
Deciphering the Functional Role of RIPK4 in Melanoma

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
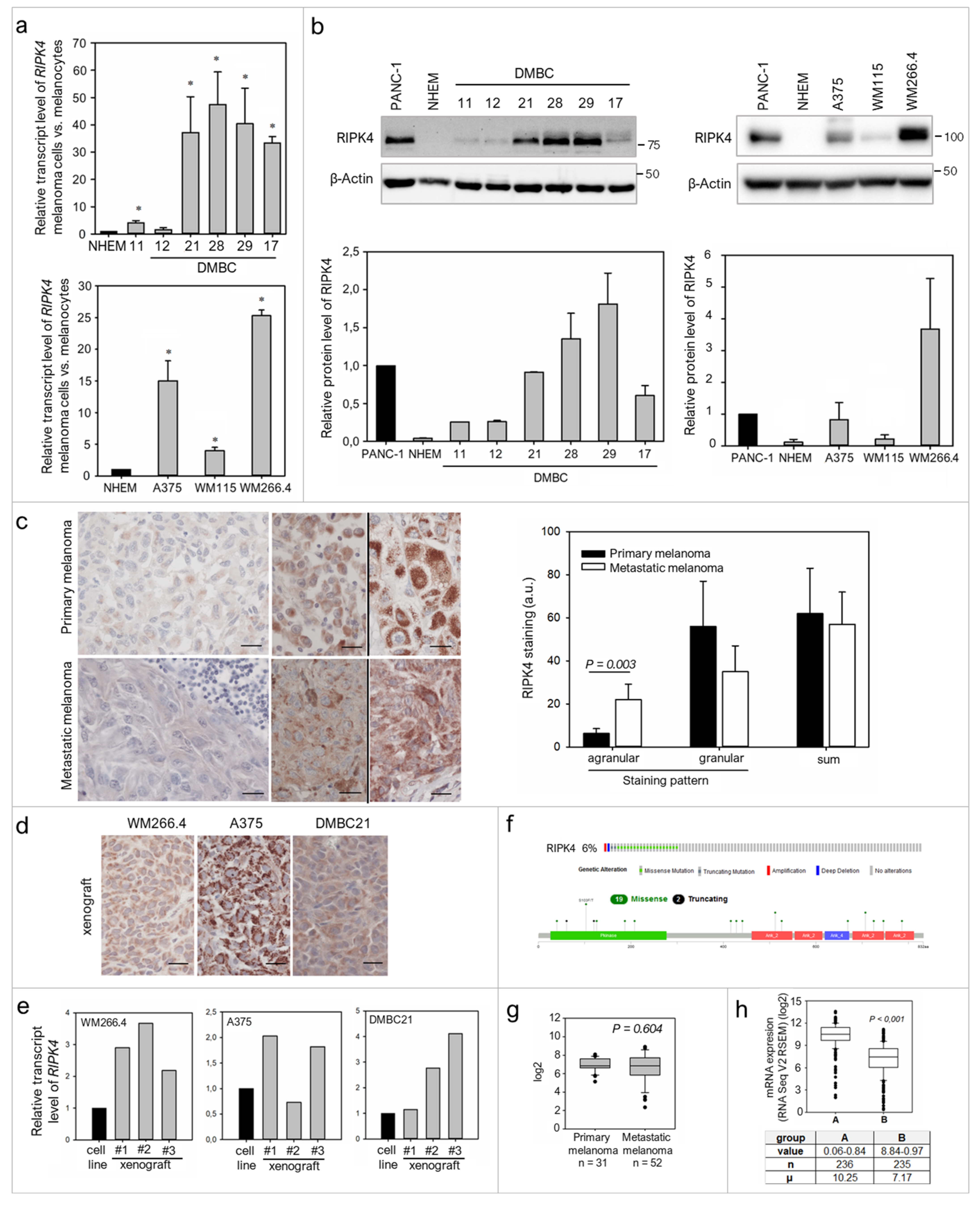
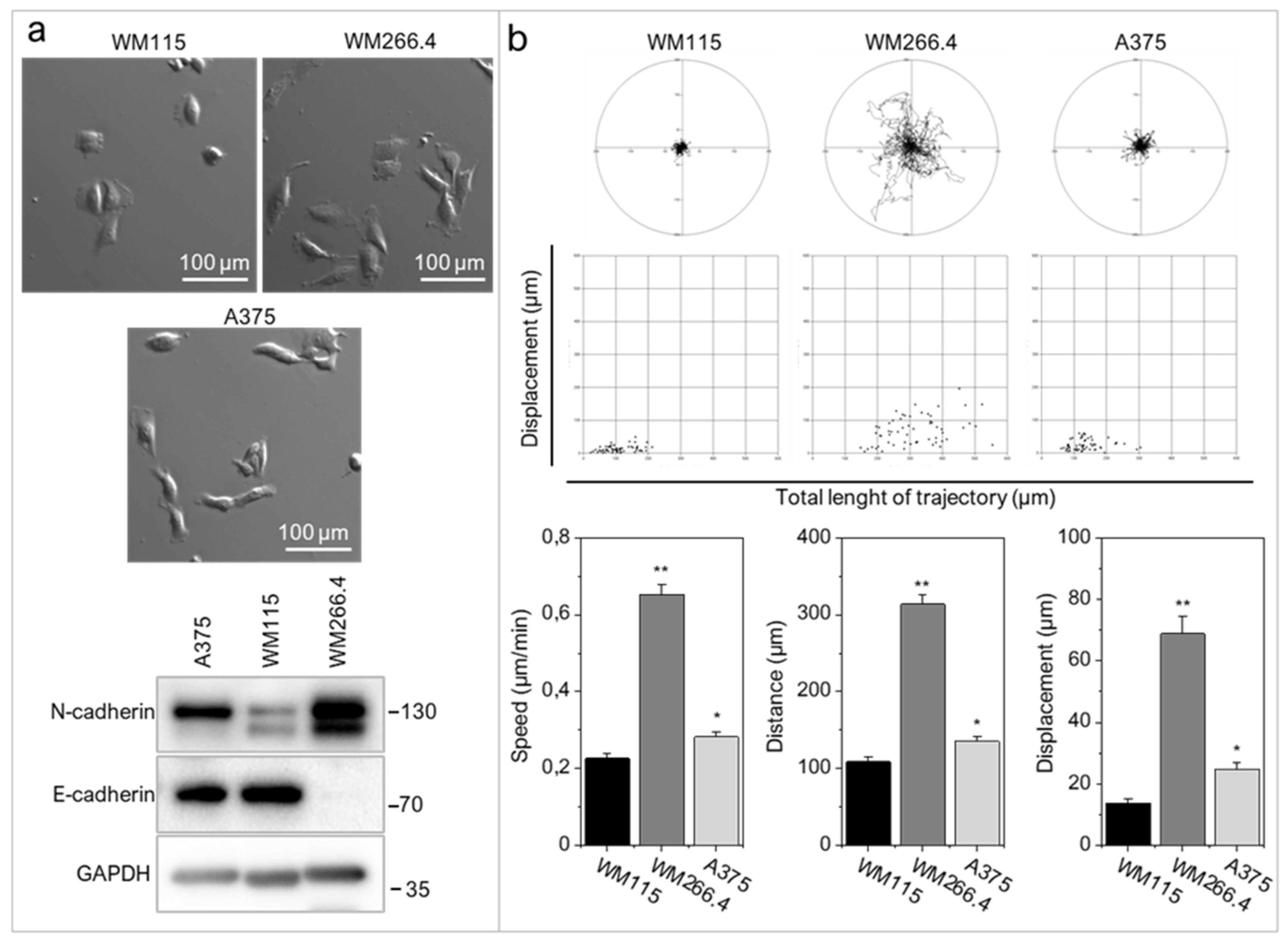
2.1. RIPK4 Is Heterogeneously Expressed in Melanoma Cells
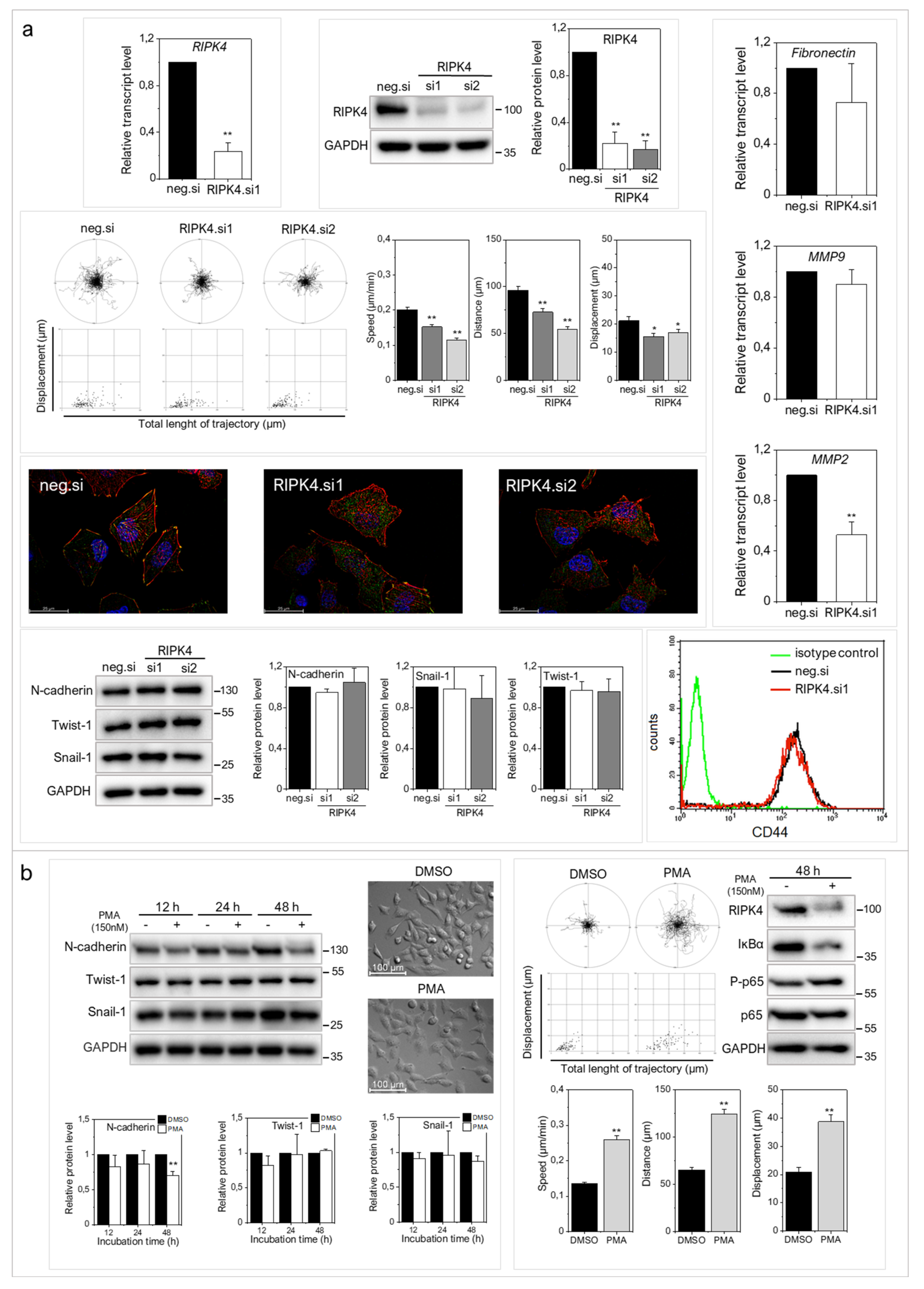
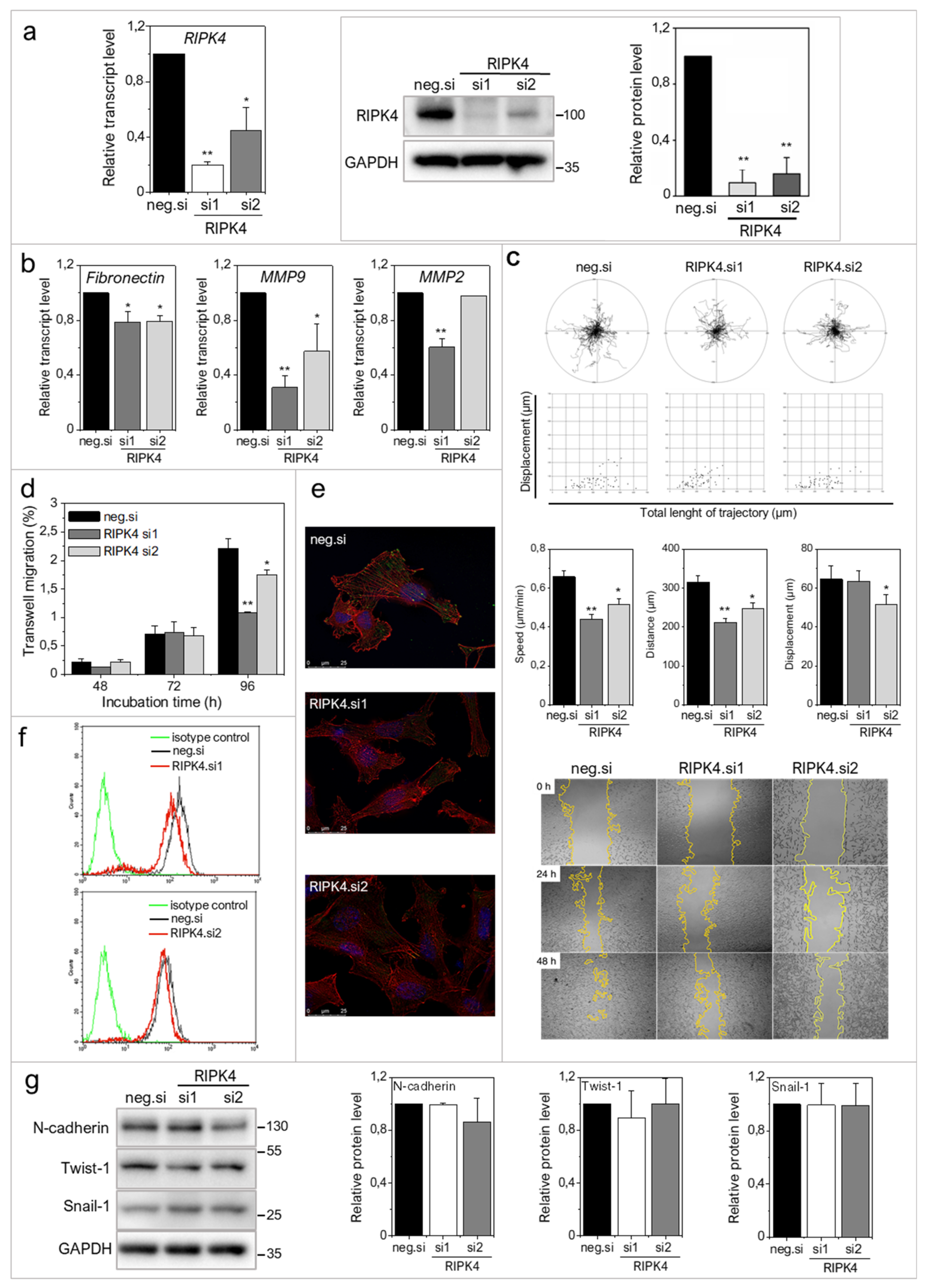
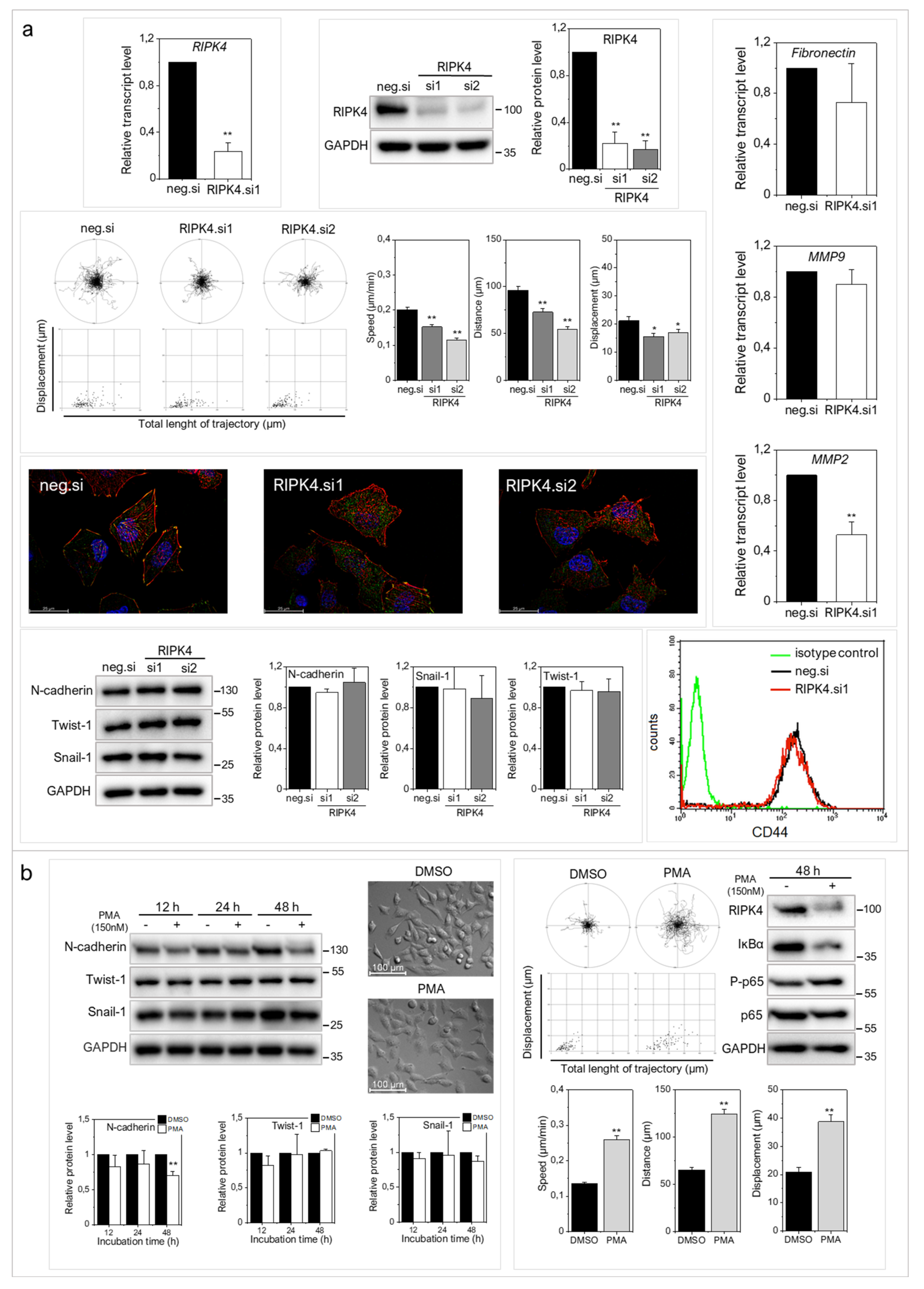
2.2. RIPK4 Down-Regulation Interferes with the Invasive Phenotype of WM266.4 Cells
2.3. RIPK4 Remains under the Control of Protein Kinase C-Dependent Signaling in WM266.4 Cells
2.4. Concerted RIPK4/PKC Signaling Regulates the Invasive Potential of A375 Cells
2.5. Immediate vs. Long-Term Effect of RIPK4 Down-Regulation on NF-κB Activity in WM266.4 and A375 Cells
3. Discussion
4. Materials and Methods
4.1. Clinical Samples
4.2. Geo Database Analysis
4.3. Xenografts
4.4. Cell Culture and Treatment
4.5. Immunostaining
4.6. Cell Transfection
4.7. Western Blot
4.8. RNA Isolation and Quantitative RT-PCR
4.9. Immunofluorescence
4.10. Time-Lapse Video Microscopy
4.11. Transmigration Tests
4.12. Wound Healing Assay
4.13. Flow Cytometry Analysis of CD44
4.14. Cell Cycle
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, L.; Haider, K.; Ponda, M.; Cariappa, A.; Rowitch, D.; Pillai, S. Protein kinase C-associated kinase (PKK), a novel membrane-associated, ankyrin repeat-containing protein kinase. J. Biol. Chem. 2001, 276, 21737–21744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.S.; Oberbeck, N.; Hsiao, Y.C.; Liu, P.; Johnson, A.R.; Dixit, V.M.; Hymowitz, S.G. Crystal Structure of Ripk4 Reveals Dimerization-Dependent Kinase Activity. Structure 2018, 26, 767–777.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meylan, E.; Tschopp, J. The RIP kinases: Crucial integrators of cellular stress. Trends Biochem. Sci. 2005, 30, 151–159. [Google Scholar] [CrossRef]
