
The Role of Centrosome Distal Appendage Proteins (DAPs) in Nephronophthisis and Ciliogenesis


Abstract
:1. Introduction
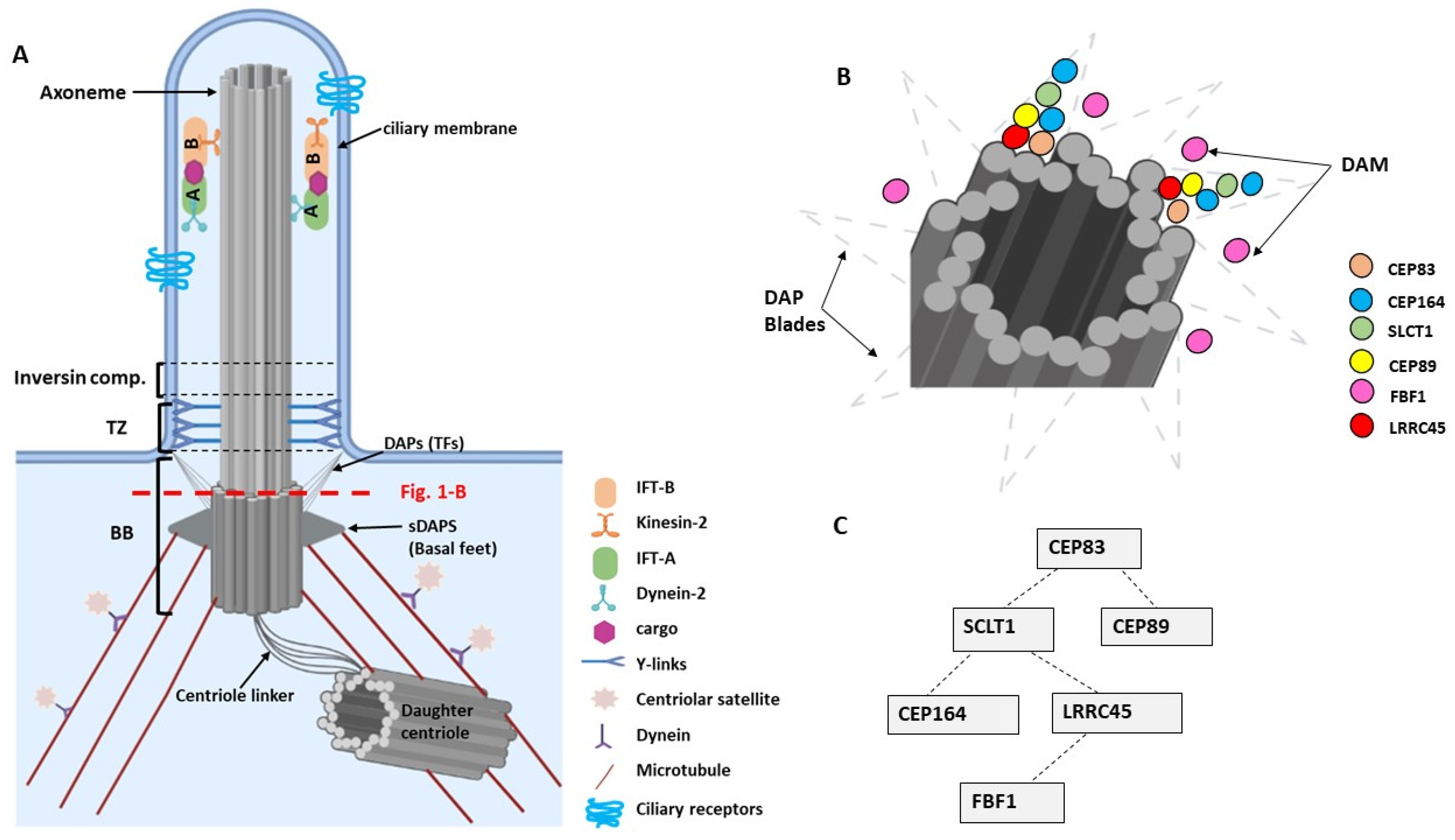
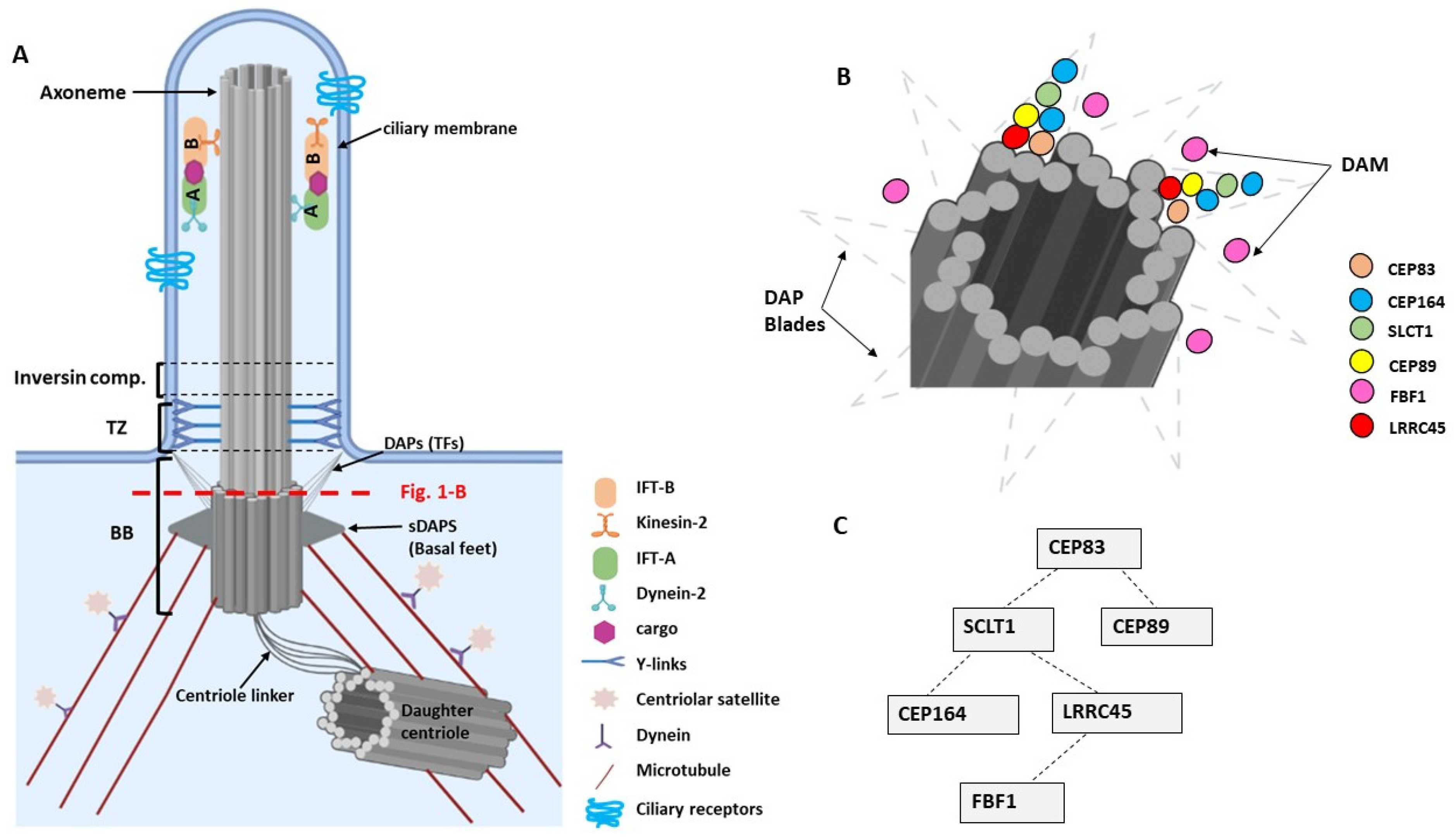
2. Primary Cilia—Basic Structure and Molecular Composition
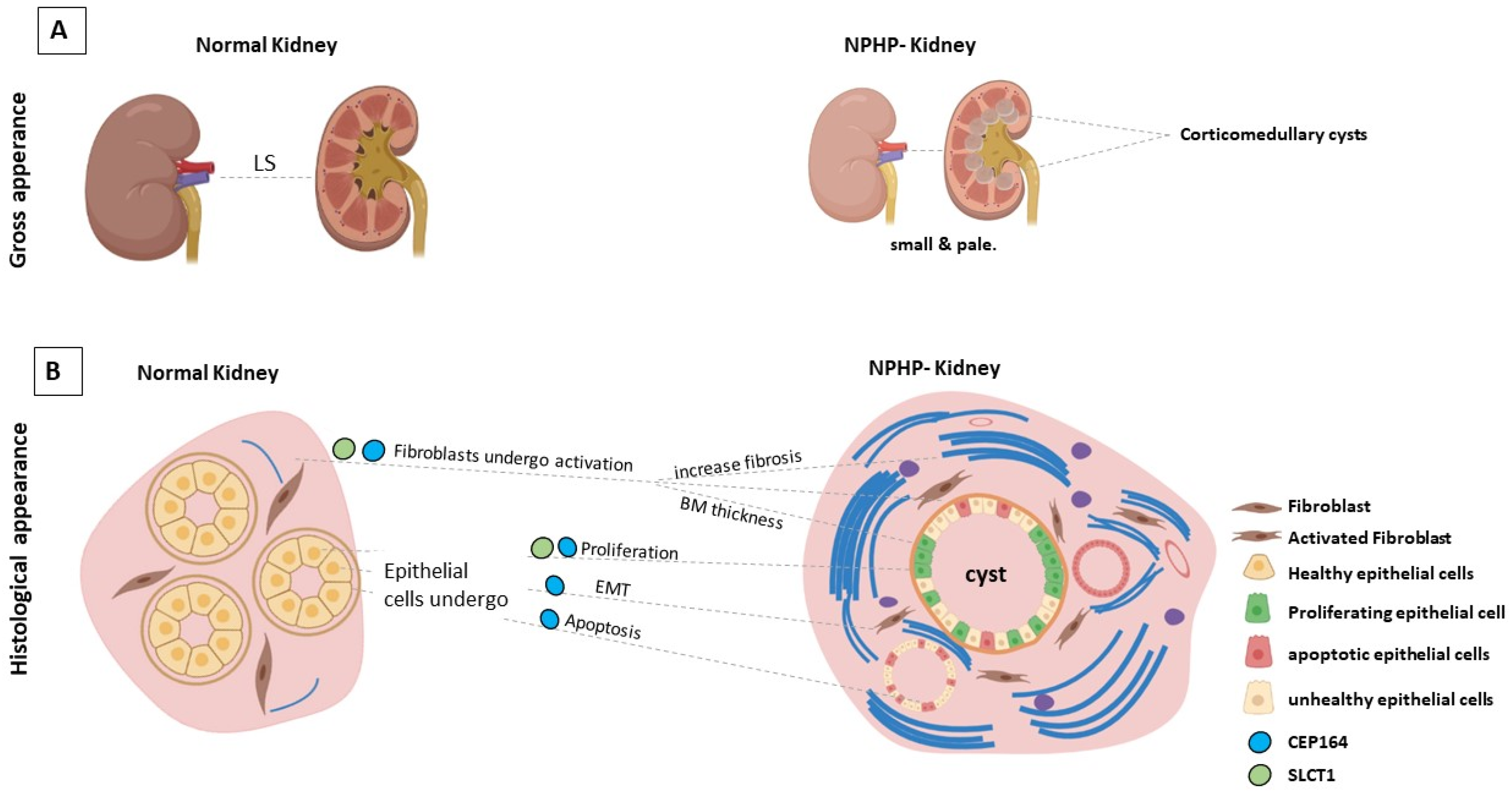
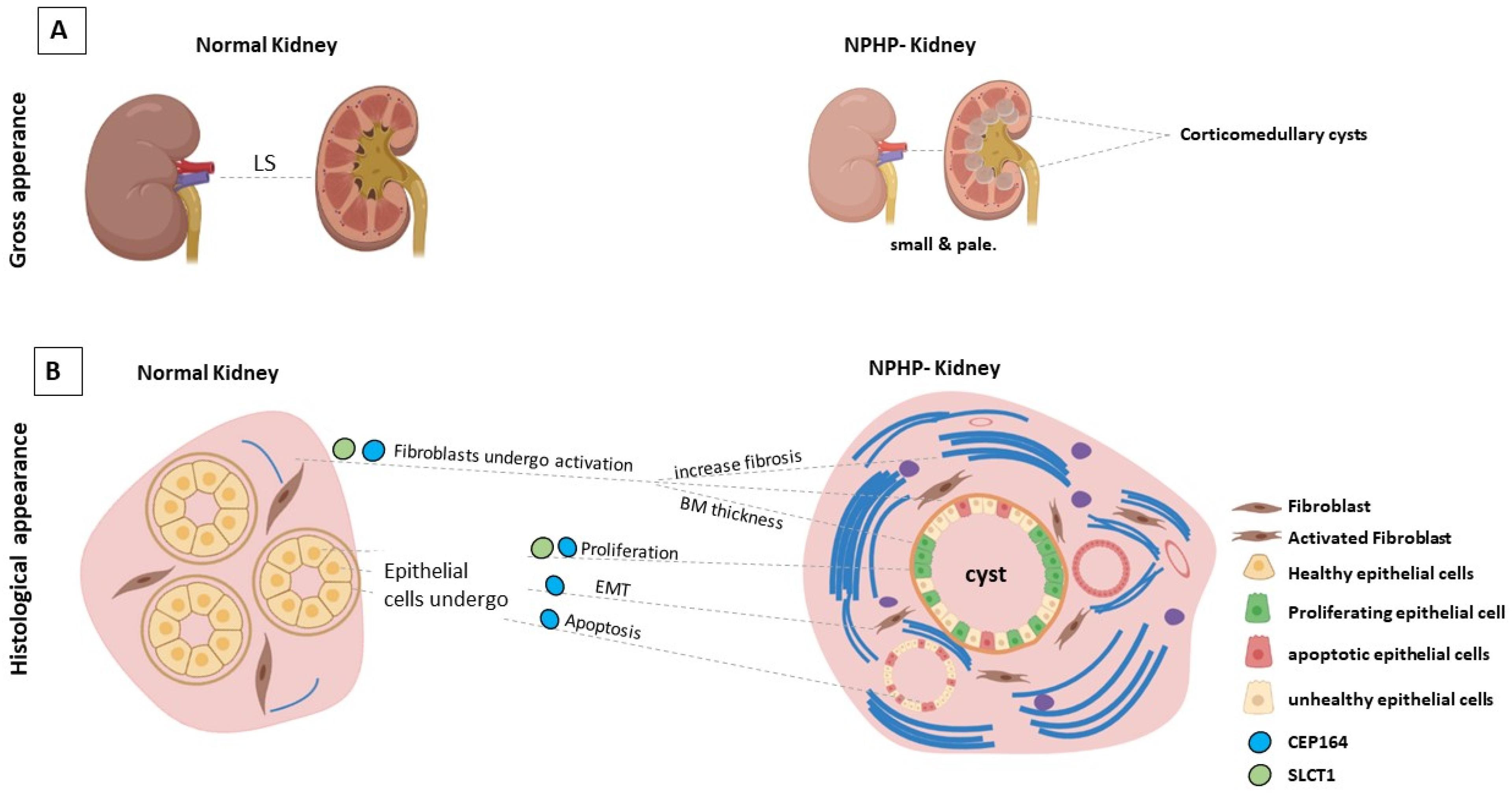
3. The Genetic Basis of Nephronophthisis (NPHP) and Related Disorders
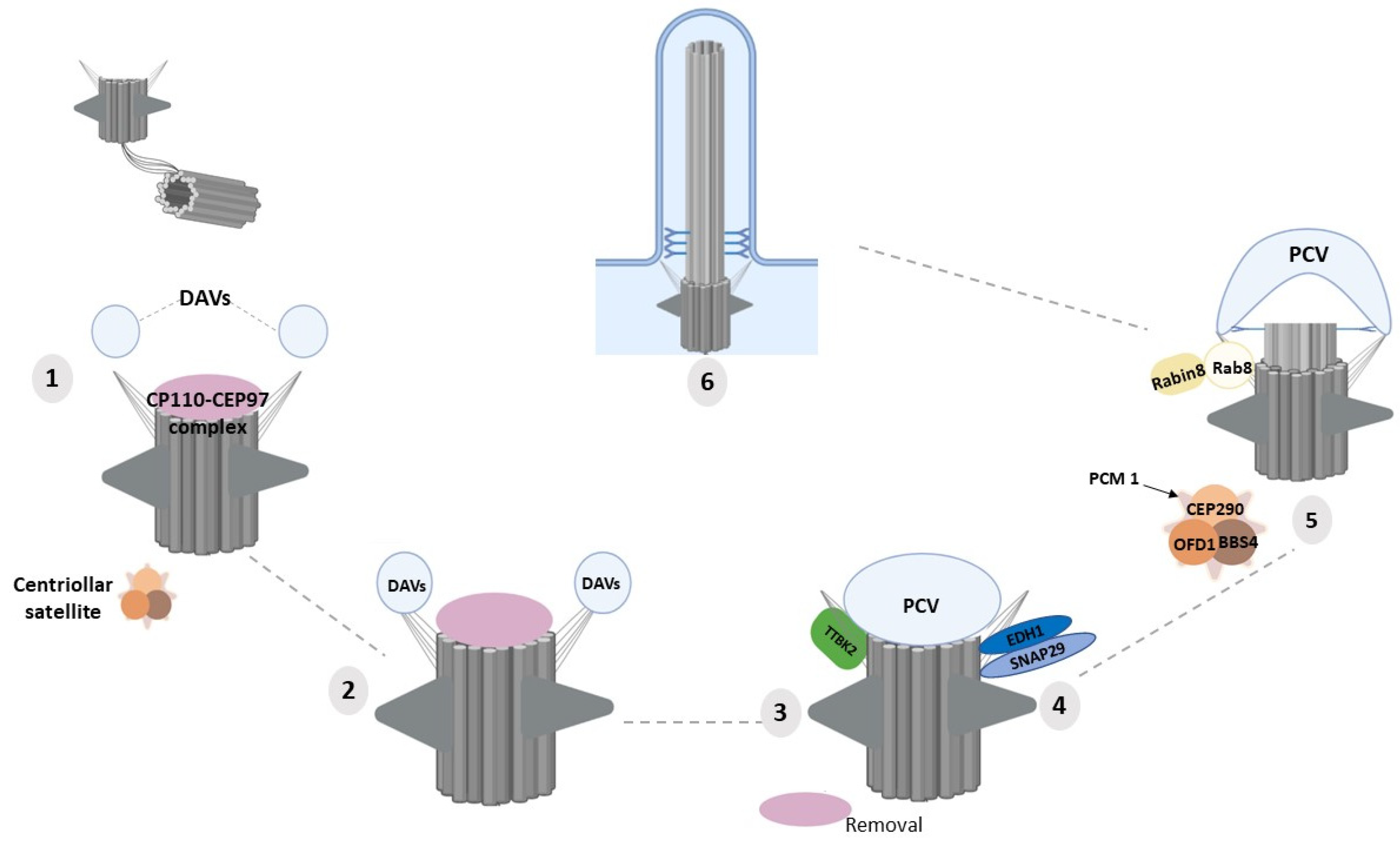
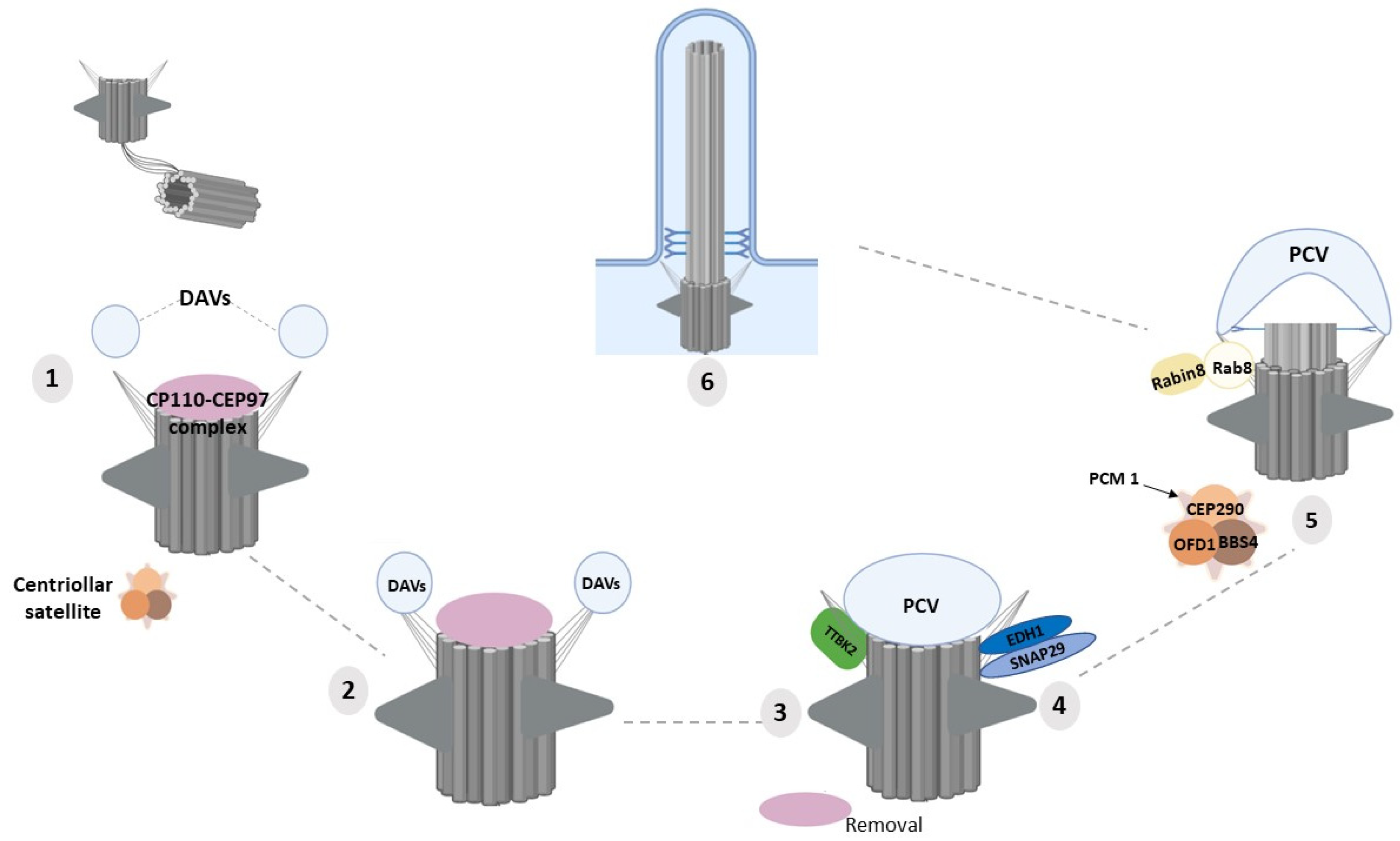
4. Molecular Functions of DAPs in Ciliogenesis
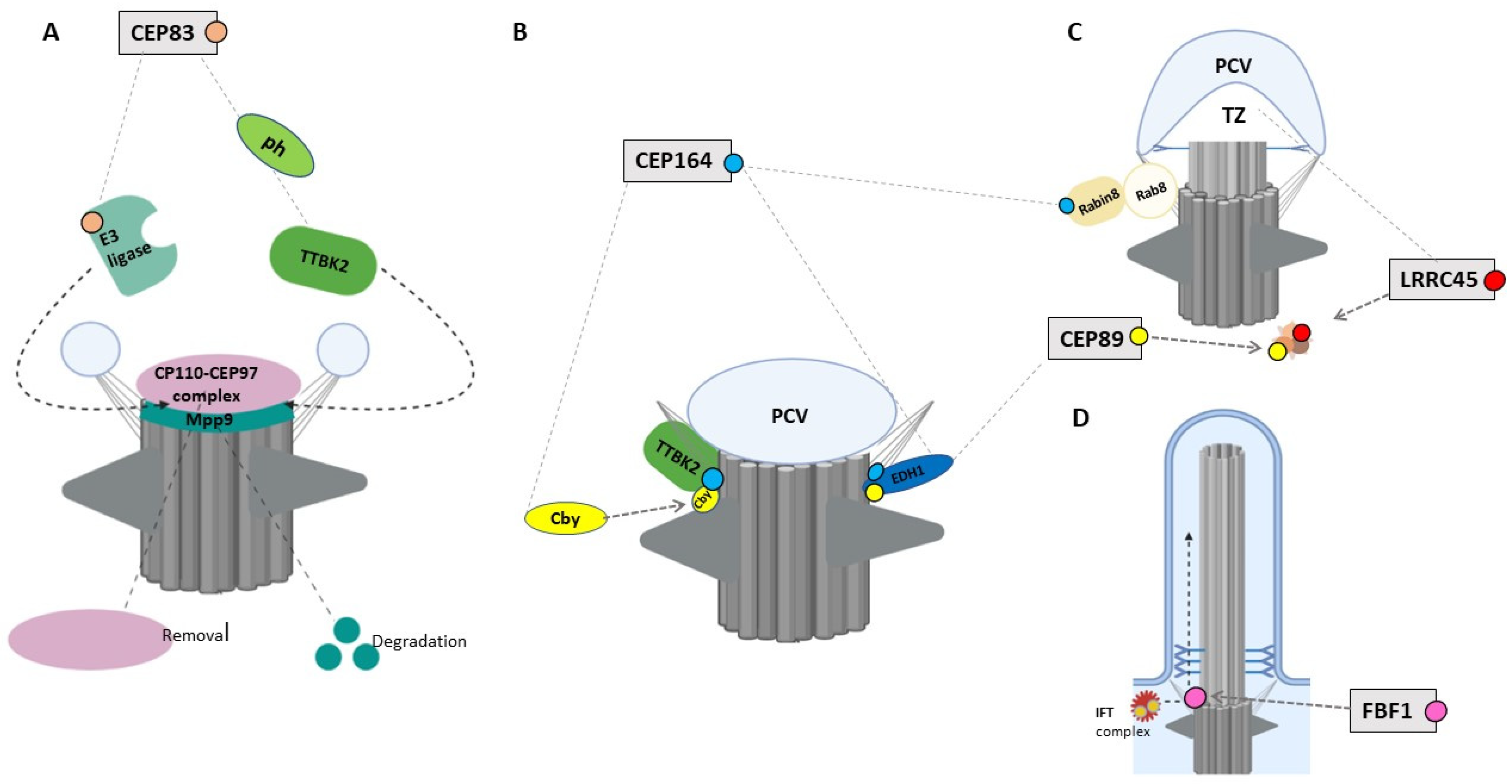
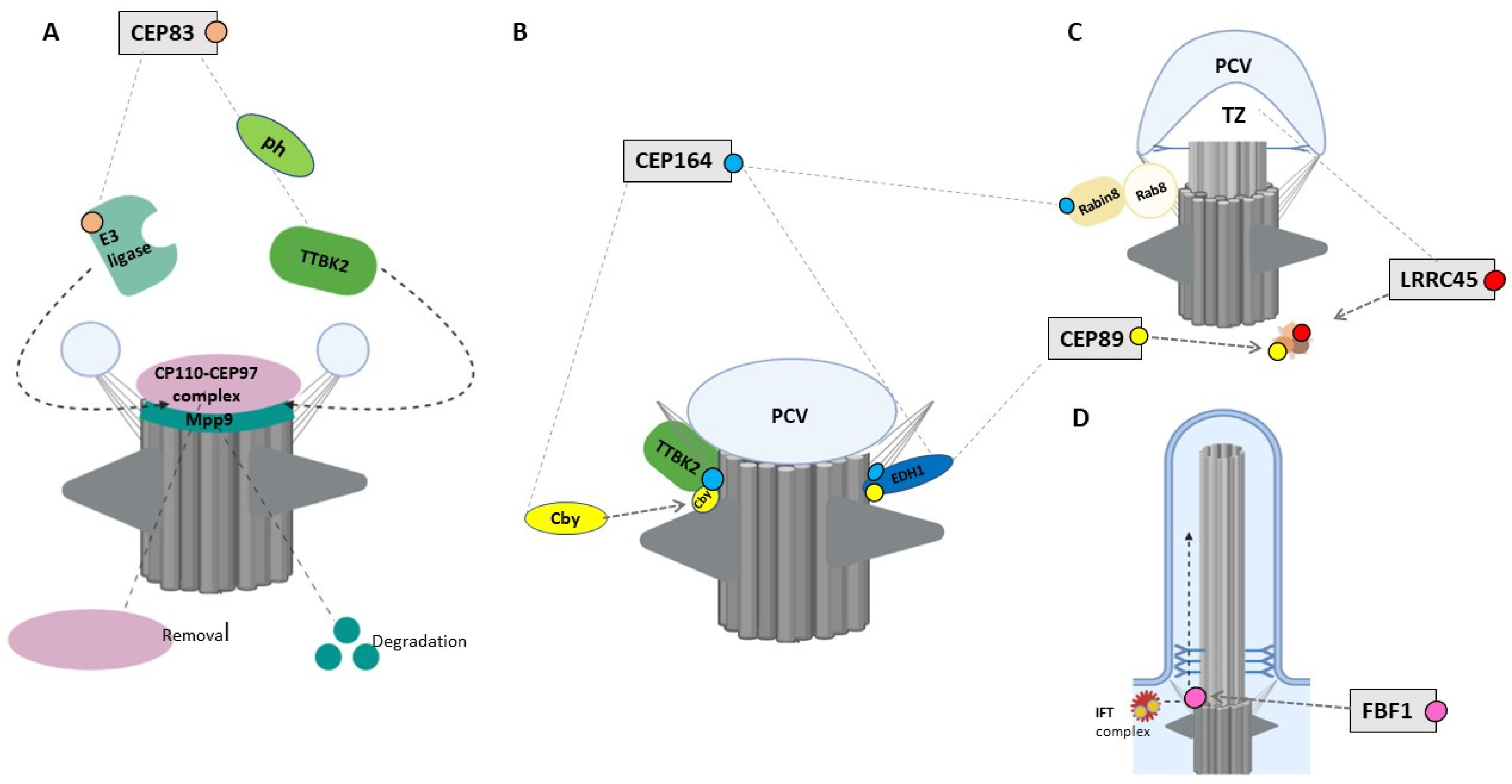
4.1. CEP83/CCDC41/NPHP18
4.2. CEP164/NPHP15
4.3. CEP89/CCDC123/CEP123
4.4. LRRC45
4.5. FBF1
4.6. SCLT1
4.7. CEP90/PIBF1
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sreekumar, V.; Norris, D.P. Cilia and development. Curr. Opin. Genet. Dev. 2019, 56, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef]
