
Mixed Transcriptome Analysis Revealed the Possible Interaction Mechanisms between Zizania latifolia and Ustilago esculenta Inducing Jiaobai Stem-Gall Formation

 ,
,
Abstract
:1. Introduction
2. Results
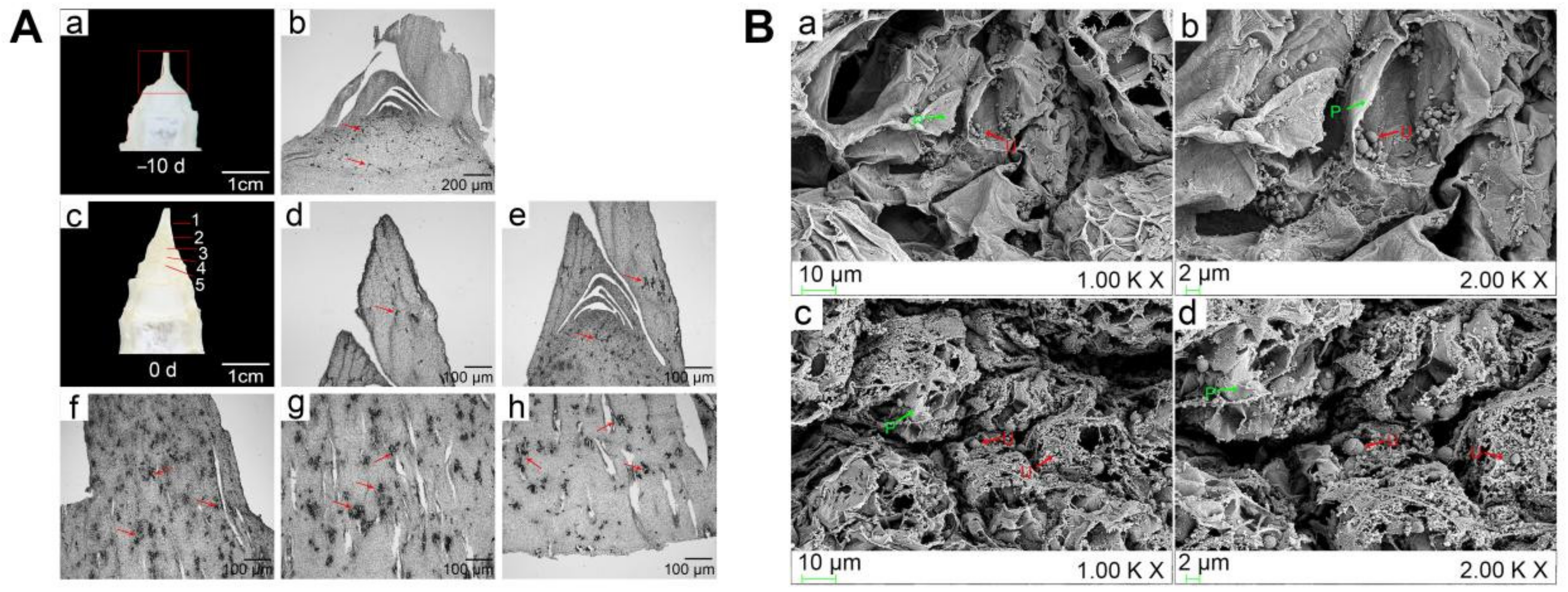
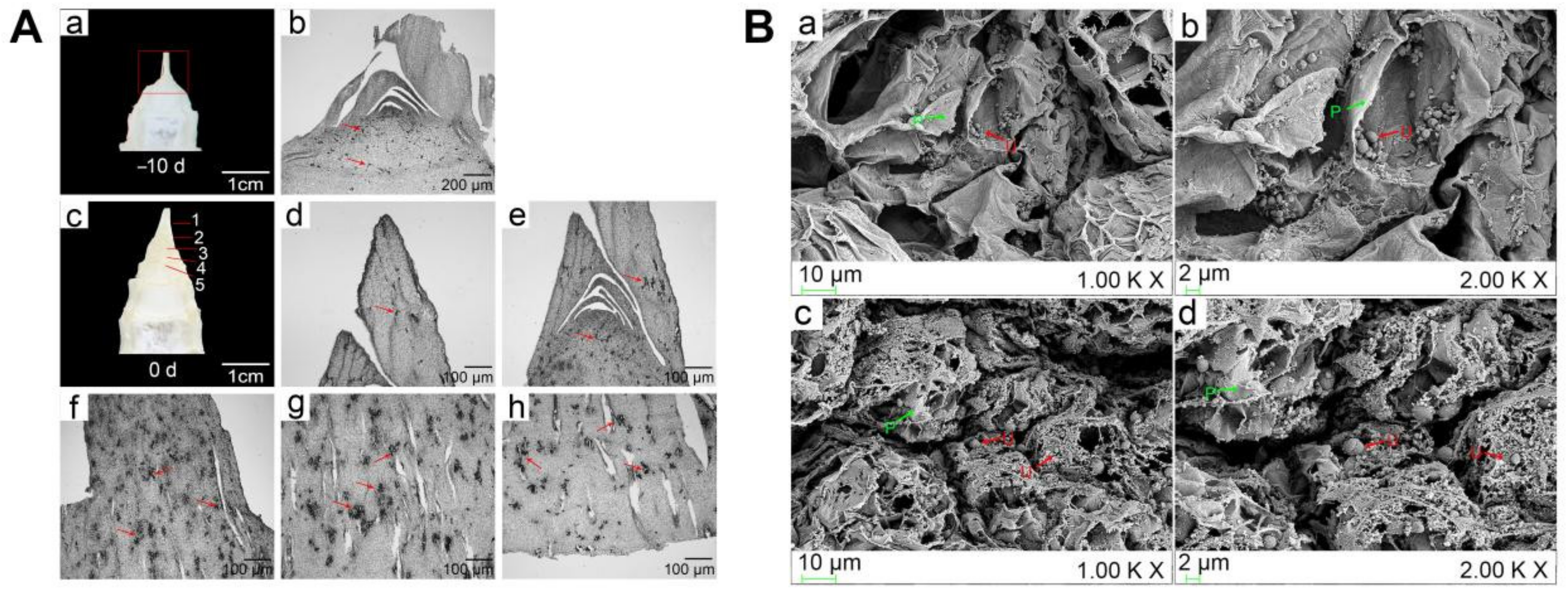
2.1. Distribution of U. esculenta in the Initial Trigger of Stem-Gall Formation of Z. latifolia
2.2. General Features of the U. esculenta and Z. latifolia Transcriptomes
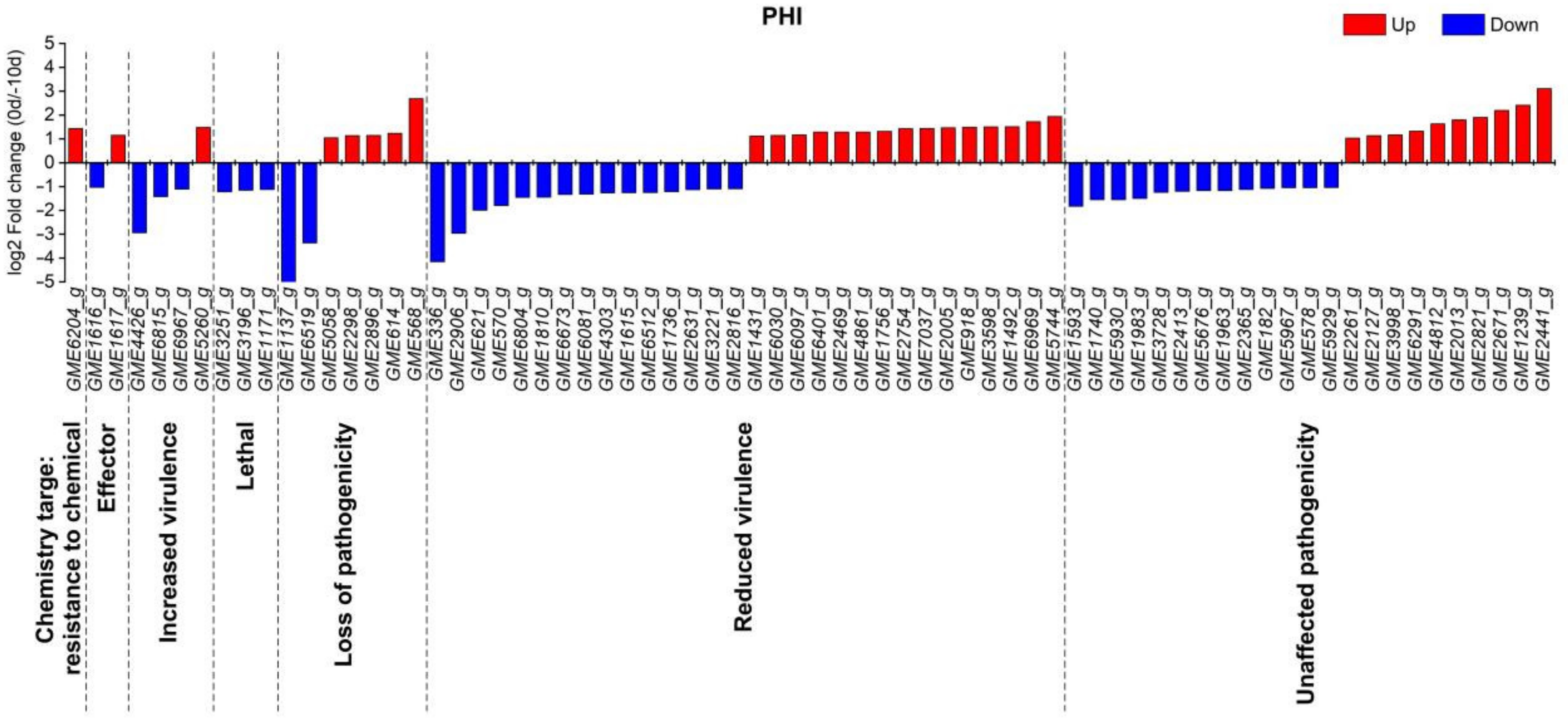
2.3. Analysis of Pathogenicity Genes in U. esculenta and Plant Resistance Genes in Z. latifolia
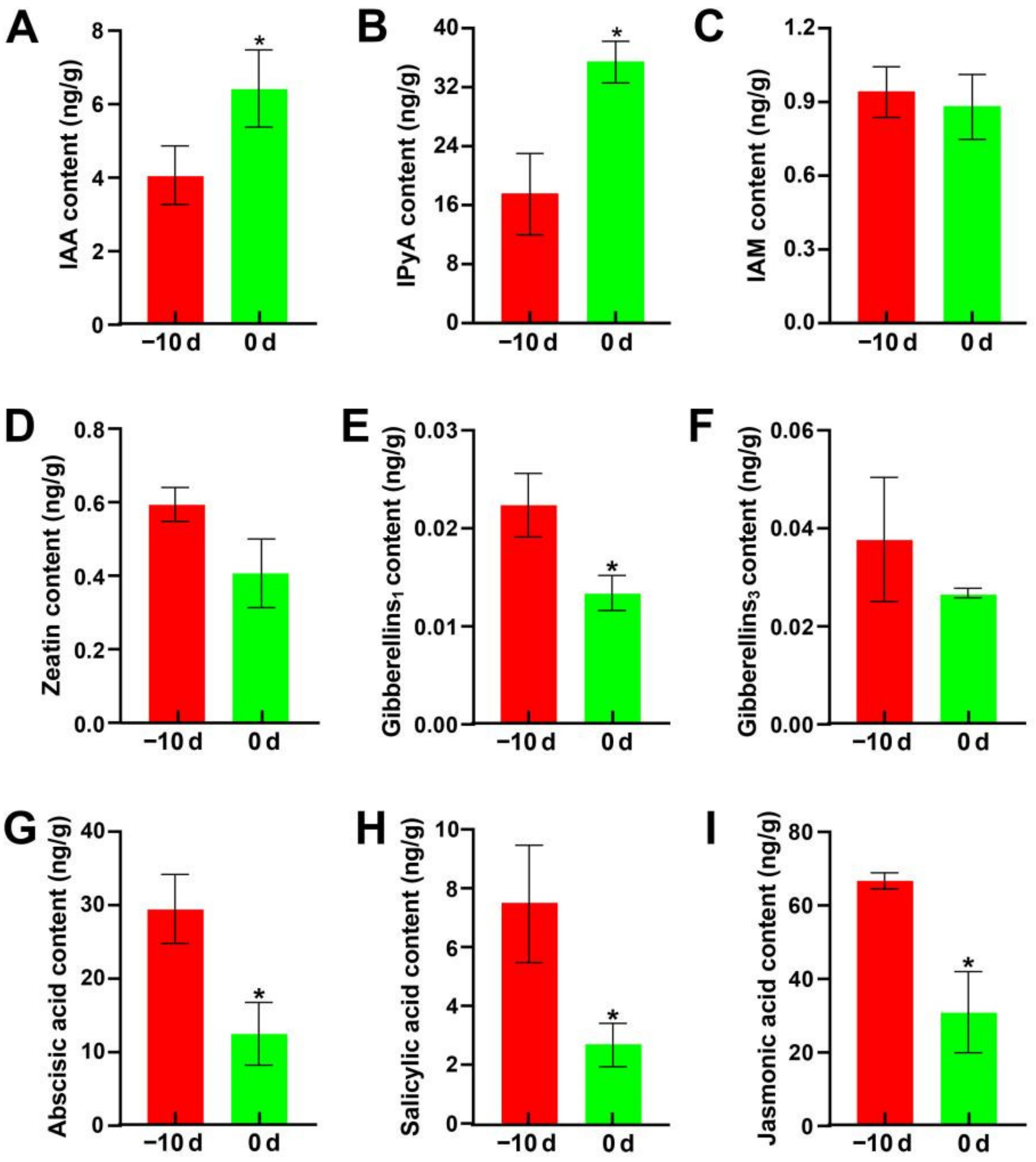
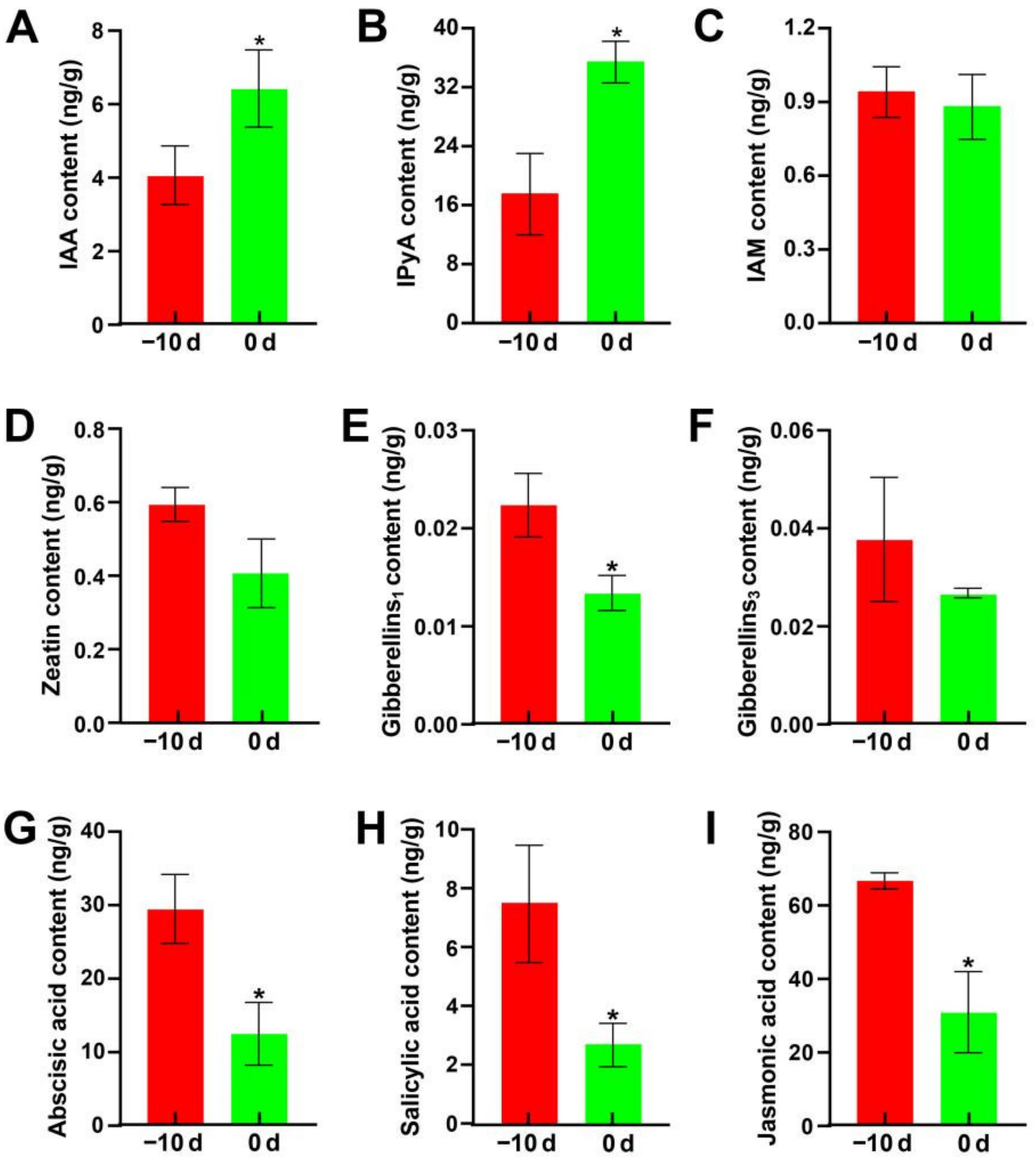
2.4. Hormonal Analysis during Stem-Gall Formation in Z. latifolia
2.4.1. Hormone Content Analysis
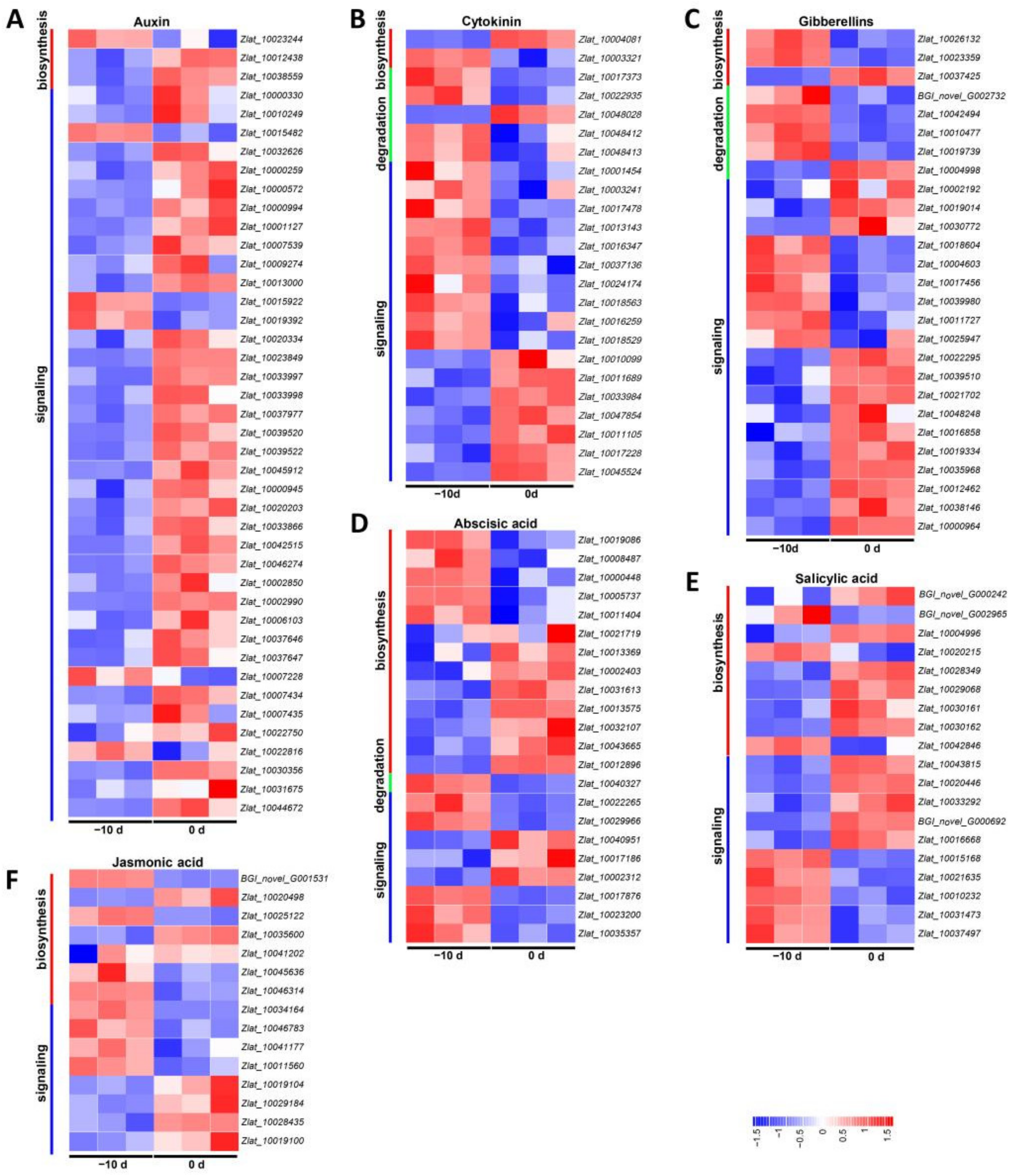
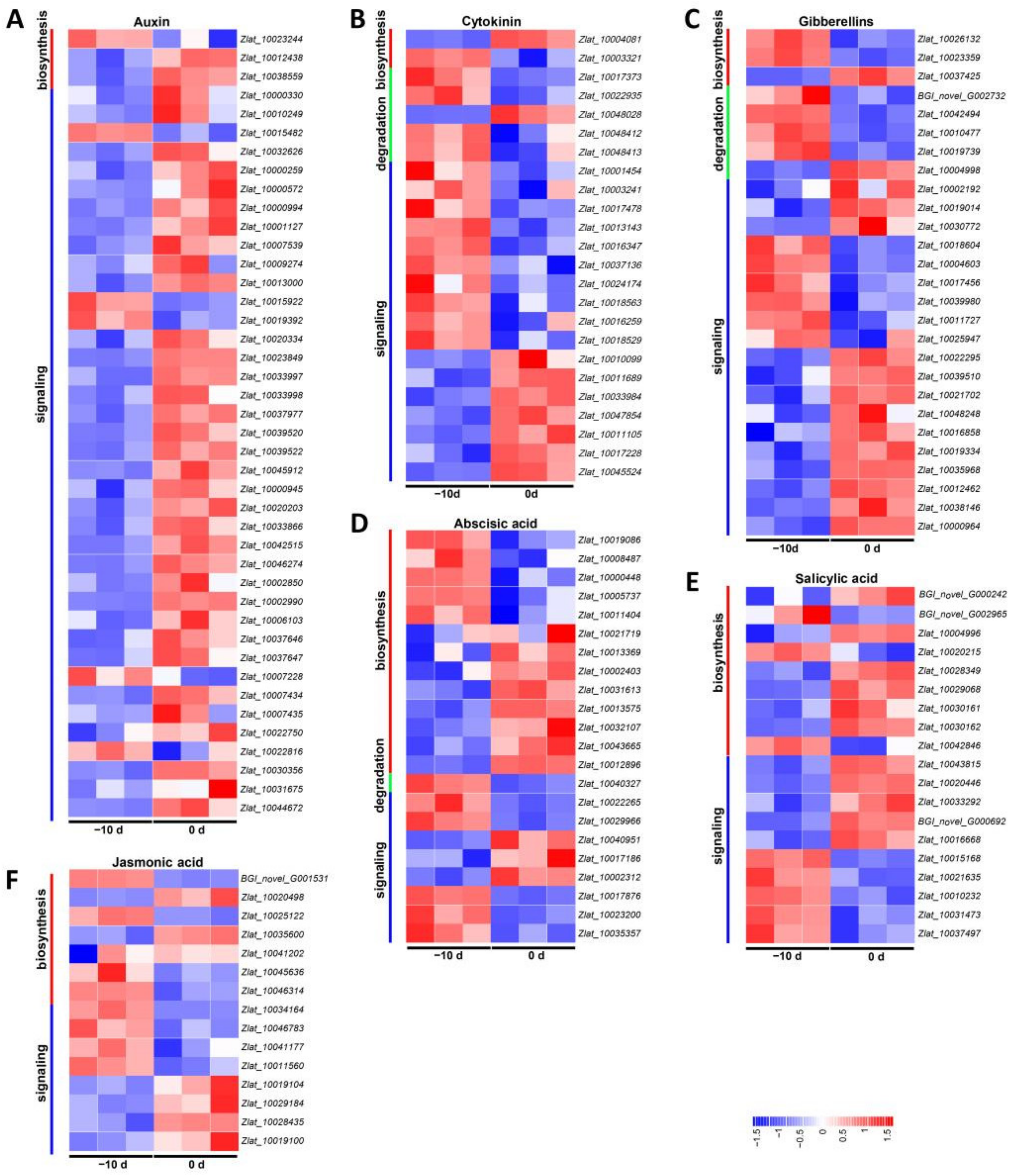
2.4.2. DEGs Involved in Hormone Metabolism and Signal Transduction
2.5. Auxin Regulation of Stem-Gall Formation in Z. latifolia
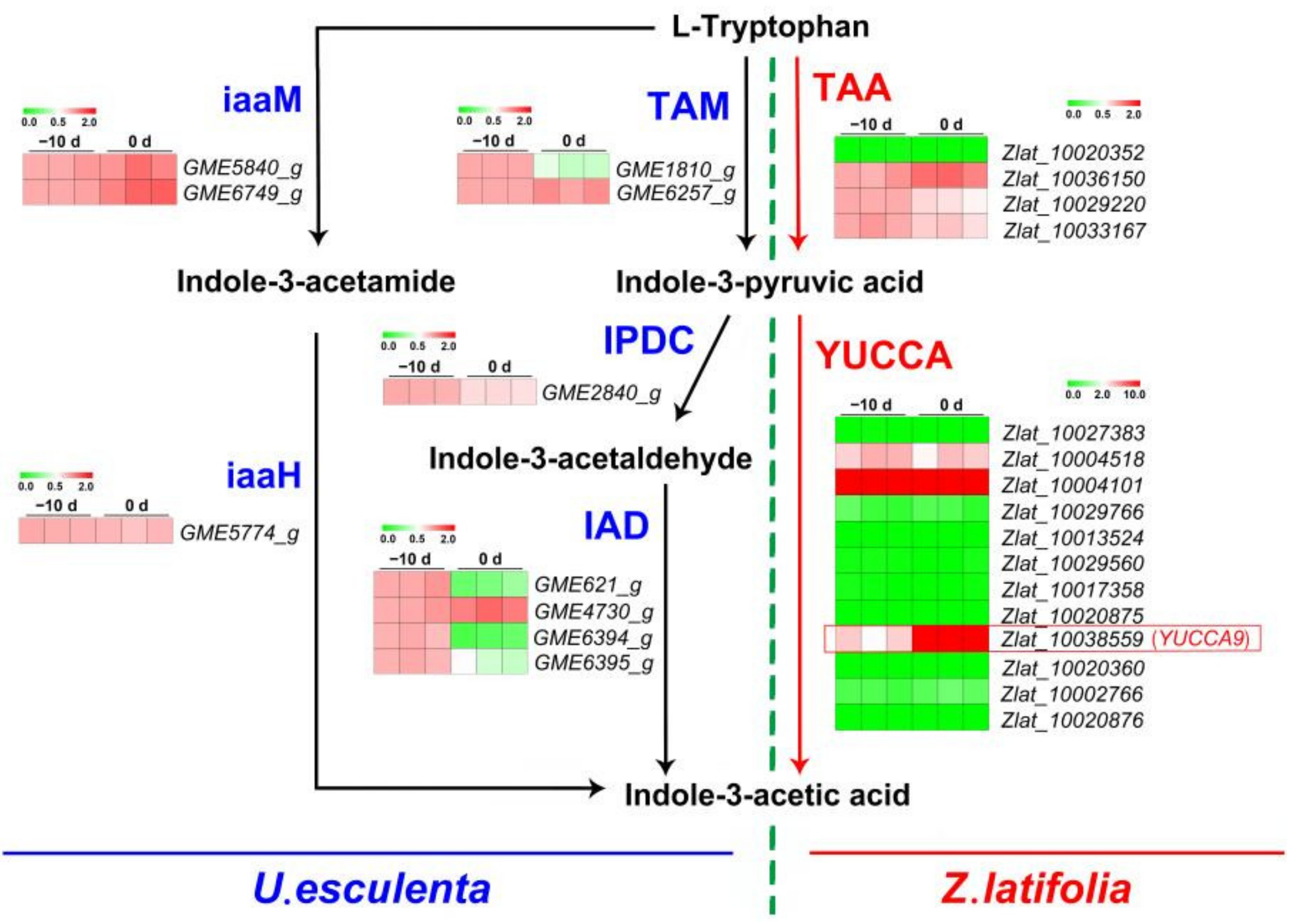
2.5.1. Genes Encoding Enzymes Involved in IAA Biosynthesis in U. esculenta and Z. latifolia
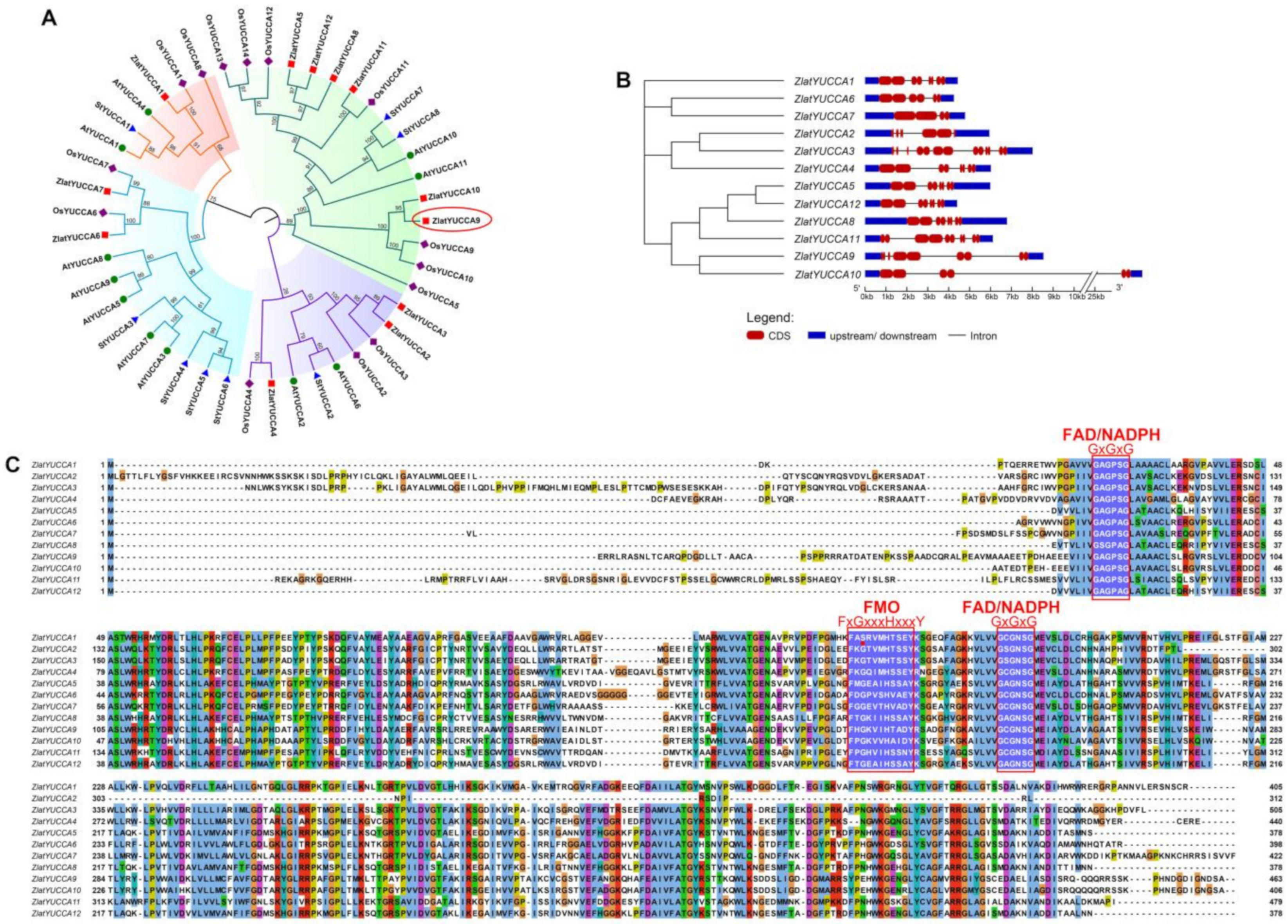
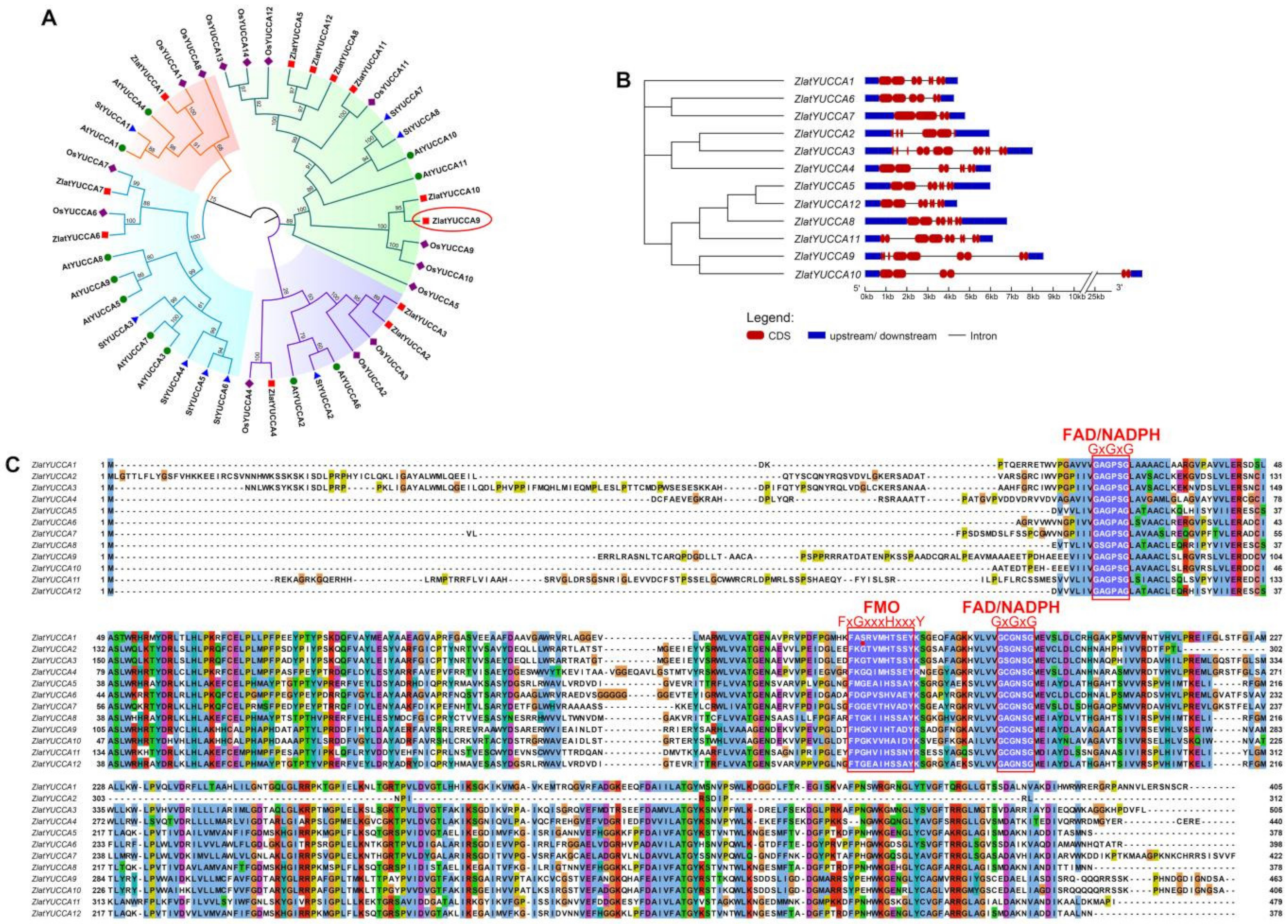
2.5.2. Structural and Functional Characteristic Analysis of the YUCCA Gene Family in Z. latifolia
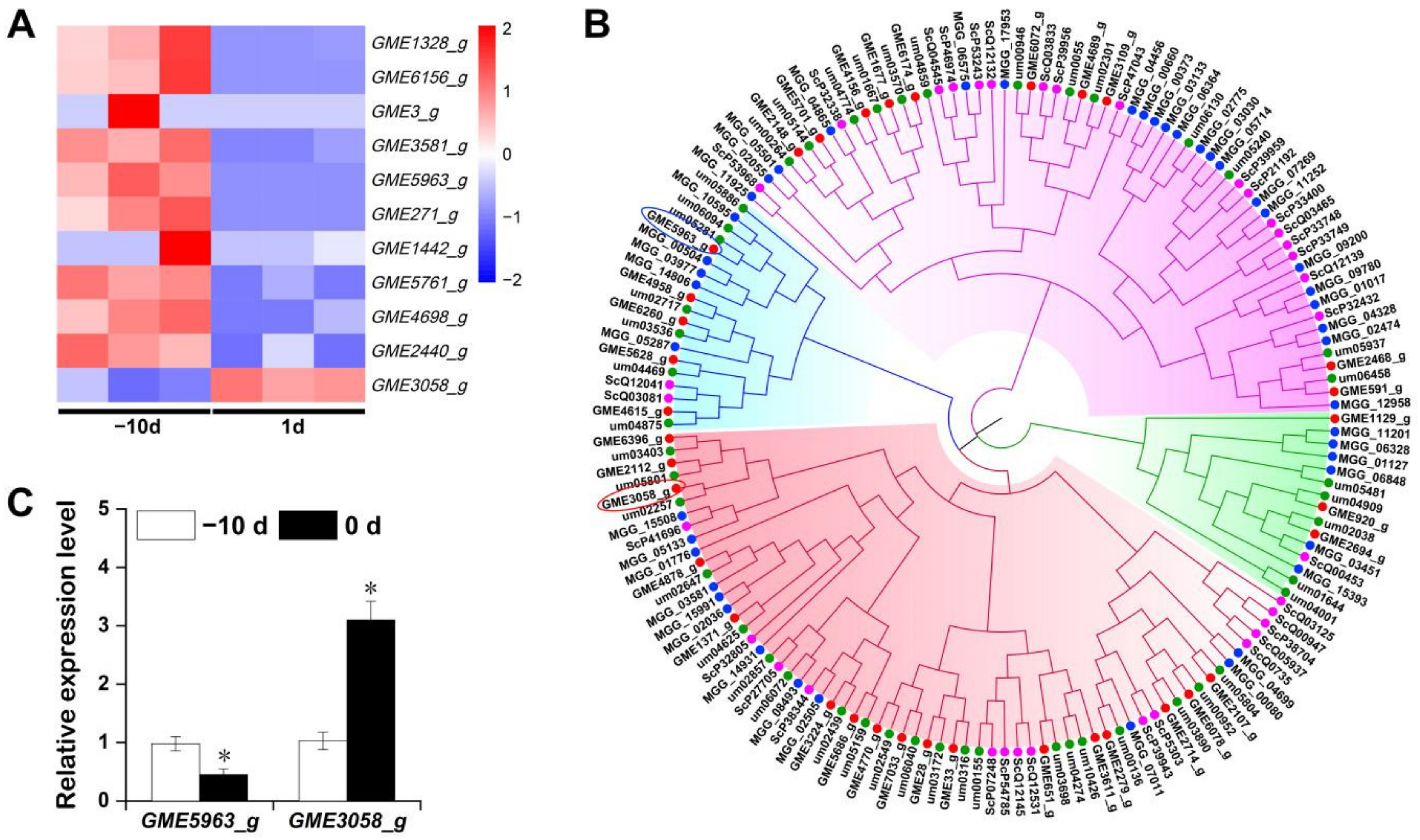
2.5.3. Gene Expression of Enzymes Related to IAA Biosynthesis at the Early Stage of Stem-Gall Formation by qRT-PCR
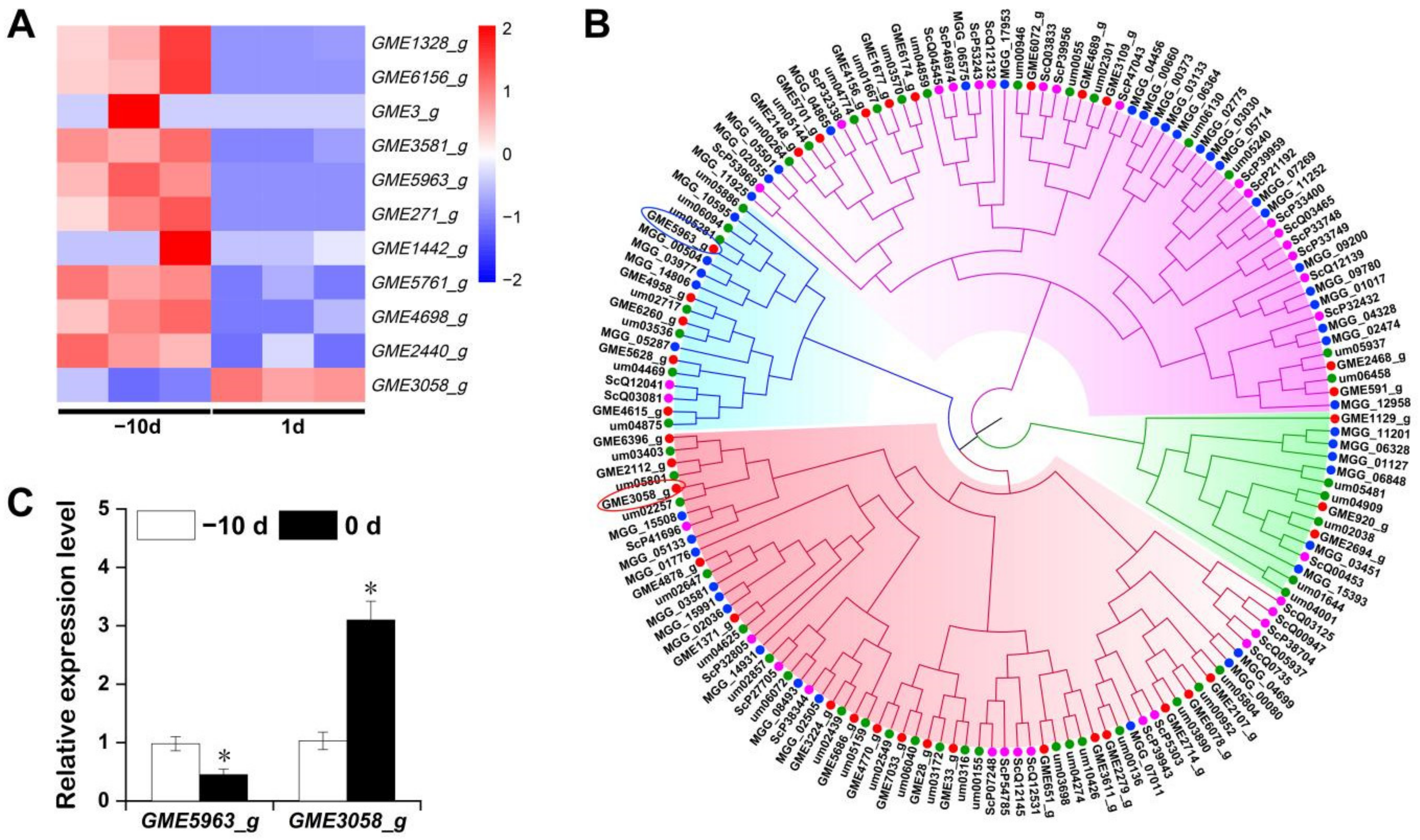
2.6. Expression Pattern of Cys2–His2 (C2H2) Zinc Finger Proteins of U. esculenta at the Early Stage of Stem-Gall Formation
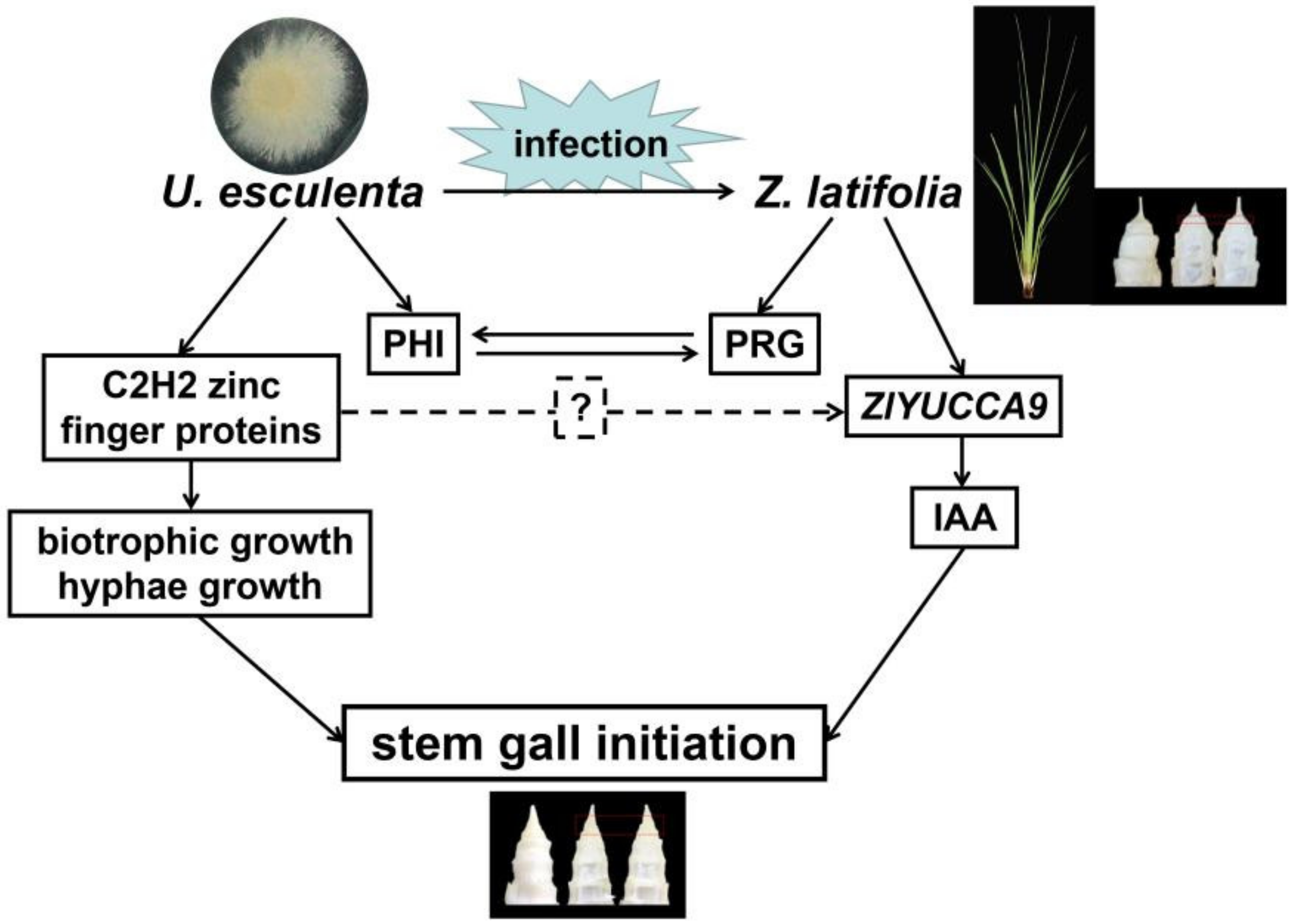
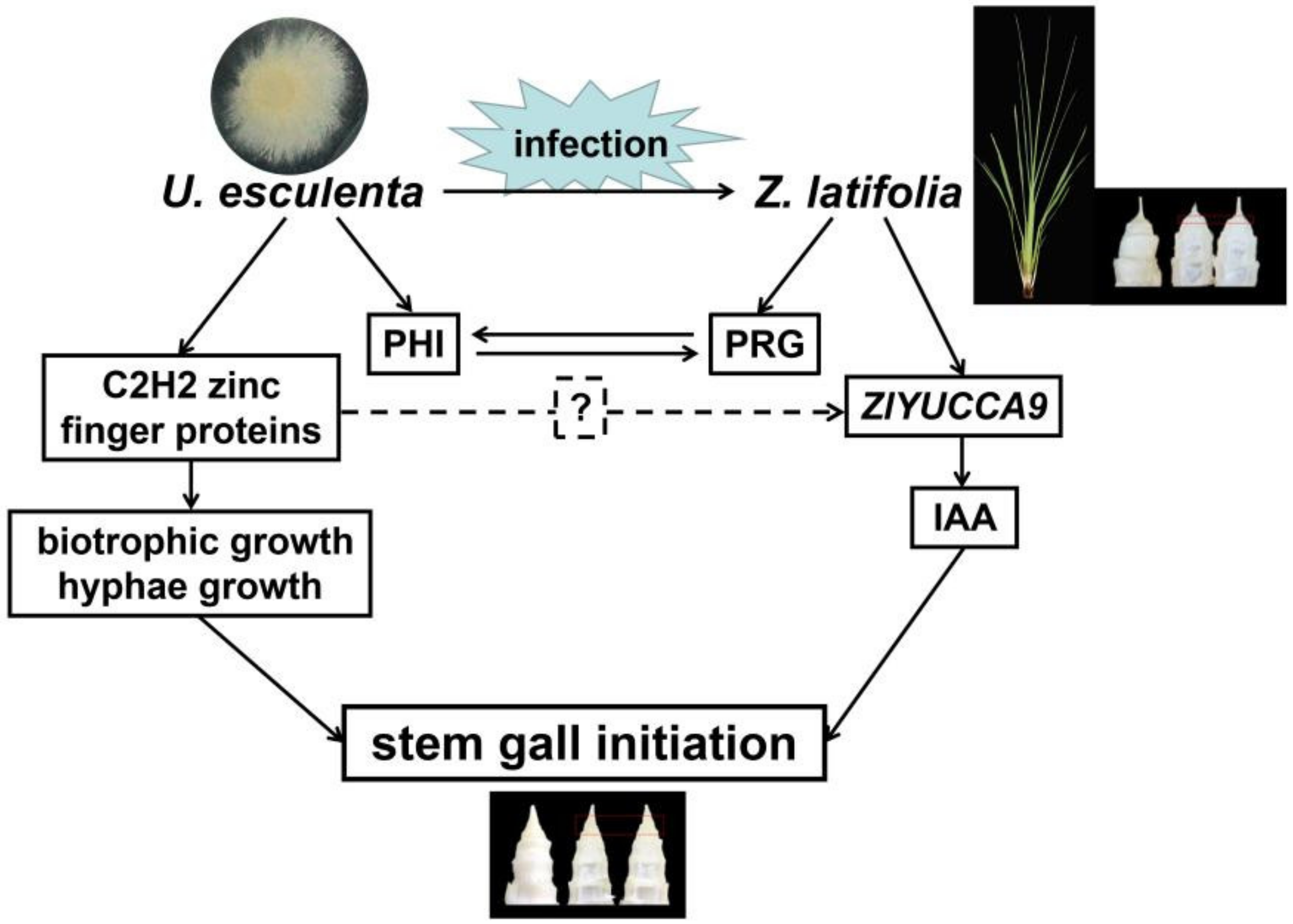
3. Discussion
4. Materials and Methods
4.1. Plant Material and Sample Collection
4.2. Distribution of Hyphae in Z. latifolia
4.3. RNA Isolation, mRNA-Seq Library Preparation and Sequencing
4.4. Preprocessing of mRNA Sequence Data
4.5. Functional Annotation of DEGs
4.6. ESI-HPLC-MS/MS Assay of Endogenous Hormones
4.7. Genome-Wide Identification of the IAA Synthesis Genes and C2H2 Zinc Finger Proteins in U. esculenta and Z. latifolia
4.8. Confirmation of DEGs by qRT-PCR