- Mitchell, K.; O’Sullivan, J.; Missero, C.; Blair, E.; Richardson, R.; Anderson, B.; Antonini, D.; Murray, J.C.; Shanske, A.L.; Schutte, B.C.; et al. Exome sequence identifies RIPK4 as the Bartsocas-Papas syndrome locus. Am. J. Hum. Genet. 2012, 90, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalay, E.; Sezgin, O.; Chellappa, V.; Mutlu, M.; Morsy, H.; Kayserili, H.; Kreiger, E.; Cansu, A.; Toraman, B.; Abdalla, E.M.; et al. Mutations in RIPK4 cause the autosomal-recessive form of popliteal pterygium syndrome. Am. J. Hum. Genet. 2012, 90, 76–85. [Google Scholar] [CrossRef] [Green Version]
- Holland, P.; Willis, C.; Kanaly, S.; Glaccum, M.; Warren, A.; Charrier, K.; Murison, J.; Derry, J.; Virca, G.; Bird, T.; et al. RIP4 is an ankyrin repeat-containing kinase essential for keratinocyte differentiation. Curr. Biol. 2002, 12, 1424–1428. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wei, Q.; He, Z. Insight Into the Function of RIPK4 in Keratinocyte Differentiation and Carcinogenesis. Front. Oncol. 2020, 10, 1562. [Google Scholar] [CrossRef]
- Heim, D.; Cornils, K.; Schulze, K.; Fehse, B.; Lohse, A.W.; Brümmendorf, T.H.; Wege, H. Retroviral insertional mutagenesis in telomerase-immortalized hepatocytes identifies RIPK4 as novel tumor suppressor in human hepatocarcinogenesis. Oncogene 2015, 34, 364–372. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, W.; Zhou, Y.; Xu, W.; Wang, H. RIPK4 is downregulated in poorly differentiated tongue cancer and is associated with migration/invasion and cisplatin-induced apoptosis. Int. J. Biol. Mark. 2014, 29, e150–e159. [Google Scholar] [CrossRef]
- Kopparam, J.; Chiffelle, J.; Angelino, P.; Piersigilli, A.; Zangger, N.; Delorenzi, M.; Meylan, E. RIP4 inhibits STAT3 signaling to sustain lung adenocarcinoma differentiation. Cell Death Differ. 2017, 24, 1761–1771. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.Q.; Li, F.F.; Zhang, J.B.; Zhou, T.J.; Xue, W.Q.; Zheng, X.H.; Chen, Y.B.; Liao, X.Y.; Zhang, L.; Zhang, S.D.; et al. Increased RIPK4 expression is associated with progression and poor prognosis in cervical squamous cell carcinoma patients. Sci. Rep. 2015, 5, 11955. [Google Scholar] [CrossRef]
