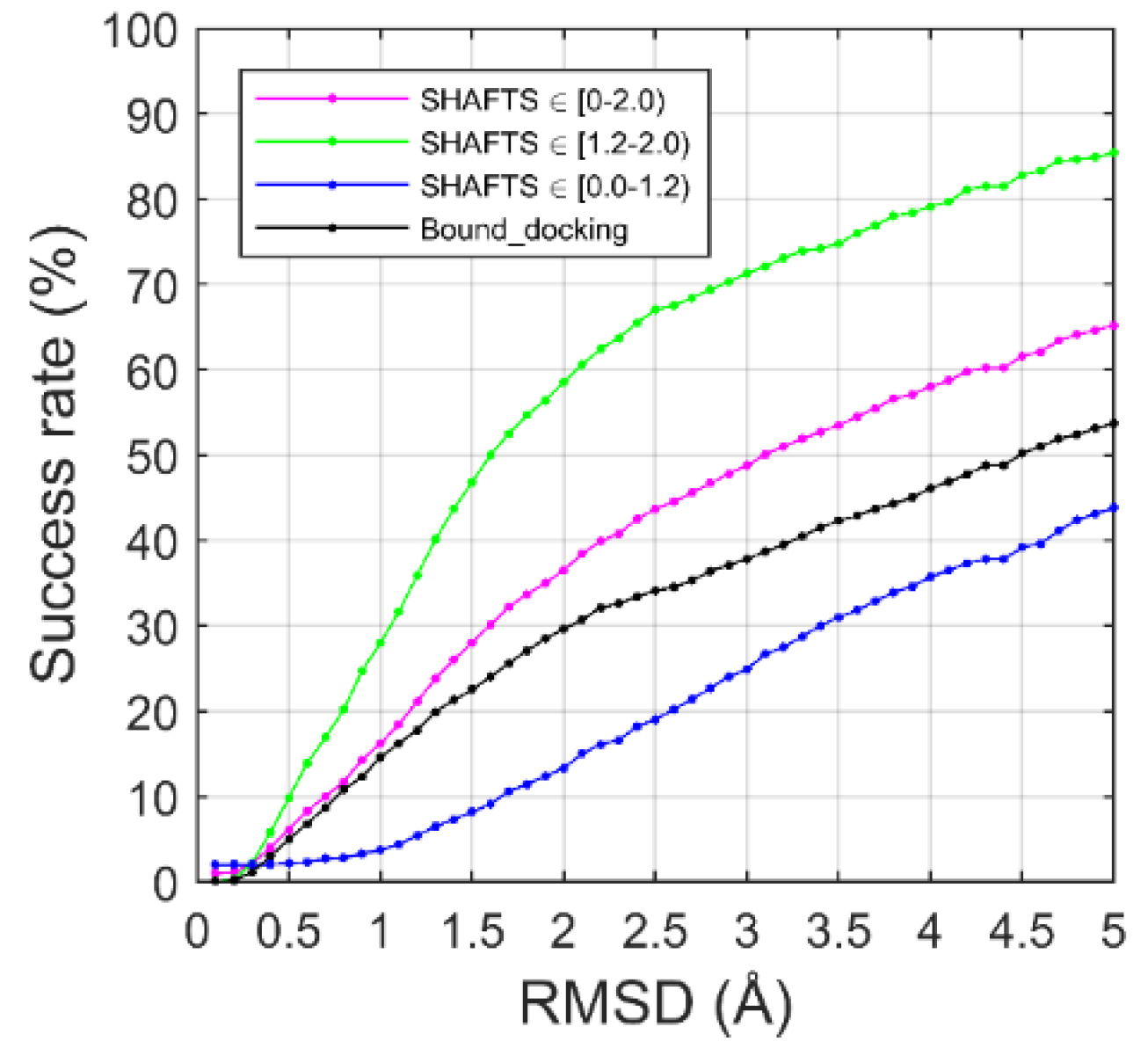
2.2. Ligand Binding-Mode Prediction
2.3. CELPP Dataset
3. Discussion
4. Materials and Methods
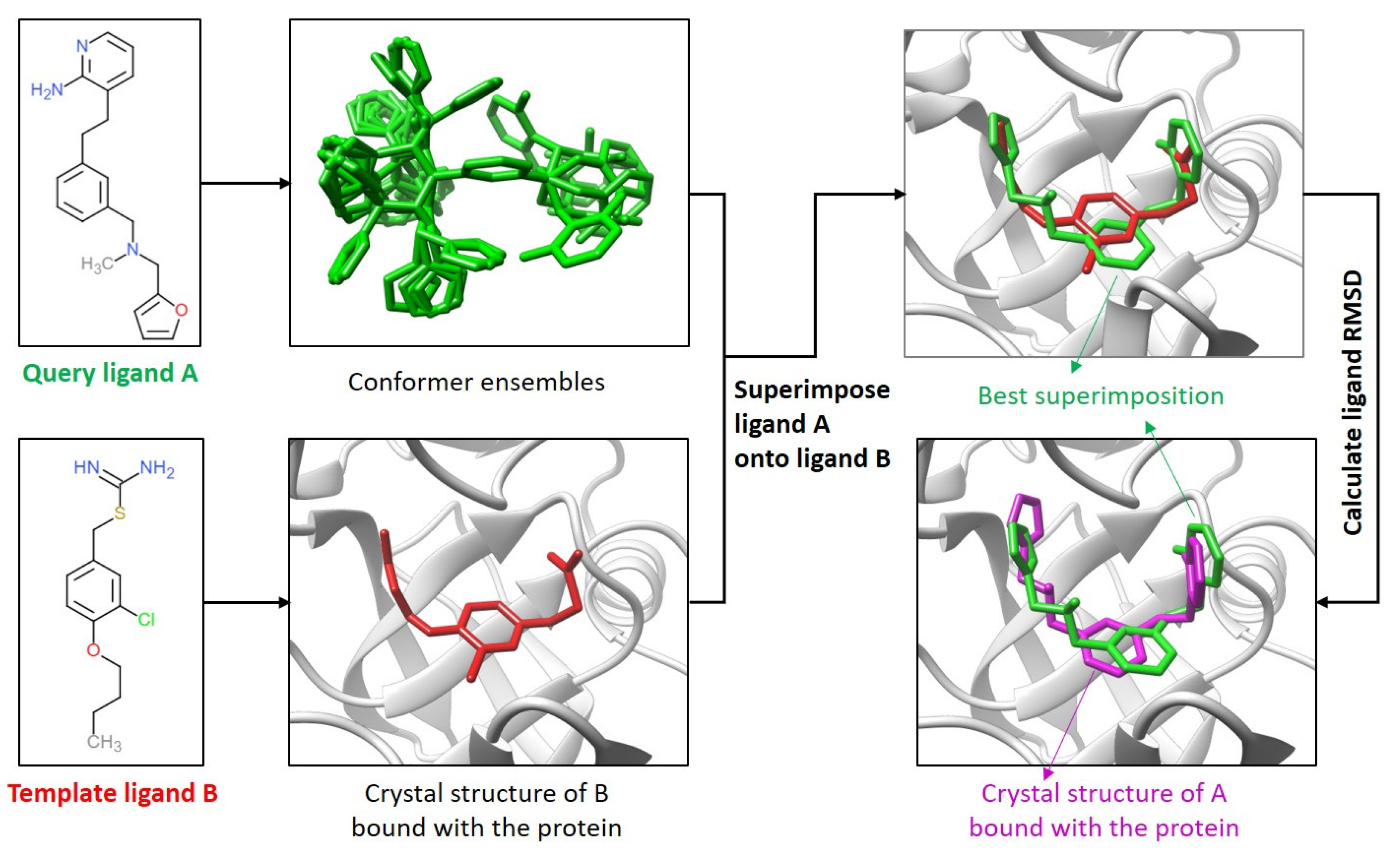
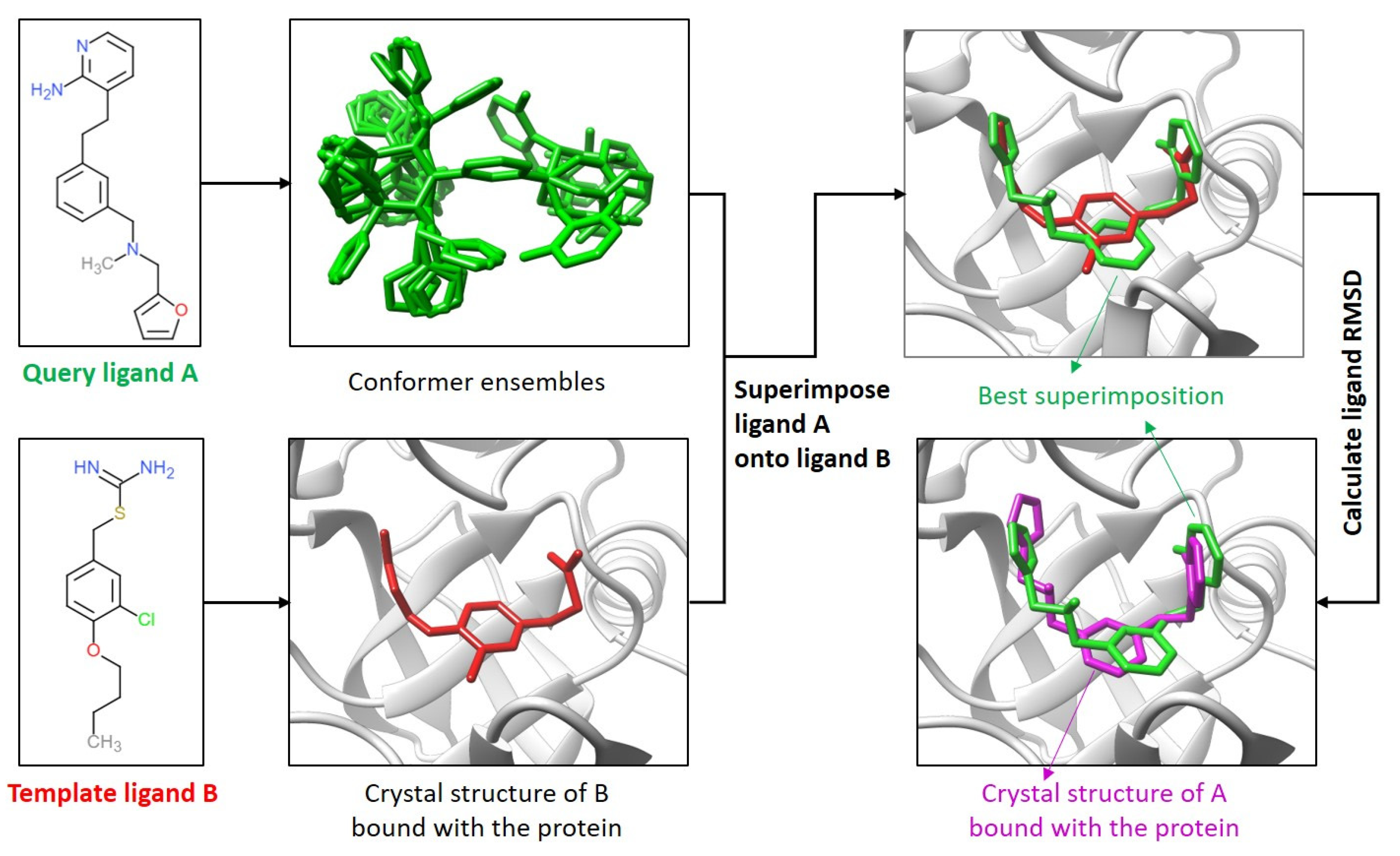
4.1. Comparison of the Ligand Binding Modes
4.2. Minimum Number of Template Ligands vs. SHAFTS Score
4.3. Construction of a Structural Dataset of Protein–Ligand Complex Structures
4.4. Template-Guiding Method for Ligand Binding-Mode Prediction
4.5. CELPP Dataset
4.6. Calculation of Ligand RMSDs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, M.A.; Maggiora, G.M. Concepts and Applications of Molecular Similarity; John Wiley & Sons: New York, NY, USA, 1990. [Google Scholar]
- Bender, A.; Glen, R.C. Molecular similarity: A key technique in molecular informatics. Org. Biomol. Chem. 2004, 2, 3204–3218. [Google Scholar] [CrossRef]
- Willett, P. Chemoinformatics—Similarity and diversity in chemical libraries. Curr. Opin. Biotechnol. 2000, 11, 85–88. [Google Scholar] [CrossRef]
- Dean, P.M. Defining molecular similarity and complementarity for drug design. In Molecular Similarity in Drug Design; Springer: Dordrecht, The Netherlands, 1995; pp. 1–23. [Google Scholar]
- Willett, P. The calculation of molecular structural similarity: Principles and practice. Mol. Inform. 2014, 33, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Boström, J.; Hogner, A.; Schmitt, S. Do structurally similar ligands bind in a similar fashion? J. Med. Chem. 2006, 49, 6716–6725. [Google Scholar] [CrossRef] [PubMed]