- Davenport, J.R.; Yoder, B.K. An incredible decade for the primary cilium: A look at a once-forgotten organelle. Am. J. Physiol. Ren. Physiol. 2005, 289, F1159–F1169. [Google Scholar] [CrossRef] [Green Version]
- Kozminski, K.G.; Johnson, K.A.; Forscher, P.; Rosenbaum, J.L. A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc. Natl. Acad. Sci. USA 1993, 90, 5519–5523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pazour, G.J.; Dickert, B.L.; Vucica, Y.; Seeley, E.S.; Rosenbaum, J.L.; Witman, G.B.; Cole, D.G. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J. Cell Biol. 2000, 151, 709–718. [Google Scholar] [CrossRef]
- Nager, A.R.; Goldstein, J.S.; Herranz-Pérez, V.; Portran, D.; Ye, F.; Garcia-Verdugo, J.M.; Nachury, M.V. An actin network dispatches ciliary GPCRs into extracellular vesicles to modulate signaling. Cell 2017, 168, 252–263.e214. [Google Scholar] [CrossRef] [Green Version]
- Labour, M.-N.; Riffault, M.; Christensen, S.T.; Hoey, D.A. TGFβ1–induced recruitment of human bone mesenchymal stem cells is mediated by the primary cilium in a SMAD3-dependent manner. Sci. Rep. 2016, 6, 35542. [Google Scholar]