- Azizmohammadi, S.; Azizmohammadi, S.; Safari, A.; Kaghazian, M.; Sadrkhanlo, M.; Behnod, V.; Seifoleslami, M. High-Level Expression of RIPK4 and EZH2 Contributes to Lymph Node Metastasis and Predicts Favorable Prognosis in Patients With Cervical Cancer. Oncol. Res. 2017, 25, 495–501. [Google Scholar] [CrossRef]
- Qi, Z.H.; Xu, H.X.; Zhang, S.R.; Xu, J.Z.; Li, S.; Gao, H.L.; Jin, W.; Wang, W.Q.; Wu, C.T.; Ni, Q.X.; et al. RIPK4/PEBP1 axis promotes pancreatic cancer cell migration and invasion by activating RAF1/MEK/ERK signaling. Int. J. Oncol. 2018, 52, 1105–1116. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Hawthorne, S.; Zhao, L.; Hanson, M.; Kanas, G.; Davis, C.; Robinson, D.; Turnure, M.; Clark, O. Treatment of Advanced/Metastatic Melanoma in the United States and Western Europe: Results of the CancerMPact Survey. Cancer Manag. Res. 2020, 12, 5633–5639. [Google Scholar] [CrossRef]
- Sacchetto, L.; Rosso, S.; Comber, H.; Bouchardy, C.; Broganelli, P.; Galceran, J.; Hackl, M.; Katalinic, A.; Louwman, M.; Robsahm, T.E.; et al. Skin melanoma deaths within 1 or 3 years from diagnosis in Europe. Int. J. Cancer 2021, 148, 2898–2905. [Google Scholar] [CrossRef]
- Bai, X.; Flaherty, K.T. Targeted and immunotherapies in BRAF mutant melanoma: Where we stand and what to expect. Br. J. Dermatol. 2021, 185, 253–262. [Google Scholar] [CrossRef]
- Atkins, M.B.; Curiel-Lewandrowski, C.; Fisher, D.E.; Swetter, S.M.; Tsao, H.; Aguirre-Ghiso, J.A.; Soengas, M.S.; Weeraratna, A.T.; Flaherty, K.T.; Herlyn, M.; et al. The State of Melanoma: Emergent Challenges and Opportunities. Clin. Cancer Res. 2021, 27, 2678–2697. [Google Scholar] [CrossRef]