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Guo, H.B.; Li, S.M.; Peng, J.; Ke, W.D. Zizania latifolia turcz. cultivated in China. Genet. Resour. Crop Evol. 2007, 54, 1211–1217. [Google Scholar] [CrossRef]
- Yang, H.C.; Leu, L.S. Formation and histopathology of galls induced by Ustilago esculenta in Zizania latifolia. Phytopathology 1978, 68, 1572–1576. [Google Scholar] [CrossRef]
- Jose, R.C.; Bengyella, L.; Handique, P.J.; Talukdar, N.C. Cellular and proteomic events associated with the localized formation of smut-gall during Zizania latifolia–Ustilago esculenta interaction. Microb. Pathog. 2018, 126, 79–84. [Google Scholar] [CrossRef]
- Brefort, T.; Doehlemann, G.; Mendoza-Mendoza, A.; Reissmann, S.; Djamei, A.; Kahmann, R. Ustilago maydis as a pathogen. Annu. Rev. Phytopathol. 2009, 47, 423–445. [Google Scholar] [CrossRef] [Green Version]
- Zuo, W.L.; Ökmen, B.; DePotter, J.R.L.; Ebert, M.K.; Redkar, A.; Villamil, J.C.M.; Doehlemann, G. Molecular interactions between smut fungi and their host plants. Annu. Rev. Phytopathol. 2019, 57, 411–430. [Google Scholar] [CrossRef]
- Wang, M.C.; Zhao, S.W.; Zhu, P.L.; Nie, C.Z.P.; Ma, S.P.; Wang, N.F.; Du, X.F.; Zhou, Y.B. Purification, characterization and immunomodulatory activity of water extractable polysaccharides from the swollen culms of Zizania latifolia. Int. J. Biol. Macromol. 2018, 107, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Yan, N.; Luo, X.; Guo, S.; Xue, S.Q.; Liu, J.Q.; Zhang, J.Z.; Guo, D.P. Gene expression in the smut fungus Ustilago esculenta governs swollen gall metamorphosis in Zizania latifolia. Microb. Pathog. 2020, 143, 104107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Chu, F.Q.; Guo, D.P.; Hyde, D.K.; Xie, G.L. Cytology and ultrastructure of interactions between Ustilago esculenta and Zizania latifolia. Mycol. Prog. 2012, 11, 499–508. [Google Scholar] [CrossRef]
- Li, J.; Lu, Z.Y.; Yang, Y.; Hou, J.F.; Yuan, L.Y.; Chen, G.H.; Wang, C.G.; Jia, S.K.; Feng, X.M.; Zhu, S.D. Transcriptome analysis reveals the symbiotic mechanism of Ustilago esculenta-Induced gall formation of Zizania latifolia. Mol. Plant-Microbe Interact. 2021, 34, 168–185. [Google Scholar] [CrossRef]