- Christensen, S.T.; Morthorst, S.K.; Mogensen, J.B.; Pedersen, L.B. Primary cilia and coordination of receptor tyrosine kinase (RTK) and transforming growth factor β (TGF-β) signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheway, G.; Nazlamova, L.; Hancock, J.T. Signaling through the primary cilium. Front. Cell Dev. Biol. 2018, 6, 8. [Google Scholar] [CrossRef]
- Haycraft, C.J.; Banizs, B.; Aydin-Son, Y.; Zhang, Q.; Michaud, E.J.; Yoder, B.K. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005, 1, e53. [Google Scholar] [CrossRef]
- Pala, R.; Alomari, N.; Nauli, S.M. Primary Cilium-Dependent Signaling Mechanisms. Int. J. Mol. Sci. 2017, 18, 2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, S.T.; Clement, C.A.; Satir, P.; Pedersen, L.B. Primary cilia and coordination of receptor tyrosine kinase (RTK) signalling. J. Pathol. 2012, 226, 172–184. [Google Scholar] [CrossRef] [Green Version]
- Schneider, L.; Clement, C.A.; Teilmann, S.C.; Pazour, G.J.; Hoffmann, E.K.; Satir, P.; Christensen, S.T. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr. Biol. 2005, 15, 1861–1866. [Google Scholar] [CrossRef] [Green Version]
- Guruharsha, K.G.; Kankel, M.W.; Artavanis-Tsakonas, S. The Notch signalling system: Recent insights into the complexity of a conserved pathway. Nat. Reviews Genet. 2012, 13, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanos, B.E.; Yang, H.J.; Soni, R.; Wang, W.J.; Macaluso, F.P.; Asara, J.M.; Tsou, M.F.B. Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev. 2013, 27, 163–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.T.; Chong, W.M.; Wang, W.J.; Mazo, G.; Tanos, B.; Chen, Z.; Liao, J.C. Super-resolution architecture of mammalian centriole distal appendages reveals distinct blade and matrix functional components. Nat. Commun. 2018, 9, 2023. [Google Scholar] [CrossRef] [Green Version]
- Bowler, M.; Kong, D.; Sun, S.; Nanjundappa, R.; Evans, L.; Farmer, V.; Loncarek, J. High-resolution characterization of centriole distal appendage morphology and dynamics by correlative STORM and electron microscopy. Nat. Commun. 2019, 10, 993. [Google Scholar] [CrossRef]