- Dumaz, N.; Lebbé, C. New perspectives on targeting RAF, MEK and ERK in melanoma. Curr. Opin. Oncol. 2021, 33, 120–126. [Google Scholar] [CrossRef]
- Madonna, G.; Ullman, C.D.; Gentilcore, G.; Palmieri, G.; Ascierto, P.A. NF-κB as potential target in the treatment of melanoma. J. Transl. Med. 2012, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, S.J.; Mijatov, B.; Gunatilake, D.; Gowrishankar, K.; Tiffen, J.; James, W.; Jin, L.; Pupo, G.; Cullinane, C.; McArthur, G.A.; et al. Control of NF-kB activity in human melanoma by bromodomain and extra-terminal protein inhibitor I-BET151. Pigment Cell Melanoma Res. 2014, 27, 1126–1137. [Google Scholar] [CrossRef]
- Ratnayake, W.S.; Apostolatos, C.A.; Apostolatos, A.H.; Schutte, R.J.; Huynh, M.A.; Ostrov, D.A.; Acevedo-Duncan, M. Oncogenic PKC-ι activates Vimentin during epithelial-mesenchymal transition in melanoma; a study based on PKC-ι and PKC-ζ specific inhibitors. Cell Adh. Migr. 2018, 12, 447–463. [Google Scholar] [CrossRef]
- Denning, M.F. Specifying protein kinase C functions in melanoma. Pigment Cell Melanoma Res. 2012, 25, 466–476. [Google Scholar] [CrossRef]
- Gajos-Michniewicz, A.; Czyz, M. WNT Signaling in Melanoma. Int. J. Mol. Sci. 2020, 21, 4852. [Google Scholar] [CrossRef]
- Muto, A.; Ruland, J.; McAllister-Lucas, L.M.; Lucas, P.C.; Yamaoka, S.; Chen, F.F.; Lin, A.; Mak, T.W.; Núñez, G.; Inohara, N. Protein kinase C-associated kinase (PKK) mediates Bcl10-independent NF-kappa B activation induced by phorbol ester. J. Biol. Chem. 2002, 277, 31871–31876. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; McGann, J.C.; Liu, B.Y.; Hannoush, R.N.; Lill, J.R.; Pham, V.; Newton, K.; Kakunda, M.; Liu, J.; Yu, C.; et al. Phosphorylation of Dishevelled by protein kinase RIPK4 regulates Wnt signaling. Science 2013, 339, 1441–1445. [Google Scholar] [CrossRef] [Green Version]