- Wang, Z.D.; Yan, N.; Wang, Z.H.; Zhang, X.H.; Zhang, J.Z.; Xue, H.M.; Wang, L.X.; Zhan, Q.; Xu, Y.P.; Guo, D.P. RNA-seq analysis provides insight into reprogramming of culm development in Zizania latifolia induced by Ustilago esculenta. Plant Mol. Biol. 2017, 95, 533–547. [Google Scholar] [CrossRef]
- Lambrecht, M.; Okon, Y.; Broek, A.V.; Vanderleyden, J. Indole-3-acetic acid: A reciprocal signalling molecule in bacteria–plant interactions. Trends Microbiol. 2000, 8, 298–300. [Google Scholar] [CrossRef]
- Jain, M.; Khurana, J.P. Transcript profiling reveals diverse roles of auxin-responsive genes during reproductive development and abiotic stress in rice. FEBS J. 2009, 276, 3148–3162. [Google Scholar] [CrossRef] [PubMed]
- Sauer, M.; Robert, S.; KleineVehn, J. Auxin: Simply complicated. J. Exp. Bot. 2013, 64, 2565–2577. [Google Scholar] [CrossRef] [Green Version]
- Vanneste, S.; Friml, J. Auxin: A trigger for change in plant development. Cell 2009, 136, 1005–1016. [Google Scholar] [CrossRef]
- Kazan, K.; Manners, J.M. Linking development to defense: Auxin in plant–pathogen interactions. Trends Plant Sci. 2009, 14, 373–382. [Google Scholar] [CrossRef]
- Yamada, T. The role of Auxin in plant-disease development. Annu. Rev. Phytopathol. 1993, 31, 253–273. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.R.; Tzeng, D.D. Biosynthesis of indole-3-acetic acid by the gall-inducing fungus Ustilago esculenta. J. Biol. Sci. 2004, 4, 744–750. [Google Scholar] [CrossRef] [Green Version]
- Reineke, G.; Heinze, B.; Schirawski, J.; Buettner, H.; Kahmann, R.; Basse, C.W. Indole-3-acetic acid (IAA) biosynthesis in the smut fungus Ustilago maydis and its relevance for increased IAA levels in infected tissue and host tumour formation. Mol. Plant Pathol. 2008, 9, 339–355. [Google Scholar] [CrossRef]
- Tivendale, N.D.; Ross, J.J.; Cohen, J.D. The shifting paradigms of auxin biosynthesis. Trends Plant Sci. 2014, 19, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Puspendu, S.; Frank, K.; Borkovich, K.A. Characterization of indole-3-pyruvic acid pathway–mediated biosynthesis of auxin in Neurospora crassa. PLoS ONE 2018, 13, 0192293. [Google Scholar] [CrossRef]