- Kurtulmus, B.; Yuan, C.; Schuy, J.; Neuner, A.; Hata, S.; Kalamakis, G.; Martin-Villalba, A.; Pereira, G. LRRC45 contributes to early steps of axoneme extension. J. Cell Sci. 2018, 131, jcs223594. [Google Scholar] [CrossRef] [Green Version]
- Griffith, E.; Walker, S.; Martin, C.A.; Vagnarelli, P.; Stiff, T.; Vernay, B.; Al Sanna, N.; Saggar, A.; Hamel, B.; Earnshaw, W.C.; et al. Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat. Genet. 2008, 40, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Kalay, E.; Yigit, G.; Aslan, Y.; Brown, K.E.; Pohl, E.; Bicknell, L.S.; Bicknell, H.; Kayserili, Y.; Li, B.; Tüysüz, G.; et al. CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat. Genet. 2011, 43, 23–26. [Google Scholar] [CrossRef] [Green Version]
- Rauch, A.; Thiel, C.T.; Schindler, D.; Wick, U.; Crow, Y.J.; Ekici, A.B.; Ekici, A.J.; Essen, T.O.; Goecke, L.; Al-Gazali, K.H.C.; et al. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science 2008, 319, 816–819. [Google Scholar] [CrossRef] [PubMed]