- Martin, Y.C.; Kofron, J.L.; Traphagen, L.M. Do structurally similar molecules have similar biological activity? J. Med. Chem. 2002, 45, 4350–4358. [Google Scholar] [CrossRef]
- Willett, P.; Barnard, J.M.; Downs, G.M. Chemical similarity searching. J. Chem. Inf. Comput. Sci. 1998, 38, 983–996. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Jiang, H.; Li, H. SHAFTS: A hybrid approach for 3D molecular similarity calculation. 1. Method and assessment of virtual screening. J. Chem. Inf. Model. 2011, 51, 2372–2385. [Google Scholar] [CrossRef]
- Lu, W.; Liu, X.; Cao, X.; Xue, M.; Liu, K.; Zhao, Z.; Shen, X.; Jiang, H.; Xu, Y.; Huang, J.; et al. SHAFTS: A hybrid approach for 3D molecular similarity calculation. 2. Prospective case study in the discovery of diverse p90 ribosomal S6 protein kinase 2 inhibitors to suppress cell migration. J. Med. Chem. 2011, 54, 3564–3574. [Google Scholar] [CrossRef]
- Ma, B.; Shatsky, M.; Wolfson, H.J.; Nussinov, R. Multiple diverse ligands binding at a single protein site: A matter of pre-existing populations. Protein Sci. 2002, 11, 184–197. [Google Scholar] [CrossRef]
- Gathiaka, S.; Liu, S.; Chiu, M.; Yang, H.; Stuckey, J.A.; Kang, Y.N.; Delproposto, J.; Kubish, G.; Dunbar, J.B.; Carlson, H.A.; et al. D3R grand challenge 2015: Evaluation of protein–ligand pose and affinity predictions. J. Comput.-Aided Mol. Des. 2016, 30, 651–668. [Google Scholar] [CrossRef] [Green Version]