- Tsavkelova, E.; Oeser, B.; Oren-Young, L.; Israeli, M.; Sasson, Y.; Tudzynski, B.; Sharon, A. Identification and functional characterization of indole-3-acetamide-mediated IAA biosynthesis in plant-associated Fusarium species. Fungal Genet. Biol. 2012, 49, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Won, C.; Shen, X.L.; Mashiguchi, K.; Zheng, Z.Y.; Dai, X.H.; Cheng, Y.F.; Kasahara, H.; Kamiya, Y.; Chory, J.; Zhao, Y.D. Conversion of tryptophan to indole-3-acetic acid by tryptophan aminotransferases of Arabidopsis and YUCCAs in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 18518–18523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.B.; Dai, X.H.; Aoi, Y.K.; Takebayashi, Y.; Yang, L.P.; Guo, X.R.; Zeng, Q.W.; Yu, H.; Kasahara, H.; Zhao, Y.D. Two homologous INDOLE-3-ACETAMIDE (IAM) HYDROLASE genes are required for the auxin effects of IAM in Arabidopsis. J. Genet. Genom. 2020, 47, 157–165. [Google Scholar] [CrossRef]
- Kendrew, S.G. YUCCA: A flavin monooxygenase in auxin biosynthesis. Trends Biochem. Sci. 2001, 26, 218. [Google Scholar] [CrossRef]
- Kim, J.L.; Baek, D.; Park, H.C.; Chun, H.J.; Oh, D.H.; Lee, M.K.; Cha, J.Y.; Kim, W.Y.; Kim, M.C.; Chung, W.S.; et al. Overexpression of Arabidopsis YUCCA6 in potato results in high-auxin developmental phenotypes and enhanced resistance to water deficit. Mol. Plant 2013, 6, 337–349. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Kamiya, N.; Morinaka, Y.; Matsuoka, M.; Sazuka, T. Auxin biosynthesis by the YUCCA genes in rice. Plant Physiol. 2007, 143, 1362–1371. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Christensen, S.K.; Fankhauser, C.; Cashman, J.R.; Cohen, J.D.; Weigel, D.; Chory, J. A role for flavin monooxygenase-like enzymes in auxin biosynthesis. Science 2001, 291, 306–309. [Google Scholar] [CrossRef]
- Doehlemann, G.; Wahl, R.; Horst, R.J.; Voll, L.M.; Usadel, B.; Poree, F.; Stitt, M.; Pons-Kühnemann, J.; Sonnewald, U.; Kahmann, R.; et al. Reprogramming a maize plant: Transcriptional and metabolic changes induced by the fungal biotroph Ustilago maydis. Plant J. 2008, 56, 181–195. [Google Scholar] [CrossRef]
