
Four-Dimensional Chromosome Structure Prediction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
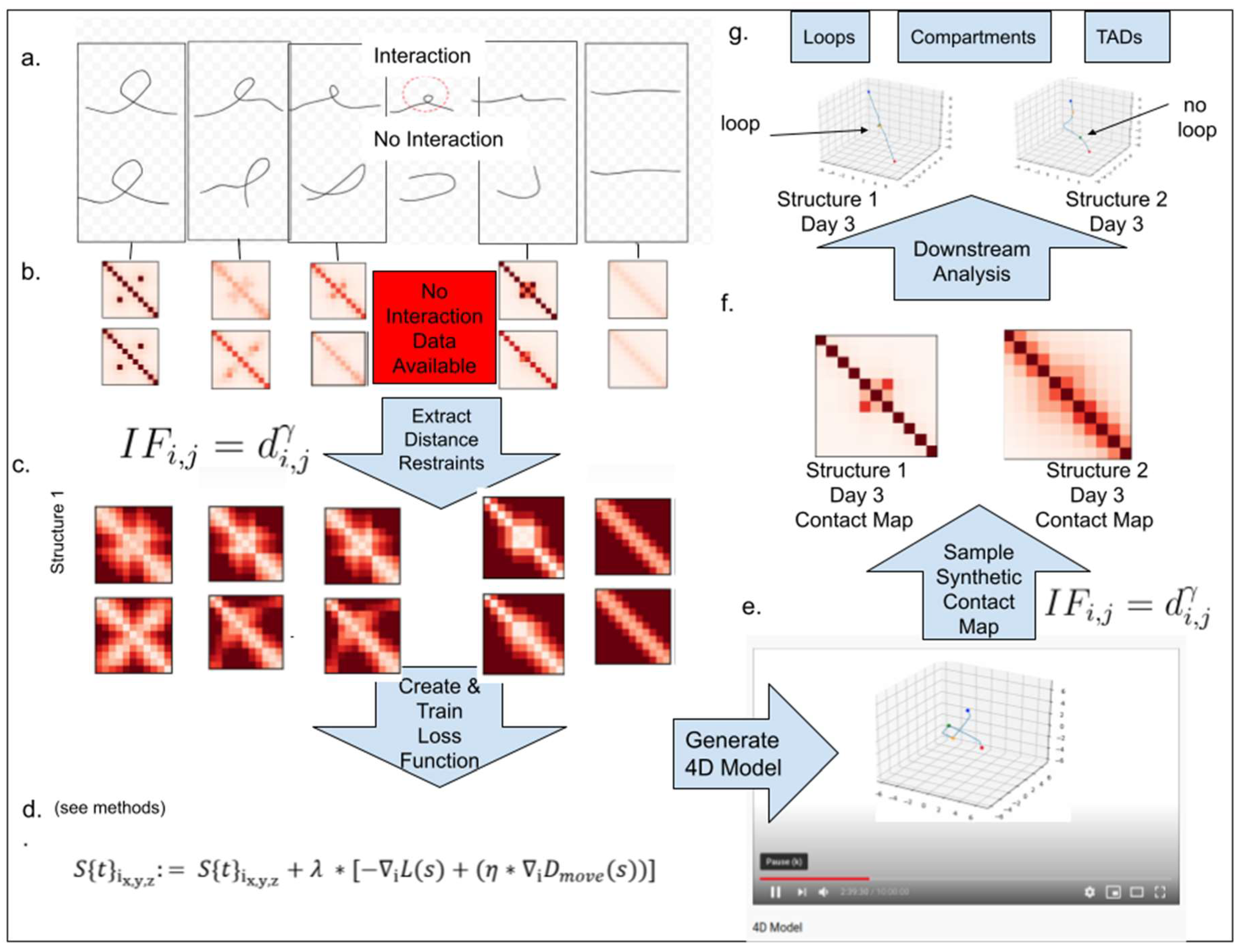
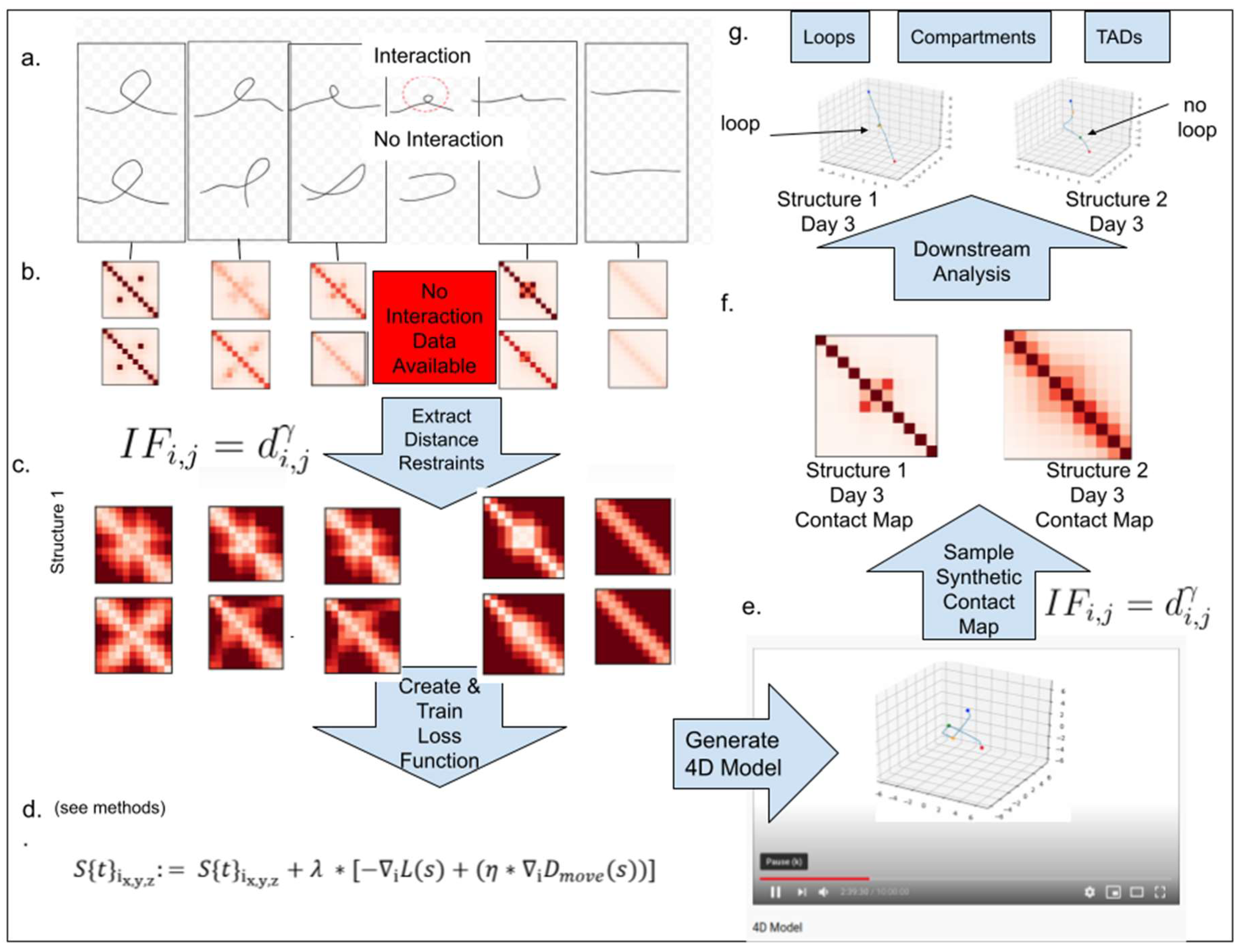
2.1. Overview of the 4DMax Approach
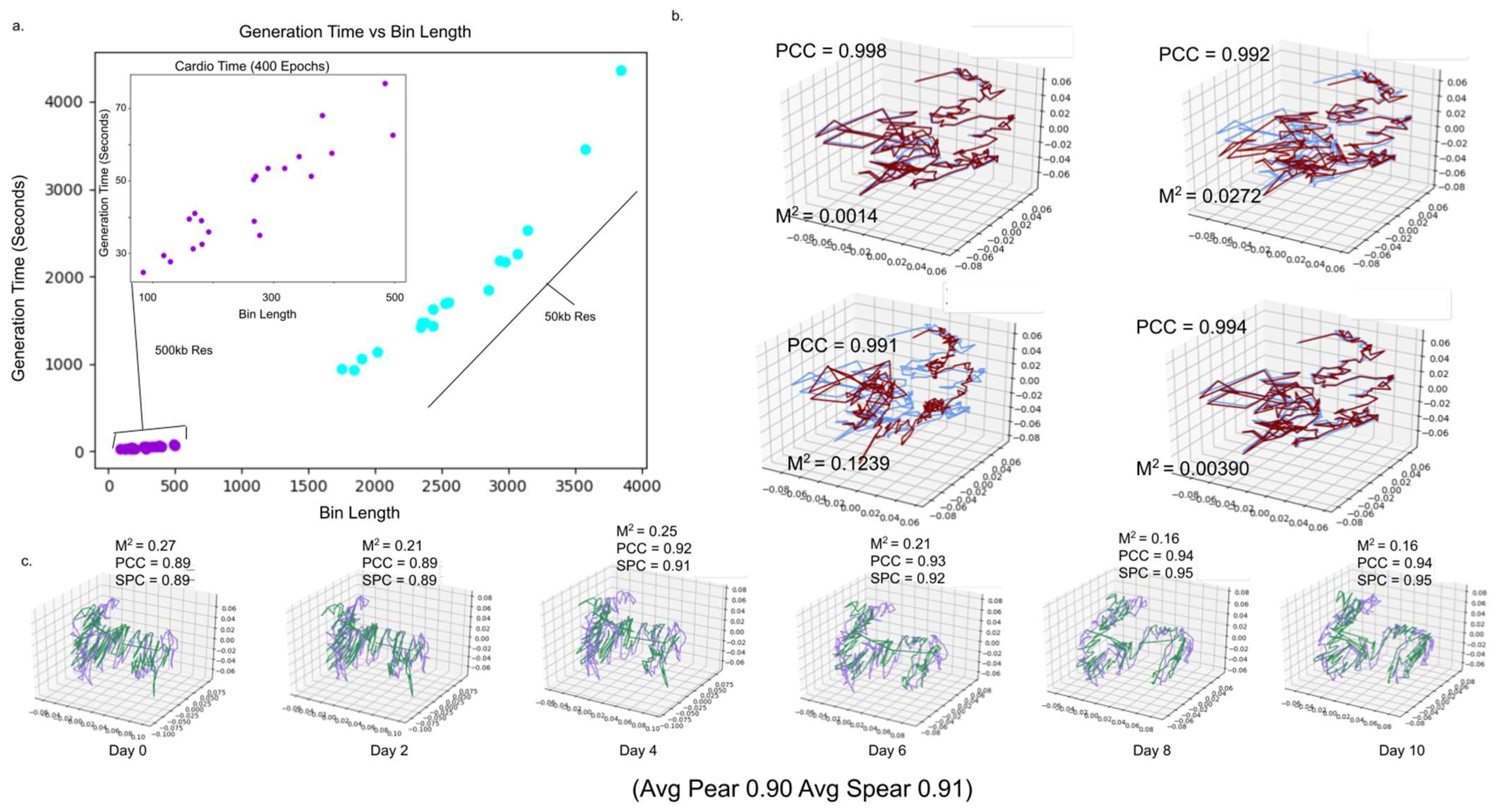
2.2. 4DMax Correctly Reconstructs Models of Synthetic Time Series Hi-C Data
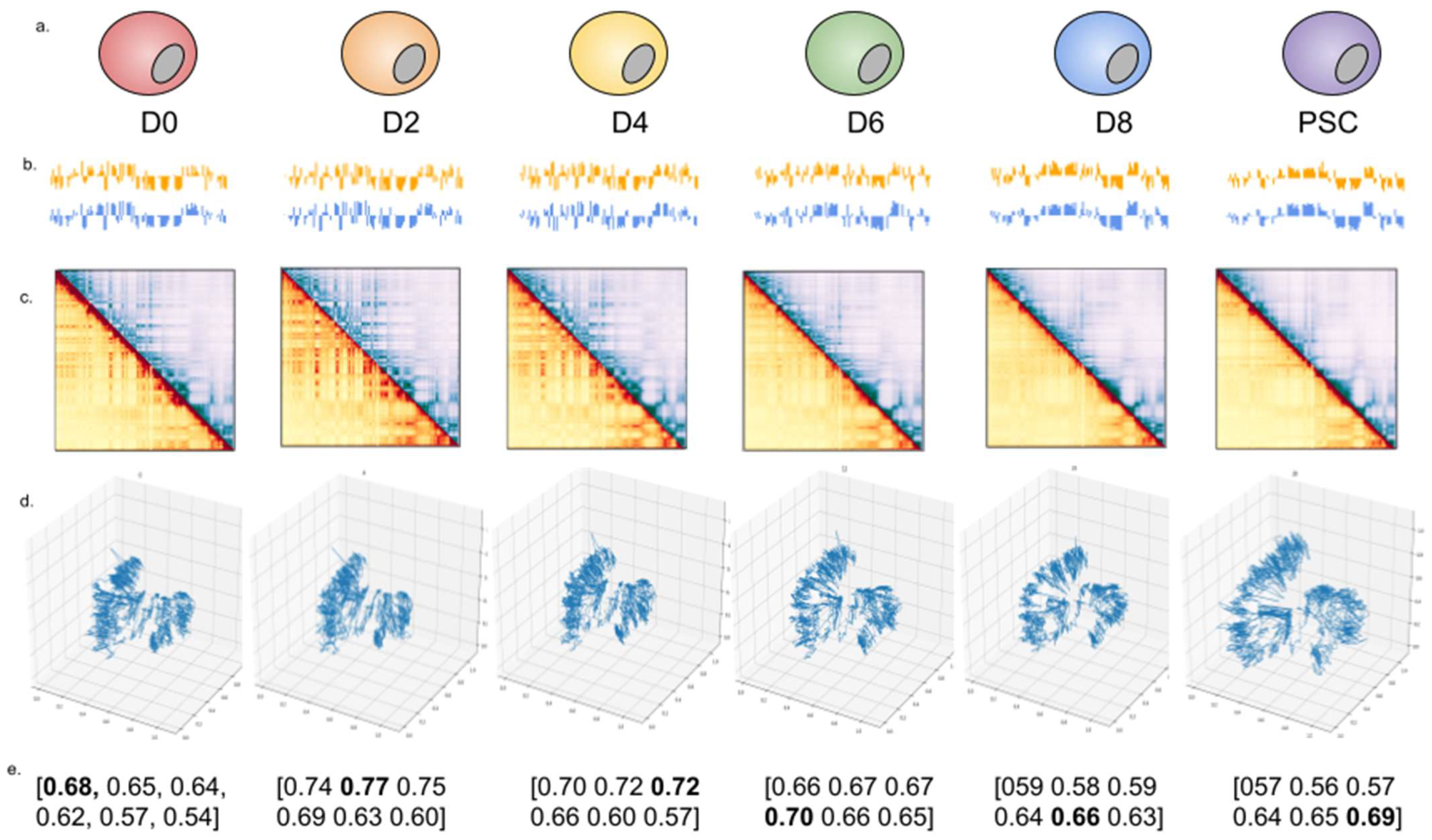
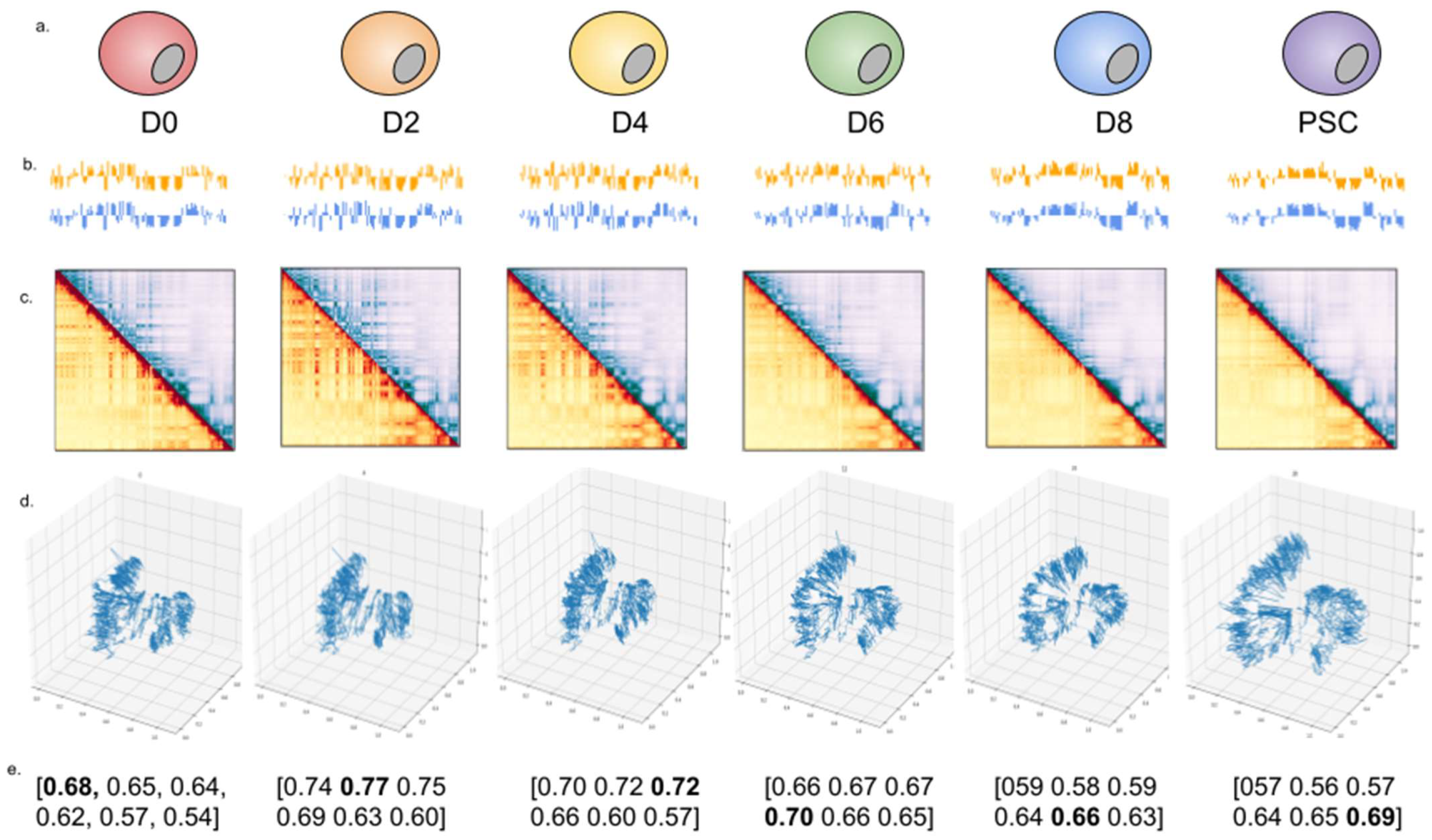
2.3. 4DMax Predicts Smooth 4D Models of Induced Pluripotent Stem Cell Differentiation in Mice
2.4. 4DMax Preicts Smooth 4D Models of Cardiomyocyte Differentiation in Humans
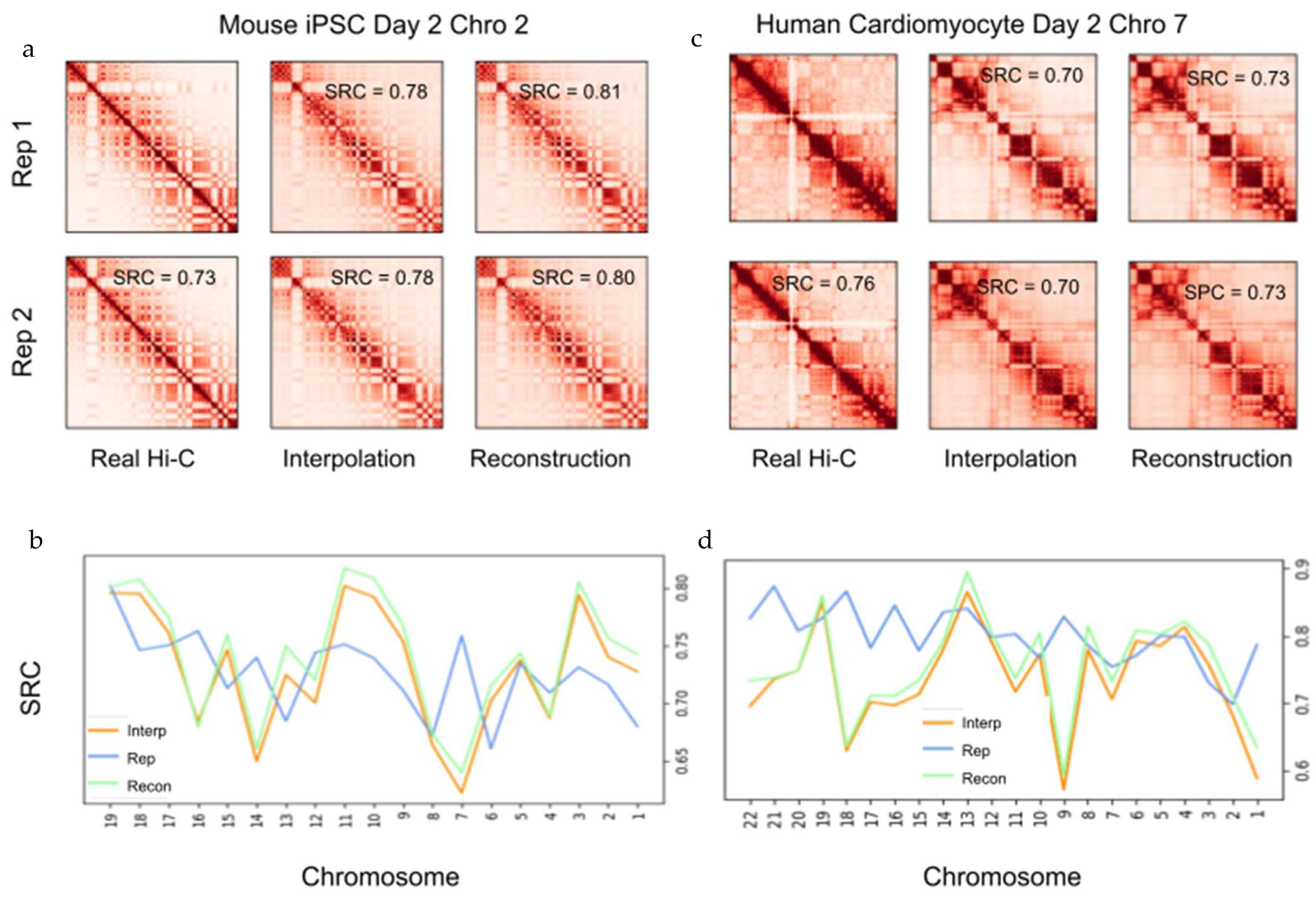
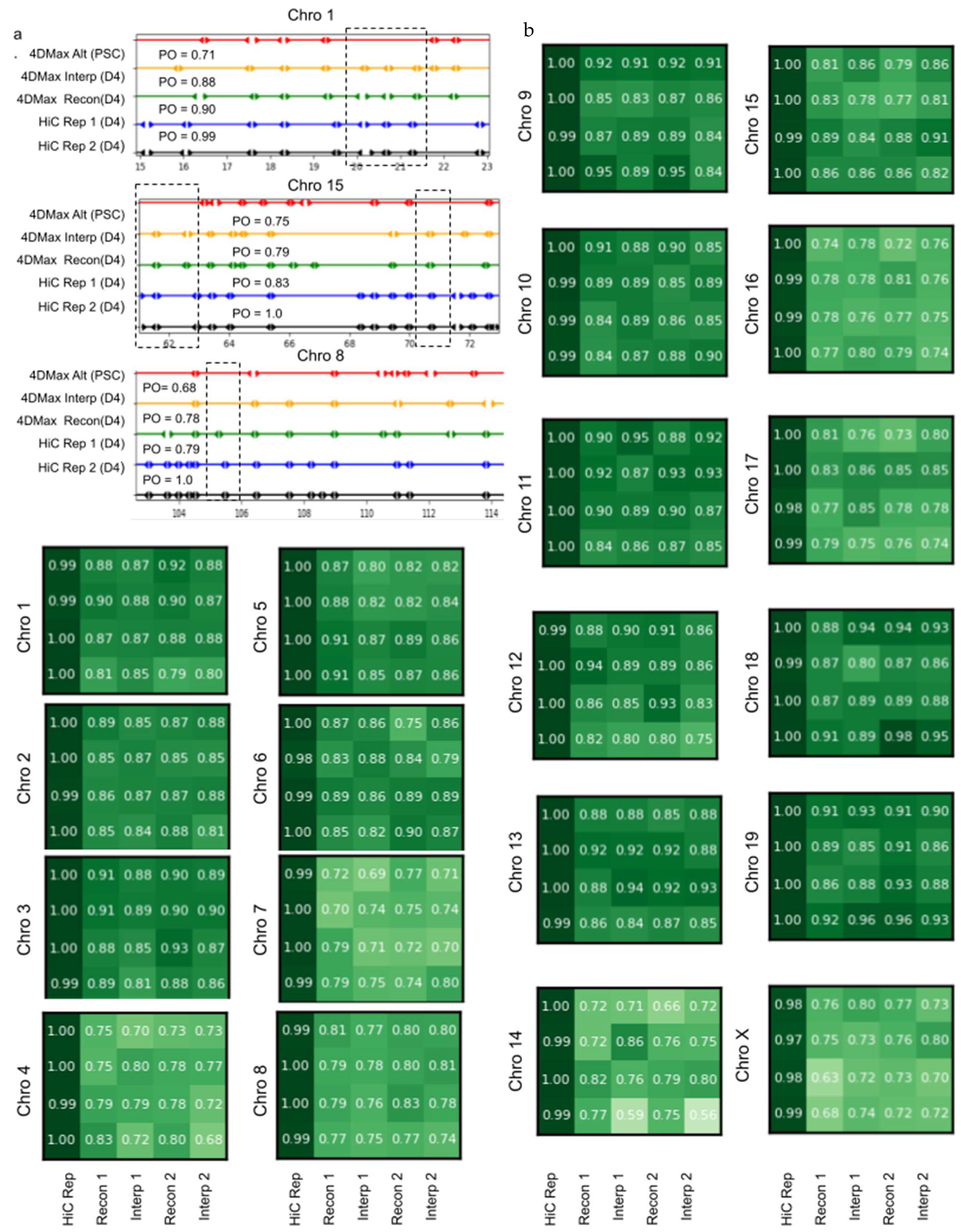
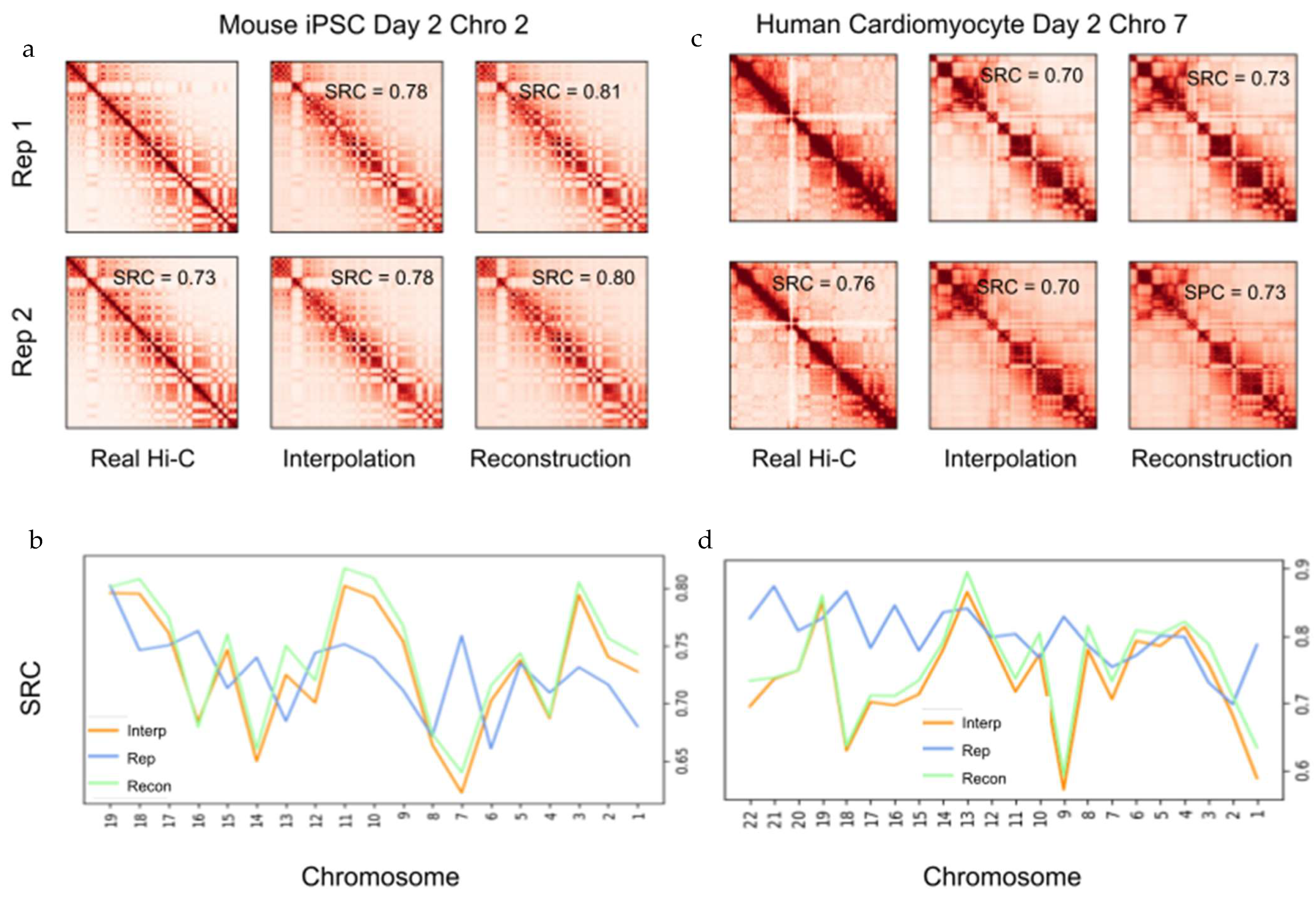
2.5. Interpolation of Time Series Hi-C Data Using 4DMax Generated Models Show High Consistency with Experimental Hi-C
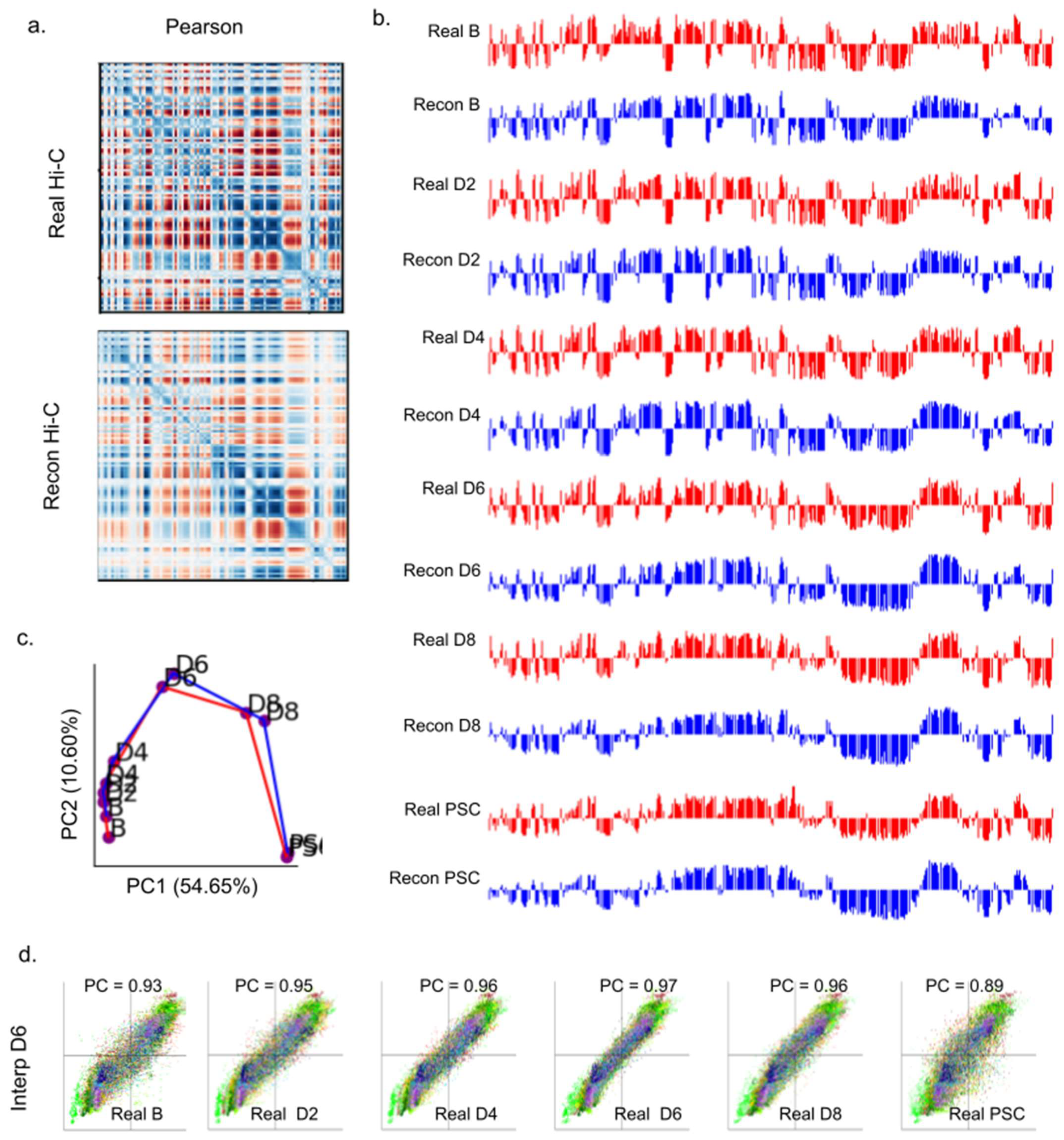
2.6. 4DMax Correctly Preserves and Predicts AB Compartment Assignment