- Cariappa, A.; Chen, L.; Haider, K.; Tang, M.; Nebelitskiy, E.; Moran, S.T.; Pillai, S. A catalytically inactive form of protein kinase C-associated kinase/receptor interacting protein 4, a protein kinase C beta-associated kinase that mediates NF-kappa B activation, interferes with early B cell development. J. Immunol. 2003, 171, 1875–1880. [Google Scholar] [CrossRef] [Green Version]
- Yi, Z.; Pu, Y.; Gou, R.; Chen, Y.; Ren, X.; Liu, W.; Dong, P. Silencing of RIPK4 inhibits epithelial-mesenchymal transition by inactivating the Wnt/β-catenin signaling pathway in osteosarcoma. Mol. Med. Rep. 2020, 21, 1154–1162. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Y.; Zeng, Q.H.; Cao, P.G.; Xie, D.; Chen, X.; Yang, F.; He, L.Y.; Dai, Y.B.; Li, J.J.; Liu, X.M.; et al. RIPK4 promotes bladder urothelial carcinoma cell aggressiveness by upregulating VEGF-A through the NF-κB pathway. Br. J. Cancer 2018, 118, 1617–1627. [Google Scholar] [CrossRef]
- De Groote, P.; Tran, H.T.; Fransen, M.; Tanghe, G.; Urwyler, C.; De Craene, B.; Leurs, K.; Gilbert, B.; Van Imschoot, G.; De Rycke, R.; et al. A novel RIPK4-IRF6 connection is required to prevent epithelial fusions characteristic for popliteal pterygium syndromes. Cell Death Differ. 2015, 22, 1012–1024. [Google Scholar] [CrossRef] [Green Version]
- Urwyler-Rösselet, C.; Tanghe, G.; Leurs, K.; Gilbert, B.; De Rycke, R.; De Bruyne, M.; Lippens, S.; Bartunkova, S.; De Groote, P.; Niessen, C.; et al. Keratinocyte-Specific Ablation of RIPK4 Allows Epidermal Cornification but Impairs Skin Barrier Formation. J. Investig. Dermatol. 2018, 138, 1268–1278. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.; Jiang, S.; Li, Y.; Yue, J.; Gou, X.; Chen, S.Y.; Zhao, Y.; Schober, M.; Tan, M.; Wu, X. Phosphorylation of Pkp1 by RIPK4 regulates epidermal differentiation and skin tumorigenesis. EMBO J. 2017, 36, 1963–1980. [Google Scholar] [CrossRef] [PubMed]