- Kawahara, Y.; Oono, Y.; Kanamori, H.; Matsumoto, T.; Itoh, T.; Minami, E. Simultaneous RNA-Seq analysis of a mixed tran-scriptome of rice and blast fungus interaction. PLoS ONE 2012, 7, e49423. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, P.J.; Thomazella, D.P.; Reis, O.; Prado, P.F.; Rio, M.C.; Fiorin, G.L.; José, J.; Costa, G.G.; Negri, V.A.; Mondego, J.M.C.; et al. High-resolution transcript profiling of the atypical biotrophic interaction between theobroma cacao and the fungal pathogen moniliophthora perniciosa. Plant Cell 2014, 26, 4245–4269. [Google Scholar] [CrossRef] [Green Version]
- Camilios-Neto, D.; Bonato, P.; Wassem, R.; Tadra-Sfeir, M.Z.; Brusamarello-Santos, L.C.; Valdameri, G.; Donatti, L.; Faoro, H.; Weiss, V.A.; Chubatsu, L.S.; et al. Dual RNA-seq transcriptional analysis of wheat roots colonized by Azospirillum brasilense reveals up-regulation of nutrient acquisition and cell cycle genes. BMC Genom. 2014, 15, 378. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhao, Q.; Yang, Q.Y.; Liu, H.; Li, Q.Y.; Yi, X.Q.; Cheng, Y.; Guo, L.; Fan, C.C.; Zhou, Y.M. Comparative transcriptomic analysis uncovers the complex genetic network for resistance to sclerotinia sclerotiorum in Brassica napus. Sci. Rep. 2016, 6, 19007. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Huang, P.; Zhang, L.L.; Shi, Y.K.; Sun, D.D.; Yan, Y.X.; Liu, X.H.; Dong, B.; Chen, G.Q.; Snyder, J.H.; et al. Characterization of 47 Cys2 -His2 zinc finger proteins required for the development and pathogenicity of the rice blast fungus Magnaporthe oryzae. New Phytol. 2016, 211, 1035–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Kief, J.; Auffarth, K.; Farfsing, J.W.; Mahlert, M.; Nieto, F.; Basse, C.W. The Ustilago maydis Cys(2)His(2)-type zinc finger transcription factor Mzr1 regulates fungal gene expression during the biotrophic growth stage. Mol. Microbiol. 2008, 68, 1450–1470. [Google Scholar] [CrossRef] [PubMed]
- Teichmann, B.; Liu, L.; Schink, K.O.; Bölker, M. Activation of the Ustilagic acid biosynthesis gene cluster in Ustilago maydis by the C2H2 zinc finger transcription factor rua1. Appl. Environ. Microbiol. 2010, 76, 2633–2640. [Google Scholar] [CrossRef] [Green Version]