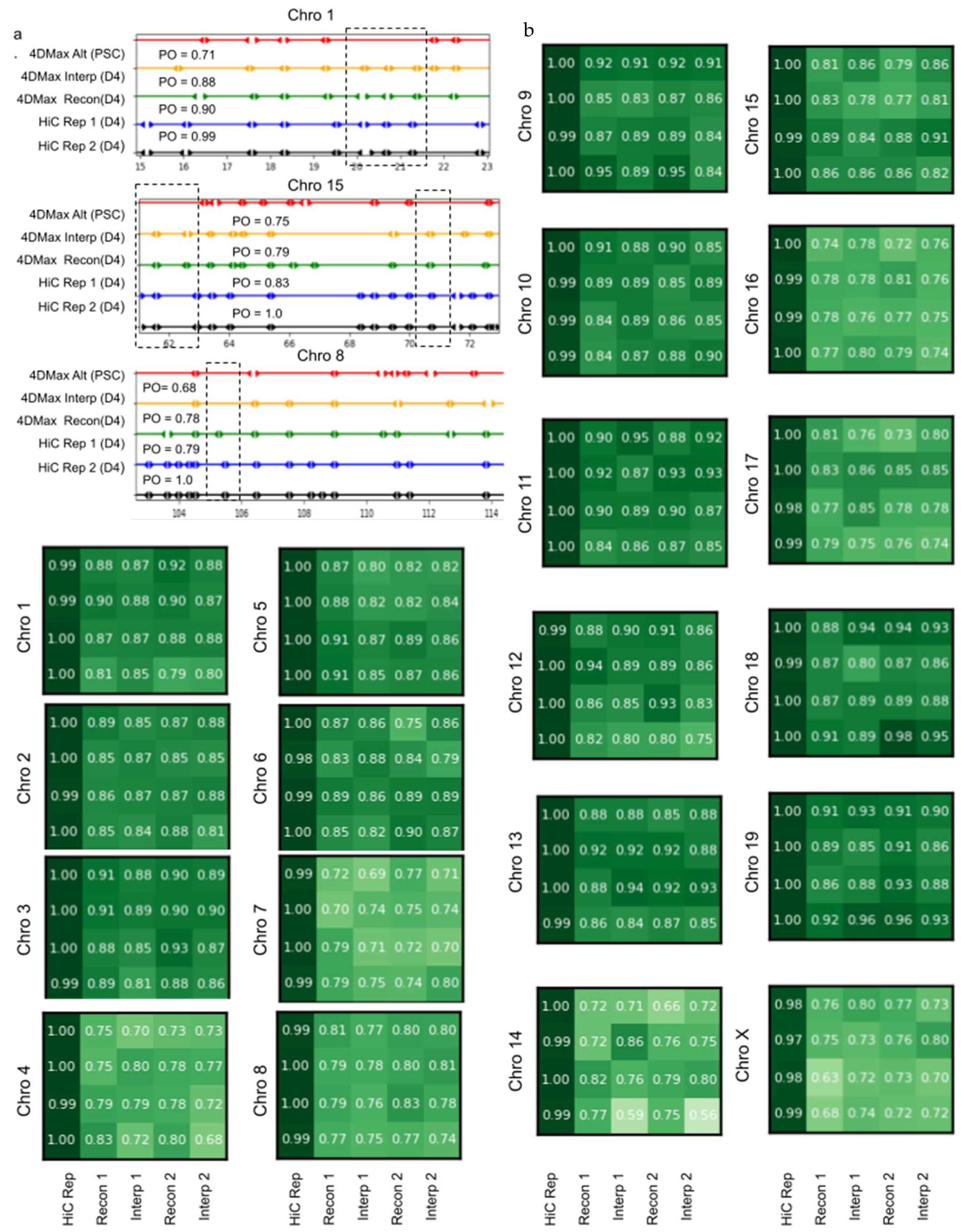
2.7. 4DMax Correctly Preserves and Predicts TAD Border Positioning
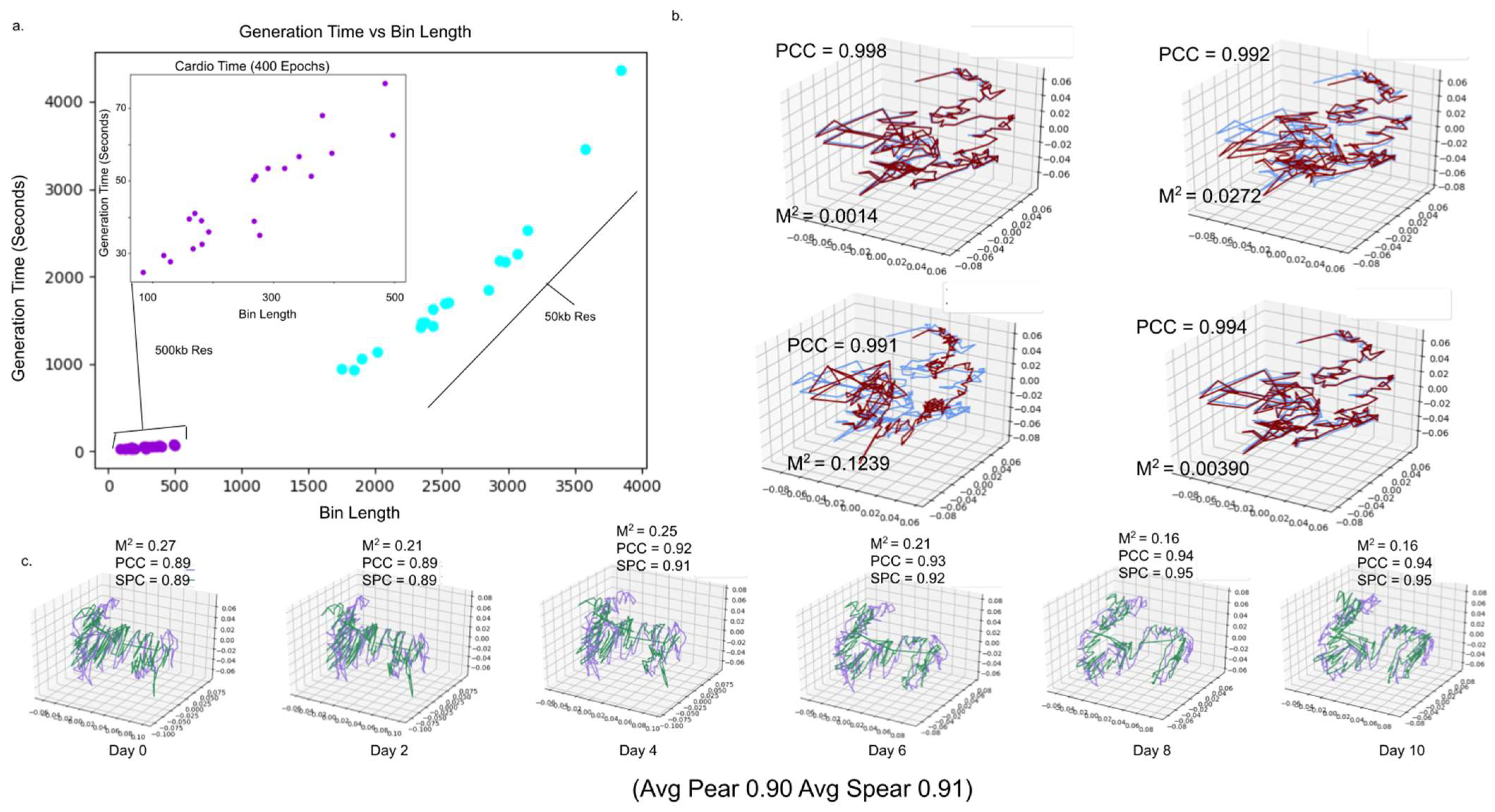
2.8. 4DMax Completes in Tractable Time for Human and Mouse Chromosome Construction
2.9. 4DMax Predictions Remain Stable against Change in Time Point Granularity
2.10. 4DMax Predictions Remain Stable to Variation in Hi-C Contact Matrix Resolution
3. Discussion
4. Materials and Methods
4.1. Description of 4DMax Algorithm
4.2. Interpolation of Contacts
4.3. AB Compartment Analysis
4.4. TAD Identification
4.5. Statistical Analysis
4.6. Notes on HiC Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dekker, J. Gene regulation in the third dimension. Science 2008, 319, 1793–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, P.; Bickmore, W. Nuclear organization of the genome and the potential for gene regulation. Nature 2007, 447, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Miele, A.; Dekker, J. Long-range chromosomal interactions and gene regulation. Mol. Biosyst. 