- Damm, S.; Koefinger, P.; Stefan, M.; Wels, C.; Mehes, G.; Richtig, E.; Kerl, H.; Otte, M.; Schaider, H. HGF-promoted motility in primary human melanocytes depends on CD44v6 regulated via NF-kappa B, Egr-1, and C/EBP-beta. J. Investig. Dermatol. 2010, 130, 1893–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.M.; Lyu, Y.L.; Cai, L. NF-κB affects proliferation and invasiveness of breast cancer cells by regulating CD44 expression. PLoS ONE. 2014, 9, e106966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.W.; Schifano, M.; Oleksyn, D.; Jordan, C.T.; Ryan, D.; Insel, R.; Zhao, J.; Chen, L. Protein kinase C-associated kinase regulates NF-κB activation through inducing IKK activation. Int. J. Oncol. 2014, 45, 1707–1714. [Google Scholar] [CrossRef]
- Adams, S.; Pankow, S.; Werner, S.; Munz, B. Regulation of NF-kappaB activity and keratinocyte differentiation by the RIP4 protein: Implications for cutaneous wound repair. J. Investig. Dermatol. 2007, 127, 538–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czyz, J. The stage-specific function of gap junctions during tumourigenesis. Cell Mol. Biol. Lett. 2008, 13, 92–102. [Google Scholar] [CrossRef]
- Czyż, J.; Szpak, K.; Madeja, Z. The role of connexins in prostate cancer promotion and progression. Nat. Rev. Urol. 2012, 9, 274–282. [Google Scholar] [CrossRef]
- Czyż, J.; Piwowarczyk, K.; Paw, M.; Luty, M.; Wróbel, T.; Catapano, J.; Madeja, Z.; Ryszawy, D. Connexin-dependent intercellular stress signaling in tissue homeostasis and tumor development. Acta Biochim. Pol. 2017, 64, 377–389. [Google Scholar] [CrossRef]
- Xu, L.; Shen, S.S.; Hoshida, Y.; Subramanian, A.; Ross, K.; Brunet, J.P.; Wagner, S.N.; Ramaswamy, S.; Mesirov, J.P.; Hynes, R.O. Gene expression changes in an animal melanoma model correlate with aggressiveness of human melanoma metastases. Mol. Cancer Res. 2008, 6, 760–769. [Google Scholar] [CrossRef] [Green Version]