- Weber, I.; Assmann, D.; Thines, E.; Steinberg, G. Polar localizing class V myosin chitin synthases are essential during early plant infection in the plant pathogenic fungus Ustilago maydis. Plant Cell 2005, 18, 225–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, M.; Yamashita, T.; Takahashi, M.; Nakano, Y.; Takeda, T. Identification, cloning, and characterization of β-glucosidase from Ustilago esculenta. Appl. Microbiol. Biotechnol. 2012, 93, 1989–1998. [Google Scholar] [CrossRef]
- Liang, S.W.; Huang, Y.H.; Chiu, J.Y.; Tseng, H.W.; Huang, J.H.; Shen, W.C. The smut fungus Ustilago esculenta has a bipolar mating system with three idiomorphs larger than 500 kb. Fungal Genet. Biol. 2019, 126, 61–74. [Google Scholar] [CrossRef]
- Scofield, S.R.; Tobias, C.M.; Rathjen, J.P.; Chang, J.H.; Lavelle, D.T.; Michelmore, R.W.; Staskawicz, B.J. Molecular basis of gene-for-gene specificity in bacterial speck disease of tomato. Science 1996, 274, 2063–2065. [Google Scholar] [CrossRef] [Green Version]
- Kourelis, J.; Van Der Hoorn, R.A.L. Defended to the nines: 25 Years of resistance gene cloning identifies nine mechanisms for R protein function. Plant Cell 2018, 30, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Roncero, C. The genetic complexity of chitin synthesis in fungi. Curr. Genet. 2002, 41, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.I.; Choi, J.H.; Park, B.C.; Park, Y.H.; Lee, M.Y.; Park, H.M.; Maeng, P.J. Differential expression of the chitin synthase genes of Aspergillus nidulans, chs A, chs B, and chs C, in response to developmental status and environmental factors. Fungal Genet. Biol. 2004, 41, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Martín-Udíroz, M.; Madrid, M.P.; Roncero, M.I.G. Role of chitin synthase genes in Fusarium oxysporum. Microbiology 2004, 150, 3175–3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaepen, S.; Vanderleyden, J. Auxin and plant-microbe interactions. Cold Spring Harb. Perspect. Biol. 2011, 3, a001438. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Wang, S.P. Insights into auxin signaling in plant–pathogen interactions. Front. Plant Sci. 2011, 2, 74. [Google Scholar] [CrossRef] [Green Version]