2008, 4, 1046–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lajoie, B.R.; Dekker, J.; Kaplan, N. The Hitchhiker’s guide to Hi-C analysis: Practical guidelines. Methods 2015, 72, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Zufferey, M.; Tavernari, D.; Oricchio, E.; Ciriello, G. Comparison of computational methods for the identification of topologically associating domains. Genome Biol. 2018, 19, 217. [Google Scholar] [CrossRef] [Green Version]
- Calandrelli, R.; Wu, Q.; Guan, J.; Zhong, S. GITAR: An open source tool for analysis and visualization of Hi-C data. Genom. Proteom. Bioinform. 2018, 16, 365–372. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Duan, Z.; Andronescu, M.; Schutz, K.; McIlwain, S.; Kim, Y.J.; Lee, C.; Shendure, J.; Fields, S.; Blau, C.A.; Noble, W.S. A three-dimensional model of the yeast genome. Nature 2010, 465, 363–367. [Google Scholar] [CrossRef]
- Oluwadare, O.; Highsmith, M.; Cheng, J. An overview of methods for reconstructing 3-D chromosome and genome structures from Hi-C data. Biol. Proced. Online 2019, 21, 7. [Google Scholar] [CrossRef]
- Oluwadare, O.; Zhang, Y.; Cheng, J. A maximum likelihood algorithm for reconstructing 3D structures of human chromosomes from chromosomal contact data. BMC Genom. 2018, 19, 161. [Google Scholar] [CrossRef] [Green Version]
- Rieber, L.; Mahony, S. miniMDS: 3D structural inference from high-resolution Hi-C data. Bioinformatics 2017, 33, i261–i266. [Google Scholar] [CrossRef] [Green Version]
- Trieu, T.; Cheng, J. 3D genome structure modeling by Lorentzian objective function. Nucleic Acids Res. 2017, 45, 1049–1058. [Google Scholar] [CrossRef] [Green Version]
- Varoquaux, N.; Ay, F.; Noble, W.S.; Vert, J.P. A statistical approach for inferring the 3D structure of the genome. Bioinformatics 2014, 30, i26–i33. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [Green Version]
