
The Role of Mitochondria in Acute Kidney Injury and Chronic Kidney Disease and Its Therapeutic Potential

Abstract
1. Introduction
2. An Overview of AKI and CKD as Well as AKI to CKD Transition
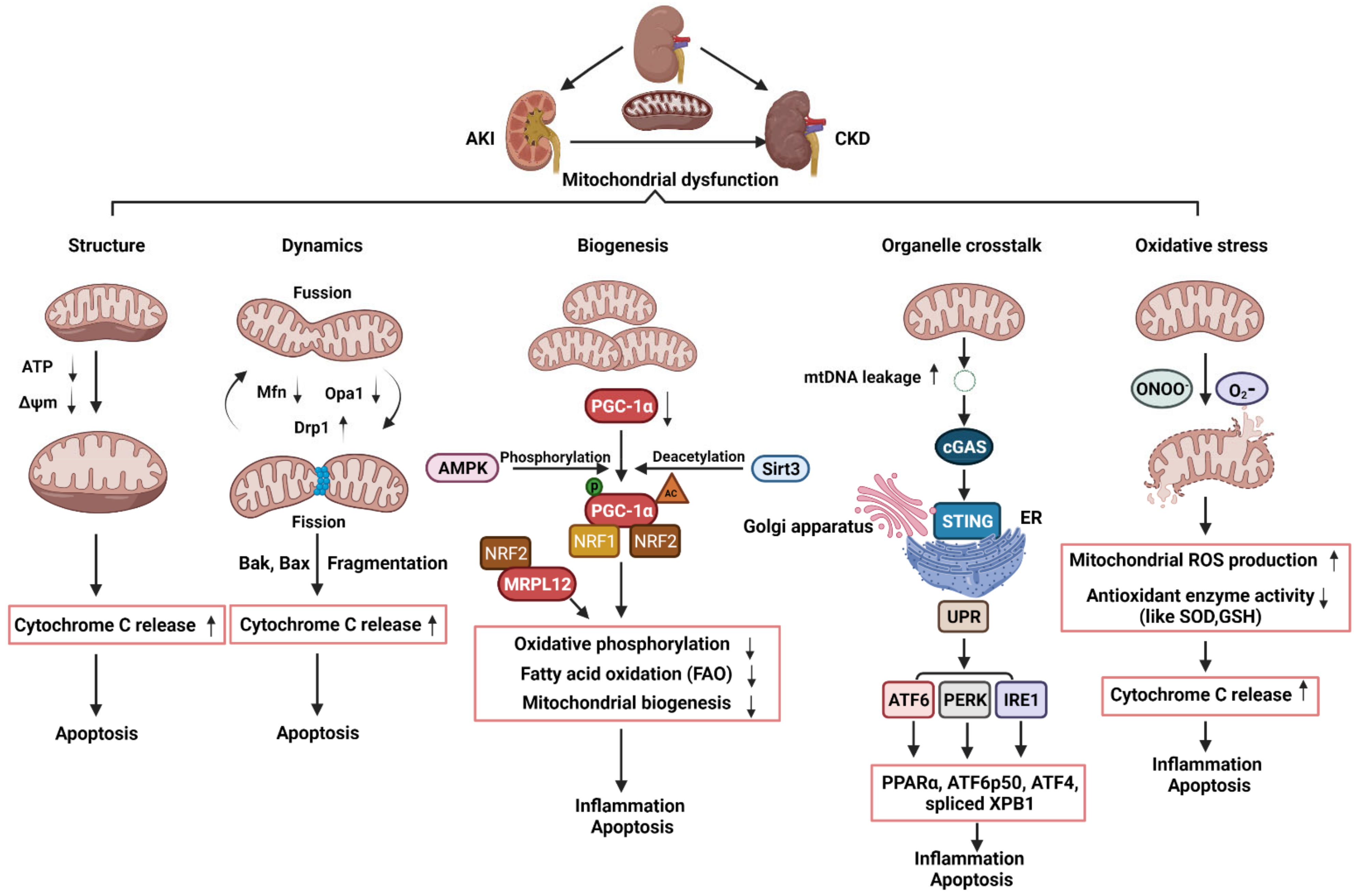
3. An Overview of Mitochondrial Structure, Biogenesis, and Dynamics
3.1. Mitochondrial Structure
3.2. Mitochondrial Dynamics
3.3. Mitochondrial Biogenesis
4. The Roles of Mitochondrial in AKI, CKD, and AKI to CKD Transition
4.1. The Roles of Mitochondrial Structure, Dynamics, and Biogenesis in AKI
4.2. The Roles of Mitochondrial Dynamics and Biogenesis in CKD
4.3. The Roles of Mitochondrial Dysfunction and Its Crosstalk with ER in the Transition of AKI to CKD
5. Mitochondrial Oxidative Stress in AKI and CKD
6. Mitochondrial Targeting for AKI and CKD Therapy
6.1. Cardiolipin Protection Agents
6.2. Modulating Mitochondrial Dynamics
6.3. Altering Mitochondrial Biogenesis
6.4. Altering Mitochondrial Biogenesis
6.5. mPTP Inhibitors
6.6. Mitochondrial-Targeted Antioxidants
7. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Makris, K.; Spanou, L. Acute Kidney Injury: Definition, Pathophysiology and Clinical Phenotypes. Clin. Biochem. Rev. 2016, 37, 85–98. [Google Scholar] [PubMed]
- Lee, S.A.; Cozzi, M.; Bush, E.L.; Rabb, H. Distant Organ Dysfunction in Acute Kidney Injury: A Review. Am. J. Kidney Dis. 2018, 72, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Lameire, N.; van Biesen, W.; Vanholder, R. Acute renal failure. Lancet 2005, 365, 417–430. [Google Scholar] [CrossRef]
- Couser, W.G.; Remuzzi, G.; Mendis, S.; Tonelli, M. The contribution of chronic kidney disease to the global burden of major noncommunicable diseases. Kidney Int. 2011, 80, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Palomba, H.; Castro, I.; Yu, L.; Burdmann, E.A. The duration of acute kidney injury after cardiac surgery increases the risk of long-term chronic kidney disease. J. Nephrol. 2017, 30, 567–572. [Google Scholar] [CrossRef] [PubMed]
- James, M.T.; Grams, M.E.; Woodward, M.; Elley, C.R.; Green, J.A.; Wheeler, D.C.; de Jong, P.; Gansevoort, R.T.; Levey, A.S.; Warnock, D.G.; et al. A Meta-analysis of the Association of Estimated GFR, Albuminuria, Diabetes Mellitus, and Hypertension with Acute Kidney Injury. Am. J. Kidney Dis. 2015, 66, 602–612. [Google Scholar] [CrossRef]
- Legouis, D.; Galichon, P.; Bataille, A.; Chevret, S.; Provenchere, S.; Boutten, A.; Buklas, D.; Fellahi, J.L.; Hanouz, J.L.; Hertig, A. Rapid Occurrence of Chronic Kidney Disease in Patients Experiencing Reversible Acute Kidney Injury after Cardiac Surgery. Anesthesiology 2017, 126, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Polichnowski, A.J.; Lan, R.; Geng, H.; Griffin, K.A.; Venkatachalam, M.A.; Bidani, A.K. Severe renal mass reduction impairs recovery and promotes fibrosis after AKI. J. Am. Soc. Nephrol. 2014, 25, 1496–1507. [Google Scholar] [CrossRef]
- Chawla, L.S.; Kimmel, P.L. Acute kidney injury and chronic kidney disease: An integrated clinical syndrome. Kidney Int. 2012, 82, 516–524. [Google Scholar] [CrossRef]
- Duann, P.; Lin, P.H. Mitochondria Damage and Kidney Disease. Adv. Exp. Med. Biol. 2017, 982, 529–551. [Google Scholar] [PubMed]
- Nourbakhsh, N.; Singh, P. Role of renal oxygenation and mitochondrial function in the pathophysiology of acute kidney injury. Nephron. Clin. Pract. 2014, 127, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Che, R.; Yuan, Y.; Huang, S.; Zhang, A. Mitochondrial Dysfunction in the Pathophysiology of Renal Diseases. Am. J. Physiol. Ren. Physiol. 2014, 306, F367–F378. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.; Dong, Z. Regulation of mitochondrial morphological dynamics during apoptosis by Bcl-2 family proteins: A key in Bak? Cell Cycle 2007, 6, 3043–3047. [Google Scholar] [CrossRef] [PubMed]
- Wirthensohn, G.; Guder, W.G. Renal substrate metabolism. Physiol. Rev. 1986, 66, 469–497. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Bellomo, R.; Ronco, C.; Kellum, J.A.; Mehta, R.L.; Palevsky, P. Acute renal failure—definition, outcome measures, animal models, fluid therapy and information technology needs: The Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care 2004, 8, R204–R212. [Google Scholar] [CrossRef]
- Section 2: AKI Definition. Kidney Int. Suppl. 2012, 2, 19–36. [CrossRef] [PubMed]
- Khwaja, A. KDIGO clinical practice guidelines for acute kidney injury. Nephron. Clin. Pract. 2012, 120, c179–c184. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Shad, F.; Smith, M.C. Acute kidney injury: A guide to diagnosis and management. Am. Fam. Physician 2012, 86, 631–639. [Google Scholar]