- Nassar, A.H.; El-Tarabily, K.A.; Sivasithamparam, K. Promotion of plant growth by an auxin-producing isolate of the yeast Williopsis saturnus endophytic in maize (Zea mays L.) roots. Biol. Fertil. Soils 2005, 42, 97–108. [Google Scholar] [CrossRef]
- Glickmann, E.; Gardan, L.; Jacquet, S.; Hussain, S.; Elasri, M.; Petit, A.; Dessaux, Y. Auxin production is a common feature of most pathovars of Pseudomonas syringae. Mol. Plant–Microbe Interact. 1998, 11, 156–162. [Google Scholar] [CrossRef] [Green Version]
- Mole, B.M.; Baltrus, D.; Dangl, J.L.; Grant, S.R. Global virulence regulation networks in phytopathogenic bacteria. Trends Microbiol. 2007, 15, 363–371. [Google Scholar] [CrossRef]
- Yan, S.S.; Che, G.; Ding, L.; Chen, Z.J.; Liu, X.F.; Wang, H.Y.; Zhao, W.S.; Ning, K.; Zhao, J.Y.; Tesfamichael, K.; et al. Different cucumber CsYUC genes regulate response to abiotic stresses and flower development. Sci. Rep. 2016, 6, 20760. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Sharma, P.; Gonzalez, D.H.; Viola, I.L.; Hannapel, D.J. The impact of the long-distance transport of a BEL1-like messenger RNA on development. Plant Physiol. 2013, 161, 760–772. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.P.; Xu, S.X.; Kong, M.Q.; Dai, H.B.; Liu, Y.C.; Miao, M.M. Isolation, identification and artificial inoculation of Ustilago esculenta on Zizania latifolia. Hortic. Plant J. 2020, 7, 347–358. [Google Scholar] [CrossRef]
- Waikhom, S.D.; Louis, B.; Roy, P.; Singh, W.M.; Bharwaj, P.K.; Talukdar, N.C. Scanning electron microscopy of pollen structure throws light on resolving bambusa–dendrocalamus complex: Bamboo flowering evidence. Plant Syst. Evol. 2014, 300, 1261–1268. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped–read alignment with Bowtie2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA–Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.W.; Zhang, X.J. DEGseq: An R package for identifying differentially expressed genes from RNA–seq data. Bioinformatics 2009, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Welti, R.; Wang, X. Quantitative analysis of major plant hormones in crude plant extracts by high-performance liquid chromatography–mass spectrometry. Nat. Protoc. 2010, 5, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. Fast Tree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistics | U. esculenta | Z. latifolia |