- Gaieb, Z.; Liu, S.; Gathiaka, S.; Chiu, M.; Yang, H.; Shao, C.; Feher, V.A.; Walters, W.P.; Kuhn, B.; Rudolph, M.G.; et al. D3R grand challenge 2: Blind prediction of protein–ligand poses, affinity rankings, and relative binding free energies. J. Comput.-Aided Mol. Des. 2018, 32, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Gaieb, Z.; Parks, C.D.; Chiu, M.; Yang, H.; Shao, C.; Walters, W.P.; Lambert, M.H.; Nevins, N.; Bembenek, S.D.; Ameriks, M.K.; et al. D3R grand challenge 3: Blind prediction of protein–ligand poses and affinity rankings. J. Comput.-Aided Mol. Des. 2019, 33, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Parks, C.D.; Gaieb, Z.; Chiu, M.; Yang, H.; Shao, C.; Walters, W.P.; Jansen, J.M.; McGaughey, G.; Lewis, R.A.; Bembenek, S.D.; et al. D3R grand challenge 4: Blind prediction of protein–ligand poses, affinity rankings, and relative binding free energies. J. Comput.-Aided Mol. Des. 2020, 34, 99–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, J.R.; Churas, C.P.; Liu, S.; Swift, R.V.; Chiu, M.; Shao, C.; Feher, V.A.; Burley, S.K.; Gilson, M.K.; Amaro, R.E. Continuous evaluation of ligand protein predictions: A weekly community challenge for drug docking. Structure 2019, 27, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.S.; Strickland, C.; Voigt, J.H.; Kennedy, M.E.; Beyer, B.M.; Senior, M.M.; Smith, E.M.; Nechuta, T.L.; Madison, V.S.; Czarniecki, M.; et al. Application of fragment-based NMR screening, X-ray crystallography, structure-based design, and focused chemical library design to identify novel μM leads for the development of nM BACE-1 (β-site APP cleaving enzyme 1) inhibitors. J. Med. Chem. 2010, 53, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, P.C.