- Linkermann, A.; Chen, G.; Dong, G.; Kunzendorf, U.; Krautwald, S.; Dong, Z. Regulated cell death in AKI. J. Am. Soc. Nephrol. 2014, 25, 2689–2701. [Google Scholar] [CrossRef]
- Agarwal, A.; Dong, Z.; Harris, R.; Murray, P.; Parikh, S.M.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Cellular and Molecular Mechanisms of AKI. J. Am. Soc. Nephrol. 2016, 27, 1288–1299. [Google Scholar] [CrossRef] [PubMed]
- Rewa, O.; Bagshaw, S.M. Acute kidney injury-epidemiology, outcomes and economics. Nat. Rev. Nephrol. 2014, 10, 193–207. [Google Scholar] [CrossRef]
- Matovinovic, M.S. 1. Pathophysiology and Classification of Kidney Diseases. EJIFCC 2009, 20, 2–11. [Google Scholar] [PubMed]
- Levey, A.S.; Coresh, J. Chronic kidney disease. Lancet 2012, 379, 165–180. [Google Scholar] [CrossRef]
- Shu, S.; Zhu, J.; Liu, Z.; Tang, C.; Cai, J.; Dong, Z. Endoplasmic reticulum stress is activated in post-ischemic kidneys to promote chronic kidney disease. EBioMedicine 2018, 37, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, J.R.; Chen, X.M.; Cai, G.Y.; Lin, L.R.; He, Y.N. Impact of ER stress-regulated ATF4/p16 signaling on the premature senescence of renal tubular epithelial cells in diabetic nephropathy. Am. J. Physiol. Cell Physiol. 2015, 308, C621–C630. [Google Scholar] [CrossRef]
- Tammaro, A.; Scantlebery, A.M.L.; Rampanelli, E.; Borrelli, C.; Claessen, N.; Butter, L.M.; Soriani, A.; Colonna, M.; Leemans, J.C.; Dessing, M.C.; et al. TREM1/3 Deficiency Impairs Tissue Repair After Acute Kidney Injury and Mitochondrial Metabolic Flexibility in Tubular Epithelial Cells. Front. Immunol 2019, 10, 1469. [Google Scholar] [CrossRef] [PubMed]
- Ko, G.J.; Grigoryev, D.N.; Linfert, D.; Jang, H.R.; Watkins, T.; Cheadle, C.; Racusen, L.; Rabb, H. Transcriptional analysis of kidneys during repair from AKI reveals possible roles for NGAL and KIM-1 as biomarkers of AKI-to-CKD transition. Am. J. Physiol. Ren. Physiol. 2010, 298, F1472–F1483. [Google Scholar] [CrossRef]
- Ellenrieder, L.; Rampelt, H.; Becker, T. Connection of Protein Transport and Organelle Contact Sites in Mitochondria. J. Mol. Biol. 2017, 429, 2148–2160. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Furusato, M.; Takasaki, S.; Ishikawa, E. Giant mitochondria in the epithelial cells of the proximal convoluted tubules of diseased human kidneys. Lab Investig. 1975, 33, 578–590. [Google Scholar] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [PubMed]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef]
- Braschi, E.; Zunino, R.; McBride, H.M. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009, 10, 748–754. [Google Scholar] [CrossRef]
- Prudent, J.; Zunino, R.; Sugiura, A.; Mattie, S.; Shore, G.C.; McBride, H.M. MAPL SUMOylation of Drp1 Stabilizes an ER/Mitochondrial Platform Required for Cell Death. Mol. Cell 2015, 59, 941–955. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Bai, M.; Lei, J.; Xie, Y.; Xu, S.; Jia, Z.; Zhang, A. Mitochondrial dysfunction and the AKI-to-CKD transition. Am. J. Physiol. Ren. Physiol. 2020, 319, F1105–F1116. [Google Scholar] [CrossRef] [PubMed]
- Lan, R.; Geng, H.; Singha, P.K.; Saikumar, P.; Bottinger, E.P.; Weinberg, J.M.; Venkatachalam, M.A. Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J. Am. Soc. Nephrol. 2016, 27, 3356–3367. [Google Scholar] [CrossRef]
- Wakabayashi, T.; Karbowski, M. Structural changes of mitochondria related to apoptosis. Biol. Signals Recept. 2001, 10, 26–56. [Google Scholar] [CrossRef] [PubMed]
- Guan, N.; Ren, Y.L.; Liu, X.Y.; Zhang, Y.; Pei, P.; Zhu, S.N.; Fan, Q. Protective role of cyclosporine A and minocycline on mitochondrial disequilibrium-related podocyte injury and proteinuria occurrence induced by adriamycin. Nephrol. Dial. Transpl. 2015, 30, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Ma, Z.; Zhu, J.; Zeng, M.; Liu, H.; Dong, Z. miR-214 represses mitofusin-2 to promote renal tubular apoptosis in ischemic acute kidney injury. Am. J. Physiol. Ren. Physiol. 2020, 318, F878–F887. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.; Wei, Q.; Cho, S.G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef]
- Brooks, C.; Cho, S.G.; Wang, C.Y.; Yang, T.; Dong, Z. Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am. J. Physiol. Cell Physiol. 2011, 300, C447–C455. [Google Scholar] [CrossRef] [PubMed]
- Saikumar, P.; Dong, Z.; Patel, Y.; Hall, K.; Hopfer, U.; Weinberg, J.M.; Venkatachalam, M.A. Role of hypoxia-induced Bax translocation and cytochrome c release in reoxygenation injury. Oncogene 1998, 17, 3401–3415. [Google Scholar] [CrossRef]