- Nagano, T.; Lubling, Y.; Stevens, T.J.; Schoenfelder, S.; Yaffe, E.; Dean, W.; Laue, E.D.; Tanay, A.; Fraser, P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 2013, 502, 59–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, N.; Kruse, K.; Erdmann, T.; Staiger, A.M.; Ott, G.; Lenz, G.; Vaquerizas, J.M. Chromatin conformation analysis of primary patient tissue using a low input Hi-C method. Nat. Commun. 2018, 9, 4938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carstens, S.; Nilges, M.; Habeck, M. Bayesian inference of chromatin structure ensembles from population-averaged contact data. Proc. Natl. Acad. Sci. USA 2020, 117, 7824–7830. [Google Scholar] [CrossRef] [PubMed]
- Stadhouders, R.; Vidal, E.; Serra, F.; Di Stefano, B.; Le Dily, F.; Quilez, J.; Gomez, A.; Collombet, S.; Berenguer, C.; Cuartero, Y.; et al. Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat. Genet. 2018, 50, 238–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertero, A.; Fields, P.A.; Ramani, V.; Bonora, G.; Yardimci, G.G.; Reinecke, H.; Pabon, L.; Noble, W.S.; Shendure, J.; Murry, C.E. Dynamics of genome reorganization during human cardiogenesis reveal an RBM20-dependent splicing factory. Nat. Commun. 2019, 10, 1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Stefano, M.; Paulsen, J.; Jost, D.; Marti-Renom, M.A. 4D nucleome modeling. Curr. Opin. Genet. Dev. 2021, 67, 25–32. [Google Scholar] [CrossRef]
- Nagano, T.; Lubling, Y.; Varnai, C.; Dudley, C.; Leung, W.; Baran, Y.; Mendelson Cohen, N.; Wingett, S.; Fraser, P.; Tanay, A. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 2017, 547, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Gibcus, J.H.; Samejima, K.; Goloborodko, A.; Samejima, I.; Naumova, N.; Nuebler, J.; Kanemaki, M.T.