- Czyz, M.; Sztiller-Sikorska, M.; Gajos-Michniewicz, A.; Osrodek, M.; Hartman, M.L. Plasticity of Drug-Naïve and Vemurafenib- or Trametinib-Resistant Melanoma Cells in Execution of Differentiation/Pigmentation Program. J. Oncol. 2019, 2019, 1697913. [Google Scholar] [CrossRef] [Green Version]
- Hartman, M.L.; Gajos-Michniewicz, A.; Talaj, J.A.; Mielczarek-Lewandowska, A.; Czyz, M. BH3 mimetics potentiate pro-apoptotic activity of encorafenib in BRAFV600E melanoma cells. Cancer Lett. 2021, 499, 122–136. [Google Scholar] [CrossRef]
- Brożyna, A.A.; Jóźwicki, W.; Jetten, A.M.; Slominski, A.T. On the relationship between VDR, RORα and RORγ receptors expression and HIF1-α levels in human melanomas. Exp. Dermatol. 2019, 28, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Brożyna, A.A.; Kim, T.K.; Zabłocka, M.; Jóźwicki, W.; Yue, J.; Tuckey, R.C.; Jetten, A.M.; Slominski, A.T. Association among Vitamin D, Retinoic Acid-Related Orphan Receptors, and Vitamin D Hydroxyderivatives in Ovarian Cancer. Nutrients 2020, 12, 3541. [Google Scholar] [CrossRef] [PubMed]
- Skalniak, L.; Smejda, M.; Cierniak, A.; Adamczyk, A.; Konieczny, P.; Madej, E.; Wolnicka-Glubisz, A. p38 but not p53 is responsible for UVA-induced MCPIP1 expression. Mech. Ageing Dev. 2018, 172, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Kuryłowicz, K.; Cierniak, A.; Madej, E.; Skalniak, Ł.; Wolnicka-Głubisz, A. Resveratrol enhances apoptosis induced by the heterocyclic aromatic amines in p53-wt LoVo cells, but not in p53-deficient HaCaT cells. Acta Biochim. Pol. 2020, 67, 605–611. [Google Scholar] [CrossRef]