- Mikhailov, V.; Mikhailova, M.; Degenhardt, K.; Venkatachalam, M.A.; White, E.; Saikumar, P. Association of Bax and Bak homo-oligomers in mitochondria. Bax requirement for Bak reorganization and cytochrome c release. J. Biol. Chem. 2003, 278, 5367–5376. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nangaku, M.; Miyata, T.; Inagi, R.; Ohse, T.; Ingelfinger, J.R.; Fujita, T. Blockade of calcium influx through L-type calcium channels attenuates mitochondrial injury and apoptosis in hypoxic renal tubular cells. J. Am. Soc. Nephrol. 2004, 15, 2320–2333. [Google Scholar] [CrossRef]
- Wei, Q.; Dong, G.; Chen, J.K.; Ramesh, G.; Dong, Z. Bax and Bak have critical roles in ischemic acute kidney injury in global and proximal tubule-specific knockout mouse models. Kidney Int. 2013, 84, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Hu, Y.; Quiros, P.M.; Wei, Q.; Lopez-Otin, C.; Dong, Z. OMA1 mediates OPA1 proteolysis and mitochondrial fragmentation in experimental models of ischemic kidney injury. Am. J. Physiol. Ren. Physiol. 2014, 306, F1318–F1326. [Google Scholar] [CrossRef]
- Perry, H.M.; Huang, L.; Wilson, R.J.; Bajwa, A.; Sesaki, H.; Yan, Z.; Rosin, D.L.; Kashatus, D.F.; Okusa, M.D. Dynamin-Related Protein 1 Deficiency Promotes Recovery from AKI. J. Am. Soc. Nephrol. 2018, 29, 194–206. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1alpha Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.R.; Tran, M.T.; Parikh, S.M. PGC1alpha in the kidney. Am. J. Physiol. Ren. Physiol. 2018, 314, F1–F8. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Portilla, D.; Dai, G.; McClure, T.; Bates, L.; Kurten, R.; Megyesi, J.; Price, P.; Li, S. Alterations of PPARalpha and its coactivator PGC-1 in cisplatin-induced acute renal failure. Kidney Int. 2002, 62, 1208–1218. [Google Scholar] [CrossRef]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.; Tam, D.; Bardia, A.; Bhasin, M.; Rowe, G.C.; Kher, A.; Zsengeller, Z.K.; Akhavan-Sharif, M.R.; Khankin, E.V.; Saintgeniez, M.; et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Investig. 2011, 121, 4003–4014. [Google Scholar] [CrossRef]
- Parikh, S.M.; Yang, Y.; He, L.; Tang, C.; Zhan, M.; Dong, Z. Mitochondrial function and disturbances in the septic kidney. Semin. Nephrol. 2015, 35, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Noh, M.R.; Kim, J.; Padanilam, B.J. Defective Mitochondrial Fatty Acid Oxidation and Lipotoxicity in Kidney Diseases. Front. Med. (Lausanne) 2020, 7, 65. [Google Scholar] [CrossRef]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef]
- Xiao, L.; Zhu, X.; Yang, S.; Liu, F.; Zhou, Z.; Zhan, M.; Xie, P.; Zhang, D.; Li, J.; Song, P.; et al. Rap1 ameliorates renal tubular injury in diabetic nephropathy. Diabetes 2014, 63, 1366–1380. [Google Scholar] [CrossRef]
- Zhan, M.; Usman, I.M.; Sun, L.; Kanwar, Y.S. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J. Am. Soc. Nephrol. 2015, 26, 1304–1321. [Google Scholar] [CrossRef]
- Galvan, D.L.; Long, J.; Green, N.; Chang, B.H.; Lin, J.S.; Schumacker, P.; Truong, L.D. Drp1S600 phosphorylation regulates mitochondrial fission and progression of nephropathy in diabetic mice. J. Clin. Investig. 2019, 129, 2807–2823. [Google Scholar] [CrossRef] [PubMed]
- Ayanga, B.A.; Badal, S.S.; Wang, Y.; Galvan, D.L.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Dynamin-Related Protein 1 Deficiency Improves Mitochondrial Fitness and Protects against Progression of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2733–2747. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Zhao, Y.; Gong, J.; Huang, W.; Su, H.; Yuan, F.; Fang, K.; Wang, D.; Li, J.; Zou, X.; et al. Berberine Protects Glomerular Podocytes via Inhibiting Drp1-Mediated Mitochondrial Fission and Dysfunction. Theranostics 2019, 9, 1698–1713. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, M.; Xiong, L.; Fan, J.; Zhou, Y.; Li, H.; Peng, X.; Zhong, Z.; Wang, Y.; Huang, F.; et al. Drp1-mediated mitochondrial fission promotes renal fibroblast activation and fibrogenesis. Cell Death Dis. 2020, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Platt, C.; Coward, R.J. Peroxisome proliferator activating receptor-gamma and the podocyte. Nephrol. Dial. Transpl. 2017, 32, 423–433. [Google Scholar]