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with omega: Algorithm and validation using high quality structures from the protein databank and Cambridge structural database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Hawkins, P.C.; Nicholls, A. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, S.J.; Thornton, J.M. NACCESS, Version 2.1.1; University College London: London, UK, 1993. [Google Scholar]
- Xu, X.; Ma, Z.; Duan, R.; Zou, X. Predicting protein–ligand binding modes for CELPP and GC3: Workflows and insight. J. Comput.-Aided Mol. Des. 2019, 33, 367–374. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Zou, X. Dissimilar Ligands Bind in a Similar Fashion: A Guide to Ligand Binding-Mode Prediction with Application to CELPP Studies. Int. J. Mol. Sci. 2021, 22, 12320. https://doi.org/10.3390/ijms222212320
Xu X, Zou X. Dissimilar Ligands Bind in a Similar Fashion: A Guide to Ligand Binding-Mode Prediction with Application to CELPP Studies. International Journal of Molecular Sciences. 2021; 22(22):12320. https://doi.org/10.3390/ijms222212320
Chicago/Turabian StyleXu, Xianjin, and Xiaoqin Zou. 2021. "Dissimilar Ligands Bind in a Similar Fashion: A Guide to Ligand Binding-Mode Prediction with Application to CELPP Studies" International Journal of Molecular Sciences 22, no. 22: 12320. https://doi.org/10.3390/ijms222212320
APA StyleXu, X., & Zou, X. (2021). Dissimilar Ligands Bind in a Similar Fashion: A Guide to Ligand Binding-Mode Prediction with Application to CELPP Studies. International Journal of Molecular Sciences, 22(22), 12320. https://doi.org/10.3390/ijms222212320