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. A pathway for mitotic chromosome formation. Science 2018, 359, eaao6135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sati, S.; Bonev, B.; Szabo, Q.; Jost, D.; Bensadoun, P.; Serra, F.; Loubiere, V.; Papadopoulos, G.L.; Rivera-Mulia, J.C.; Fritsch, L.; et al. 4D Genome Rewiring during Oncogene-Induced and Replicative Senescence. Mol. Cell 2020, 78, 522–538. [Google Scholar] [CrossRef]
- Di Stefano, M.; Stadhouders, R.; Farabella, I.; Castillo, D.; Serra, F.; Graf, T.; Marti-Renom, M.A. Transcriptional activation during cell reprogramming correlates with the formation of 3D open chromatin hubs. Nat. Commun. 2020, 11, 2564. [Google Scholar] [CrossRef] [PubMed]
- Abramo, K.; Valton, A.L.; Venev, S.V.; Ozadam, H.; Fox, A.N.; Dekker, J. A chromosome folding intermediate at the condensin-to-cohesin transition during telophase. Nat. Cell Biol. 2019, 21, 1393–1402. [Google Scholar] [CrossRef] [Green Version]
- Ay, F.; Bailey, T.L.; Noble, W.S. Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts. Genome Res. 2014, 24, 999–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy, CSciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Highsmith, M.; Cheng, J. Four-Dimensional Chromosome Structure Prediction. Int. J. Mol. Sci. 2021, 22, 9785. https://doi.org/10.3390/ijms22189785
Highsmith M, Cheng J. Four-Dimensional Chromosome Structure Prediction. International Journal of Molecular Sciences. 2021; 22(18):9785. https://doi.org/10.3390/ijms22189785
Chicago/Turabian StyleHighsmith, Max, and Jianlin Cheng. 2021. "Four-Dimensional Chromosome Structure Prediction" International Journal of Molecular Sciences 22, no. 18: 9785. https://doi.org/10.3390/ijms22189785
APA StyleHighsmith, M., & Cheng, J. (2021). Four-Dimensional Chromosome Structure Prediction. International Journal of Molecular Sciences, 22(18), 9785. https://doi.org/10.3390/ijms22189785