- Yuan, Y.; Huang, S.; Wang, W.; Wang, Y.; Zhang, P.; Zhu, C.; Ding, G.; Liu, B.; Yang, T.; Zhang, A. Activation of peroxisome proliferator-activated receptor-gamma coactivator 1alpha ameliorates mitochondrial dysfunction and protects podocytes from aldosterone-induced injury. Kidney Int. 2012, 82, 771–789. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, Y.; Inagi, R.; Yoshihara, D.; Kugita, M.; Nagao, S.; Shimizu, A.; Takeda, N.; Wake, M.; Honda, K.; Zhou, J.; et al. Mitochondrial Abnormality Facilitates Cyst Formation in Autosomal Dominant Polycystic Kidney Disease. Mol. Cell Biol. 2017, 37, e00337-17. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Karl, B.; Mathew, A.V.; Gangoiti, J.A.; Wassel, C.L.; Saito, R.; Pu, M.; Sharma, S.; You, Y.H.; Wang, L.; et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J. Am. Soc. Nephrol. 2013, 24, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Han, S.H.; Wu, M.Y.; Nam, B.Y.; Park, J.T.; Yoo, T.H.; Kang, S.W.; Park, J.; Chinga, F.; Li, S.Y.; Susztak, K. PGC-1alpha Protects from Notch-Induced Kidney Fibrosis Development. J. Am. Soc. Nephrol. 2017, 28, 3312–3322. [Google Scholar] [CrossRef] [PubMed]
- Brinkkoetter, P.T.; Bork, T.; Salou, S.; Liang, W.; Mizi, A.; Ozel, C.; Koehler, S.; Hagmann, H.H.; Ising, C.; Kuczkowski, A.; et al. Anaerobic Glycolysis Maintains the Glomerular Filtration Barrier Independent of Mitochondrial Metabolism and Dynamics. Cell Rep. 2019, 27, 1551.e5–1566.e5. [Google Scholar] [CrossRef]
- Zhang, T.; Chi, Y.; Kang, Y.; Lu, H.; Niu, H.; Liu, W.; Li, Y. Resveratrol ameliorates podocyte damage in diabetic mice via SIRT1/PGC-1alpha mediated attenuation of mitochondrial oxidative stress. J. Cell Physiol. 2019, 234, 5033–5043. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Tiranti, V.; Savoia, A.; Forti, F.; D’Apolito, M.F.; Centra, M.; Rocchi, M.; Zeviani, M. Identification of the gene encoding the human mitochondrial RNA polymerase (h-mtRPOL) by cyberscreening of the Expressed Sequence Tags database. Hum. Mol. Genet. 1997, 6, 615–625. [Google Scholar] [CrossRef]
- Falkenberg, M.; Gaspari, M.; Rantanen, A.; Trifunovic, A.; Larsson, N.G.; Gustafsson, C.M. Mitochondrial transcription factors B1 and B2 activate transcription of human mtDNA. Nat. Genet. 2002, 31, 289–294. [Google Scholar] [CrossRef]
- Surovtseva, Y.V.; Shutt, T.E.; Cotney, J.; Cimen, H.; Chen, S.Y.; Koc, E.C.; Shadel, G.S. Mitochondrial ribosomal protein L12 selectively associates with human mitochondrial RNA polymerase to activate transcription. Proc. Natl. Acad. Sci. USA 2011, 108, 17921–17926. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Liu, Y.; Wang, N.; Zhen, J.; Zhang, B.; Hou, S.; Cui, Z.; Wan, Q.; Feng, H. Transcription of MRPL12 regulated by Nrf2 contributes to the mitochondrial dysfunction in diabetic kidney disease. Free. Radic. Biol. Med. 2021, 164, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Thome, T.; Kumar, R.A.; Burke, S.K.; Khattri, R.B.; Salyers, Z.R.; Kelley, R.C.; Coleman, M.D.; Christou, D.D.; Hepple, R.T.; Scali, S.T.; et al. Impaired muscle mitochondrial energetics is associated with uremic metabolite accumulation in chronic kidney disease. JCI Insight 2020, 6, e139826. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Liu, S.; Soong, Y.; Seshan, S.V.; Cohen-Gould, L.; Manichev, V.; Feldman, L.C.; Gustafsson, T. Mitochondria Protection after Acute Ischemia Prevents Prolonged Upregulation of IL-1beta and IL-18 and Arrests CKD. J. Am. Soc. Nephrol. 2017, 28, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chen, Y.; Chen, Y.H.; Ta, A.P.; Lee, H.C.; MacGregor, G.R.; Vaziri, N.D.; Wang, P.H. Tubular mitochondrial AKT1 is activated during ischemia reperfusion injury and has a critical role in predisposition to chronic kidney disease. Kidney Int. 2021, 99, 870–884. [Google Scholar] [CrossRef]
- Bartoszewska, S.; Collawn, J.F. Unfolded protein response (UPR) integrated signaling networks determine cell fate during hypoxia. Cell Mol. Biol. Lett. 2020, 25, 18. [Google Scholar] [CrossRef]
- Walter, F.; O’Brien, A.; Concannon, C.G.; Dussmann, H.; Prehn, J.H.M. ER stress signaling has an activating transcription factor 6alpha (ATF6)-dependent “off-switch”. J. Biol. Chem. 2018, 293, 18270–18284. [Google Scholar] [CrossRef] [PubMed]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Pan, H.; Wang, J.; Wang, T.; Huo, X.; Ma, Y.; Lu, Z.; Sun, B.; Jiang, H. Unfolded protein response in colorectal cancer. Cell Biosci. 2021, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Noh, M.R.; Kim, K.Y.; Han, S.J.; Kim, J.I.; Kim, H.Y.; Park, K.M. Methionine Sulfoxide Reductase A Deficiency Exacerbates Cisplatin-Induced Nephrotoxicity via Increased Mitochondrial Damage and Renal Cell Death. Antioxid. Redox Signal 2017, 27, 727–741. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.K.; Han, Y.C.; He, J.R.; Yang, M.; Zhang, W.; Zhan, M.; Li, A.M.; Li, L.; Na, S.; Liu, Y.T.; et al. Mitochondria targeted peptide SS-31 prevent on cisplatin-induced acute kidney injury via regulating mitochondrial ROS-NLRP3 pathway. Biomed Pharm. 2020, 130, 110521. [Google Scholar] [CrossRef] [PubMed]