- Ryszawy, D.; Pudełek, M.; Kochanowski, P.; Janik-Olchawa, N.; Bogusz, J.; Rąpała, M.; Koczurkiewicz, P.; Mikołajczyk, J.; Borek, I.; Kędracka-Krok, S.; et al. High bisphenol A concentrations augment the invasiveness of tumor cells through Snail-1/Cx43/ERRγ-dependent epithelial-mesenchymal transition. Toxicol. In Vitro 2020, 62, 104676. [Google Scholar] [CrossRef]
- Ryszawy, D.; Pudełek, M.; Catapano, J.; Ciarach, M.; Setkowicz, Z.; Konduracka, E.; Madeja, Z.; Czyż, J. High doses of sodium ascorbate interfere with the expansion of glioblastoma multiforme cells in vitro and in vivo. Life Sci. 2019, 232, 116657. [Google Scholar] [CrossRef]
- Wolnicka-Glubisz, A.; King, W.; Noonan, F.P. SCA-1+ cells with an adipocyte phenotype in neonatal mouse skin. J. Investig. Dermatol. 2005, 125, 383–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, M.L.; Sztiller-Sikorska, M.; Czyz, M. Whole-exome sequencing reveals novel genetic variants associated with diverse phenotypes of melanoma cells. Mol. Carcinog. 2019, 58, 588–602. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madej, E.; Ryszawy, D.; Brożyna, A.A.; Czyz, M.; Czyz, J.; Wolnicka-Glubisz, A. Deciphering the Functional Role of RIPK4 in Melanoma. Int. J. Mol. Sci. 2021, 22, 11504. https://doi.org/10.3390/ijms222111504
Madej E, Ryszawy D, Brożyna AA, Czyz M, Czyz J, Wolnicka-Glubisz A. Deciphering the Functional Role of RIPK4 in Melanoma. International Journal of Molecular Sciences. 2021; 22(21):11504. https://doi.org/10.3390/ijms222111504
Chicago/Turabian StyleMadej, Ewelina, Damian Ryszawy, Anna A. Brożyna, Malgorzata Czyz, Jaroslaw Czyz, and Agnieszka Wolnicka-Glubisz. 2021. "Deciphering the Functional Role of RIPK4 in Melanoma" International Journal of Molecular Sciences 22, no. 21: 11504. https://doi.org/10.3390/ijms222111504
APA StyleMadej, E., Ryszawy, D., Brożyna, A. A., Czyz, M., Czyz, J., & Wolnicka-Glubisz, A. (2021). Deciphering the Functional Role of RIPK4 in Melanoma. International Journal of Molecular Sciences, 22(21), 11504. https://doi.org/10.3390/ijms222111504