|---|---|---|
| Total clean reads (Gb) | 15.25 | 15.25 |
| Average mapping ratio of genome sequence (%) | 4.76 | 84.03 |
| Average mapping ratio of gene set (%) | 3.56 | 66.82 |
| Total gene number | 6330 | 40,856 |
| Known gene number | 6319 | 37,622 |
| Novel gene number | 11 | 3234 |
| Total novel transcript | 1345 | 36,704 |
| Coding transcript | 1013 | 28,273 |
| Noncoding transcript | 332 | 8431 |
| Novel isoform | 1002 | 24,993 |
| Novel transcript | 11 | 3280 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.-P.; Song, S.-X.; Liu, Y.-C.; Zhu, X.-R.; Jiang, Y.-F.; Shi, L.-T.; Jiang, J.-Z.; Miao, M.-M. Mixed Transcriptome Analysis Revealed the Possible Interaction Mechanisms between Zizania latifolia and Ustilago esculenta Inducing Jiaobai Stem-Gall Formation. Int. J. Mol. Sci. 2021, 22, 12258. https://doi.org/10.3390/ijms222212258
Zhang Z-P, Song S-X, Liu Y-C, Zhu X-R, Jiang Y-F, Shi L-T, Jiang J-Z, Miao M-M. Mixed Transcriptome Analysis Revealed the Possible Interaction Mechanisms between Zizania latifolia and Ustilago esculenta Inducing Jiaobai Stem-Gall Formation. International Journal of Molecular Sciences. 2021; 22(22):12258. https://doi.org/10.3390/ijms222212258
Chicago/Turabian StyleZhang, Zhi-Ping, Si-Xiao Song, Yan-Cheng Liu, Xin-Rui Zhu, Yi-Feng Jiang, Ling-Tong Shi, Jie-Zeng Jiang, and Min-Min Miao. 2021. "Mixed Transcriptome Analysis Revealed the Possible Interaction Mechanisms between Zizania latifolia and Ustilago esculenta Inducing Jiaobai Stem-Gall Formation" International Journal of Molecular Sciences 22, no. 22: 12258. https://doi.org/10.3390/ijms222212258
APA StyleZhang, Z.-P., Song, S.-X., Liu, Y.-C., Zhu, X.-R., Jiang, Y.-F., Shi, L.-T., Jiang, J.-Z., & Miao, M.-M. (2021). Mixed Transcriptome Analysis Revealed the Possible Interaction Mechanisms between Zizania latifolia and Ustilago esculenta Inducing Jiaobai Stem-Gall Formation. International Journal of Molecular Sciences, 22(22), 12258. https://doi.org/10.3390/ijms222212258