- Kruidering, M.; Van de Water, B.; de Heer, E.; Mulder, G.J.; Nagelkerke, J.F. Cisplatin-induced nephrotoxicity in porcine proximal tubular cells: Mitochondrial dysfunction by inhibition of complexes I to IV of the respiratory chain. J. Pharm. Exp. 1997, 280, 638–649. [Google Scholar]
- Szeto, H.H.; Liu, S.; Soong, Y.; Wu, D.; Darrah, S.F.; Cheng, F.Y.; Zhao, Z.; Ganger, M.; Tow, C.Y.; Seshan, S.V. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J. Am. Soc. Nephrol. 2011, 22, 1041–1052. [Google Scholar] [CrossRef]
- Oberg, B.P.; McMenamin, E.; Lucas, F.L.; McMonagle, E.; Morrow, J.; Ikizler, T.A.; Himmelfarb, J. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int. 2004, 65, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Galvan, D.L.; Badal, S.S.; Long, J.; Chang, B.H.; Schumacker, P.T.; Overbeek, P.A.; Danesh, F.R. Real-time in vivo mitochondrial redox assessment confirms enhanced mitochondrial reactive oxygen species in diabetic nephropathy. Kidney Int. 2017, 92, 1282–1287. [Google Scholar] [CrossRef] [PubMed]
- Granata, S.; Zaza, G.; Simone, S.; Villani, G.; Latorre, D.; Pontrelli, P.; Carella, M.; Schena, F.P.; Grandaliano, G.; Pertosa, G. Mitochondrial dysregulation and oxidative stress in patients with chronic kidney disease. BMC Genom. 2009, 10, 388. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.K.; Harris, R.C. EGF receptor deletion in podocytes attenuates diabetic nephropathy. J. Am. Soc. Nephrol. 2015, 26, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Mittwede, P.N.; Xiang, L.; Lu, S.; Clemmer, J.S.; Hester, R.L. Oxidative stress contributes to orthopedic trauma-induced acute kidney injury in obese rats. Am. J. Physiol. Ren. Physiol. 2015, 308, F157–F163. [Google Scholar] [CrossRef]
- Hou, Y.; Li, S.; Wu, M.; Wei, J.; Ren, Y.; Du, C.; Wu, H.; Han, C.; Duan, H.; Shi, Y. Mitochondria-targeted peptide SS-31 attenuates renal injury via an antioxidant effect in diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2016, 310, F547–F559. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, Y.; Chen, J.; Seshan, S.V.; Poppas, D.P.; Szeto, H.H.; Felsen, D. A novel cell-permeable antioxidant peptide decreases renal tubular apoptosis and damage in unilateral ureteral obstruction. Am. J. Physiol. Ren. Physiol. 2008, 295, F1545–F1553. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Liu, S.; Soong, Y.; Alam, N.; Prusky, G.T.; Seshan, S.V. Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int. 2016, 90, 997–1011. [Google Scholar] [CrossRef]
- Szeto, H.H.; Liu, S.; Soong, Y.; Birk, A.V. Improving mitochondrial bioenergetics under ischemic conditions increases warm ischemia tolerance in the kidney. Am. J. Physiol. Ren. Physiol. 2015, 308, F11–F21. [Google Scholar] [CrossRef]
- Tang, W.X.; Wu, W.H.; Qiu, H.Y.; Bo, H.; Huang, S.M. Amelioration of rhabdomyolysis-induced renal mitochondrial injury and apoptosis through suppression of Drp-1 translocation. J. Nephrol. 2013, 26, 1073–1082. [Google Scholar] [CrossRef]
- Cassina, L.; Chiaravalli, M.; Boletta, A. Increased mitochondrial fragmentation in polycystic kidney disease acts as a modifier of disease progression. FASEB J. 2020, 34, 6493–6507. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lin, Q.; Shao, X.; Zhu, X.; Wu, J.; Wu, B.; Zhang, M.; Zhou, W.; Zhou, Y.; Jin, H.; et al. Drp1-regulated PARK2-dependent mitophagy protects against renal fibrosis in unilateral ureteral obstruction. Free. Radic. Biol. Med. 2020, 152, 632–649. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.A.; Schnellmann, R.G. Accelerated recovery of renal mitochondrial and tubule homeostasis with SIRT1/PGC-1alpha activation following ischemia-reperfusion injury. Toxicol. Appl. Pharm. 2013, 273, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Du, C.; Shi, Y.; Wei, J.; Wu, H.; Cui, H. The Sirt1 activator, SRT1720, attenuates renal fibrosis by inhibiting CTGF and oxidative stress. Int. J. Mol. Med. 2017, 39, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Lim, J.H.; Youn, H.H.; Hong, Y.A.; Yang, K.S.; Park, H.S.; Chung, S.; Ko, S.H.; Shin, S.J.; Choi, B.S.; et al. Resveratrol prevents renal lipotoxicity and inhibits mesangial cell glucotoxicity in a manner dependent on the AMPK-SIRT1-PGC1alpha axis in db/db mice. Diabetologia 2013, 56, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Palsamy, P.; Subramanian, S. Resveratrol protects diabetic kidney by attenuating hyperglycemia-mediated oxidative stress and renal inflammatory cytokines via Nrf2-Keap1 signaling. Biochim. Biophys. Acta 2011, 1812, 719–731. [Google Scholar] [CrossRef]
- Ding, D.F.; Ding, D.F.; You, N.; Wu, X.M.; Xu, J.R.; Hu, A.P.; Ye, X.L.; Zhu, Q.; Jiang, X.Q.; Miao, H.; et al. Resveratrol attenuates renal hypertrophy in early-stage diabetes by activating AMPK. Am. J. Nephrol. 2010, 31, 363–374. [Google Scholar] [CrossRef]
- Lempiainen, J.; Finckenberg, P.; Levijoki, J.; Mervaala, E. AMPK activator AICAR ameliorates ischaemia reperfusion injury in the rat kidney. Br. J. Pharm. 2012, 166, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Hsu, H.H.; Lee, C.C.; Yen, T.H.; Ko, Y.C.; Yang, C.W.; Hung, C.C. The AMPK agonist AICAR inhibits TGF-beta1 induced activation of kidney myofibroblasts. PLoS ONE 2014, 9, e106554. [Google Scholar]
- Wills, L.P.; Trager, R.E.; Beeson, G.C.; Lindsey, C.C.; Peterson, Y.K.; Beeson, C.C.; Schnellmann, R.G. The beta2-adrenoceptor agonist formoterol stimulates mitochondrial biogenesis. J. Pharm. Exp. 2012, 342, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Jesinkey, S.R.; Funk, J.A.; Stallons, L.J.; Wills, L.P.; Megyesi, J.K.; Beeson, C.C.; Schnellmann, R.G. Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2014, 25, 1157–1162. [Google Scholar] [CrossRef]
- Cleveland, K.; Cameron, R.B.; Brosius, F.C.; Schnellmann, R.G. The β2-Adrenergic Receptor Agonist Formoterol Decreases Fibrotic and Mitochondrial Fusion/Fission Proteins in a Mouse Model of Diabetic Nephropathy. FASEB J. 2019, 33 (Suppl. S1), 514. [Google Scholar] [CrossRef]
- Stadler, K.; Goldberg, I.J.; Susztak, K. The evolving understanding of the contribution of lipid metabolism to diabetic kidney disease. Curr. Diab. Rep. 2015, 15, 40. [Google Scholar] [CrossRef]
- Singh, D.; Chander, V.; Chopra, K. Cyclosporine protects against ischemia/reperfusion injury in rat kidneys. Toxicology 2005, 207, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Zhuang, S.; Dworkin, L.D.; Liu, Z.; Gong, R. Inhibition of glycogen synthase kinase-3beta prevents NSAID-induced acute kidney injury. Kidney Int. 2012, 81, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Bao, H.; Ge, Y.; Zhuang, S.; Peng, A.; Gong, R. Pharmacological targeting of GSK3beta confers protection against podocytopathy and proteinuria by desensitizing mitochondrial permeability transition. Br. J. Pharm. 2015, 172, 895–909. [Google Scholar] [CrossRef]
- Singh, S.P.; Tao, S.; Fields, T.A.; Webb, S.; Harris, R.C.; Rao, R. Glycogen synthase kinase-3 inhibition attenuates fibroblast activation and development of fibrosis following renal ischemia-reperfusion in mice. Dis. Model. Mech. 2015, 8, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Sourris, K.C.; Harcourt, B.E.; Tang, P.H.; Morley, A.L.; Huynh, K.; Penfold, S.A.; Coughlan, M.T.; Cooper, M.E.; Nguyen, T.V.; Ritchie, R.H.; et al. Ubiquinone (coenzyme Q10) prevents renal mitochondrial dysfunction in an experimental model of type 2 diabetes. Free. Radic. Biol. Med. 2012, 52, 716–723. [Google Scholar] [CrossRef]
- Chacko, B.K.; Reily, C.; Srivastava, A.; Johnson, M.S.; Ye, Y.; Ulasova, E.; Agarwal, A.; Zinn, K.R.; Murphy, M.P.; Kalyanaraman, B.; et al. Prevention of diabetic nephropathy in Ins2(+/)(−)(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochem. J. 2010, 432, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.Y.; Silachev, D.N.; Jankauskas, S.S.; Rokitskaya, T.I.; Chupyrkina, A.A.; Pevzner, I.B.; Zorova, L.D.; Isaev, N.K.; Antonenko, Y.N.; Skulachev, V.P.; et al. Mild uncoupling of respiration and phosphorylation as a mechanism providing nephro- and neuroprotective effects of penetrating cations of the SkQ family. Biochemistry (Mosc) 2012, 77, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Jankauskas, S.S.; Plotnikov, E.Y.; Morosanova, M.A.; Pevzner, I.B.; Zorova, L.D.; Skulachev, V.P.; Zorov, D.B. Mitochondria-targeted antioxidant SkQR1 ameliorates gentamycin-induced renal failure and hearing loss. Biochemistry (Mosc) 2012, 77, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Soetikno, V.; Sari, F.R.; Lakshmanan, A.P.; Arumugam, S.; Harima, M.; Suzuki, K.; Kawachi, H.; Watanabe, K. Curcumin alleviates oxidative stress, inflammation, and renal fibrosis in remnant kidney through the Nrf2-keap1 pathway. Mol. Nutr. Food Res. 2013, 57, 1649–1659. [Google Scholar] [CrossRef]
- Tapia, E.; Zatarain-Barron, Z.L.; Hernandez-Pando, R.; Zarco-Marquez, G.; Molina-Jijon, E.; Cristobal-Garcia, M.; Santamaria, J.; Pedraza-Chaverri, J. Curcumin reverses glomerular hemodynamic alterations and oxidant stress in 5/6 nephrectomized rats. Phytomedicine 2013, 20, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Yin, N.; Liu, W.; Cui, X.; Chen, S.; Wang, E. Curcumin Ameliorates Diabetic Nephropathy by Suppressing NLRP3 Inflammasome Signaling. Biomed. Res. Int. 2017, 2017, 1516985. [Google Scholar] [CrossRef] [PubMed]
- Pangborn, M.C. Isolation and Purification of a Serologically Active Phospholipid from Beef Heart. J. Biol. Chem. 1942, 143, 247–256. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol. 2017, 5, 90. [Google Scholar] [CrossRef]
- Kaasik, A.; Safiulina, D.; Zharkovsky, A.; Veksler, V. Regulation of mitochondrial matrix volume. Am. J. Physiol. Cell Physiol. 2007, 292, C157–C163. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Eirin, A.; Ebrahimi, B.; Kwon, S.H.; Fiala, J.A.; Williams, B.J.; Woollard, J.R.; He, Q.; Gupta, R.C.; Sabbah, H.N.; Prakash, Y.S.; et al. Restoration of Mitochondrial Cardiolipin Attenuates Cardiac Damage in Swine Renovascular Hypertension. J. Am. Heart Assoc. 2016, 5, e003118. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Soong, Y.; Seshan, S.V.; Szeto, H.H. Novel cardiolipin therapeutic protects endothelial mitochondria during renal ischemia and mitigates microvascular rarefaction, inflammation, and fibrosis. Am. J. Physiol. Ren. Physiol. 2014, 306, F970–F980. [Google Scholar] [CrossRef] [PubMed]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wang, S.C.; Li, M.; Ma, X.H.; Jia, X.N.; Bu, Y.; Sun, L.; Yu, K.J. An Inhibitor of DRP1 (Mdivi-1) Alleviates LPS-Induced Septic AKI by Inhibiting NLRP3 Inflammasome Activation. Biomed Res. Int. 2020, 2020, 2398420. [Google Scholar]
- Wang, H.; Guan, Y.; Karamercan, M.A.; Ye, L.; Bhatti, T.; Becker, L.B.; Baur, J.A.; Sims, C.A. Resveratrol Rescues Kidney Mitochondrial Function Following Hemorrhagic Shock. Shock 2015, 44, 173–180. [Google Scholar] [CrossRef]
- Kim, J.S.; He, L.; Lemasters, J.J. Mitochondrial permeability transition: A common pathway to necrosis and apoptosis. Biochem. Biophys. Res. Commun. 2003, 304, 463–470. [Google Scholar] [CrossRef]
- Kalani, K.; Yan, S.F.; Yan, S.S. Mitochondrial permeability transition pore: A potential drug target for neurodegeneration. Drug. Discov. Today 2018, 23, 1983–1989. [Google Scholar] [CrossRef] [PubMed]
- De Arriba, G.; Calvino, M.; Benito, S.; Parra, T. Cyclosporine A-induced apoptosis in renal tubular cells is related to oxidative damage and mitochondrial fission. Toxicol. Lett. 2013, 218, 30–38. [Google Scholar] [CrossRef]
- Serkova, N.; Klawitter, J.; Niemann, C.U. Organ-specific response to inhibition of mitochondrial metabolism by cyclosporine in the rat. Transpl. Int. 2003, 16, 748–755. [Google Scholar] [CrossRef]
- Puigmule, M.; Lopez-Hellin, J.; Sune, G.; Tornavaca, O.; Camano, S.; Tejedor, A.; Meseguer, A. Differential proteomic analysis of cyclosporine A-induced toxicity in renal proximal tubule cells. Nephrol. Dial. Transpl. 2009, 24, 2672–2686. [Google Scholar] [CrossRef] [PubMed]
- Lindblom, R.S.J.; Higgins, G.C.; Nguyen, T.V.; Arnstein, M.; Henstridge, D.C.; Granata, C.; Snelson, M.; Thallas-Bonke, V.; Cooper, M.E.; Forbes, J.M.; et al. Delineating a role for the mitochondrial permeability transition pore in diabetic kidney disease by targeting cyclophilin D. Clin. Sci. (Lond) 2020, 134, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.V.; Hajizadeh, S.; Holme, E.; Jonsson, I.M.; Tarkowski, A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J. Leukoc. Biol. 2004, 75, 995–1000. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Loor, G.; Kondapalli, J.; Iwase, H.; Chandel, N.S.; Waypa, G.B.; Guzy, R.D.; Vanden Hoek, T.L.; Schumacker, P.T. Mitochondrial oxidant stress triggers cell death in simulated ischemia-reperfusion. Biochim. Biophys. Acta. 2011, 1813, 1382–1394. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021, 11, 1845–1863. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.T.; Kolesar, J.E.; Kaufman, B.A. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim. Biophys Acta 2012, 1819, 921–929. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Ding, W.; Wang, Y. Mito-TEMPO Alleviates Renal Fibrosis by Reducing Inflammation, Mitochondrial Dysfunction, and Endoplasmic Reticulum Stress. Oxid. Med. Cell Longev. 2018, 2018, 5828120. [Google Scholar] [CrossRef]
- Dare, A.J.; Bolton, E.A.; Pettigrew, G.J.; Bradley, J.A.; Saeb-Parsy, K.; Murphy, M.P. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox. Biol. 2015, 5, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Trejo, O.E.; Tapia, E.; Molina-Jijon, E.; Medina-Campos, O.N.; Macias-Ruvalcaba, N.A.; Leon-Contreras, J.C.; Hernandez-Pando, R.; Garcia-Arroyo, F.E.; Cristobal, M.; Sanchez-Lozada, L.G.; et al. Curcumin prevents mitochondrial dynamics disturbances in early 5/6 nephrectomy: Relation to oxidative stress and mitochondrial bioenergetics. Biofactors 2017, 43, 293–310. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Therapeutics | Mechanism(s) of Action | Experimental Model | Clinical Trial | References |
|---|---|---|---|---|
| Cardiolipin protection | ||||
| SS-31 | Binds to cardiolipin, prevents peroxidase activity and improves mitochondrial respiration and ATP production; Inhibits cytochrome c release; Normalizes mitochondrial potential (ΔΨm) | IRI-AKI, UUO, and DKD | Mitochondrial myopathy (NCT02367014) Age-related skeletal muscle mitochondrial dysfunction (NCT02245620) | [80,96,97,98] |
| SS-20 | Reduces mitochondrial matrix swelling and preserves cristae membranes; Increases ATP and reduces ROS | IRI-AKI | [99] | |
| Fission inhibitor | ||||
| Mdivi-1 | Selectively inhibits Drp1; Induces mitochondria fusion and increases ATP production | IRI-AKI, ADPKD, and UUO | [100,101,102] | |
| Biogenesis activator | ||||
| SRT1720 | Sirt-1 activator; Restores renal expression of PGC-1α, mitochondrial mass, ATP levels | IRI-AKI and UUO | [103,104] | |
| Resveratrol | Sirt-1 activator; Restores mitochondrial respiratory capacity and decreases the production of mtROS and lipid peroxidation | Hemorrhagic shock induces AKI and db/db mice | [20,71,105,106,107] | |
| AICAR | AMPK activator; Attenuates nitrosative stress and monocyte/macrophage infiltration | IRI-AKI and UUO | [13,108,109] | |
| Formoterol | Agonist of β2-adrenoreceptor, increase PGC-1α synthesis and induces mitochondrial biogenesis by increasing mtDNA copy numbers, oxygen consumption rate | IRI-AKI and db/db mice | [110,111,112] | |
| Fenofibrate/Bezafibrate | Peroxisomal proliferator-activated receptor (PPAR)-α agonist, lipid lowering, increases mitochondrial biogenesis | CKD and DKD | Diabetic complications (CVD and DKD) (FIELD, ISRCTN 64783481) | [113] |
| mPTP inhibitor | ||||
| CSA | Interacts with cyclophilin D; Suppresses mPTP opening and mitochondria swelling | IRI-AKI | Acute myocardial infarction (NCT01595958) Severe traumatic brain injury (NCT01825044) | [114] |
| TDZD-8 | Selectively inhibits GSK-3β; Diminishes MPT | Drug-induced AKI and IRI-AKI | [115,116,117] | |
| Antioxidants | ||||
| CoQ10 | Normalizes ATP production, attenuates mtROS and decreases mitochondrial ΔΨm | db/db mice | Mitochondrial disorders (NCT00432744) Parkinson disease (NCT00740714) | [118] |
| MitoQ | ROS scavenger; Antioxidant concentrates at matrix in a ΔΨm-dependent manner | IRI-AKI Ins2(+/)⁻(AkitaJ) mice | Parkinson’s disease (NCT00329056) Fatty acid disease (NCT01167088) Hepatitis C (NCT00433108) CKD (NCT02364648) | [66,119] |
| SkQR1 | Antioxidant and decreased mitochondrial ΔΨm | IRI-AKI and gentamycin-induced renal failure | [120,121]. | |
| Curcumin | Antioxidant; Alteration of mitochondrial dynamics and bioenergetics. | 5/6NX mice and db/db mice | Leber hereditary optic neuropathy (NCT00528151) | [122,123,124] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Agborbesong, E.; Li, X. The Role of Mitochondria in Acute Kidney Injury and Chronic Kidney Disease and Its Therapeutic Potential. Int. J. Mol. Sci. 2021, 22, 11253. https://doi.org/10.3390/ijms222011253
Zhang X, Agborbesong E, Li X. The Role of Mitochondria in Acute Kidney Injury and Chronic Kidney Disease and Its Therapeutic Potential. International Journal of Molecular Sciences. 2021; 22(20):11253. https://doi.org/10.3390/ijms222011253
Chicago/Turabian StyleZhang, Xiaoqin, Ewud Agborbesong, and Xiaogang Li. 2021. "The Role of Mitochondria in Acute Kidney Injury and Chronic Kidney Disease and Its Therapeutic Potential" International Journal of Molecular Sciences 22, no. 20: 11253. https://doi.org/10.3390/ijms222011253
APA StyleZhang, X., Agborbesong, E., & Li, X. (2021). The Role of Mitochondria in Acute Kidney Injury and Chronic Kidney Disease and Its Therapeutic Potential. International Journal of Molecular Sciences, 22(20), 11253. https://doi.org/10.3390/ijms222011253