- McConnachie, D.J.; Stow, J.L.; Mallett, A.J. Ciliopathies and the Kidney: A Review. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2021, 77, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jana, S.C.; Mendonça, S.; Machado, P.; Werner, S.; Rocha, J.; Pereira, A.; Maiato, H.; Bettencourt-Dias, M. Differential regulation of transition zone and centriole proteins contributes to ciliary base diversity. Nat. Cell Biol. 2018, 20, 928–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves, J.; Pelletier, L. The ciliary transition zone: Finding the pieces and assembling the gate. Mol. Cells 2017, 40, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef]
- Meunier, A.; Azimzadeh, J. Multiciliated cells in animals. Cold Spring Harb. Perspect. Biol. 2016, 8, a028233. [Google Scholar] [CrossRef] [Green Version]
- Deane, J.A.; Cole, D.G.; Seeley, E.S.; Diener, D.R.; Rosenbaum, J.L. Localization of intraflagellar transport protein IFT52 identifies basal body transitional fibers as the docking site for IFT particles. Curr. Biol. 2001, 11, 1586–1590. [Google Scholar] [CrossRef] [Green Version]
- Joukov, V.; De Nicolo, A. The centrosome and the primary cilium: The Yin and Yang of a hybrid organelle. Cells 2019, 8, 701. [Google Scholar] [CrossRef] [Green Version]
- Campa, C.C.; Hirsch, E. Rab11 and phosphoinositides: A synergy of signal transducers in the control of vesicular trafficking. Adv. Biol. Regul. 2017, 63, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Campa, C.C.; Franco, I.; Hirsch, E. PI3K-C2α: One enzyme for two products coupling vesicle trafficking and signal transduction. FEBS Lett. 2015, 589, 1552–1558. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, L.B.; Rosenbaum, J.L. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr. Top. Dev. Biol. 2008, 85, 23–61. [Google Scholar] [CrossRef] [PubMed]
- Taschner, M.; Lorentzen, E. The intraflagellar transport machinery. Cold Spring Harb. Perspect. Biol. 2016, 8, a028092. [Google Scholar] [CrossRef] [Green Version]
- Prevo, B.; Scholey, J.M.; Peterman, E.J. Intraflagellar transport: Mechanisms of motor action, cooperation, and cargo delivery. FEBS J. 2017, 284, 2905–2931. [Google Scholar] [CrossRef]
- Čajánek, L.; Nigg, E.A. CEP164 triggers ciliogenesis by recruiting Tau tubulin kinase 2 to the mother centriole. Proc. Natl. Acad. Sci. USA 2014, 111, E2841–E2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Failler, M.; Gee, H.Y.; Krug, P.; Joo, K.; Halbritter, J.; Belkacem, L.; Filhol, E.; Porath, J.D.; Braun, D.A.; Schueler, M.; et al. Mutations of CEP83 cause infantile nephronophthisis and intellectual disability. Am. J. Hum. Genet. 2014, 94, 905–914. [Google Scholar] [CrossRef] [Green Version]
- Katagiri, S.; Hayashi, T.; Yoshitake, K.; Murai, N.; Matsui, Z.; Kubo, H.; Satoh, H.; Matsufuji, S.; Takamura, T.; Yokoo, T.; et al. Compound heterozygous splice site variants in the SCLT1 gene highlight an additional candidate locus for Senior-Løken syndrome. Sci. Rep. 2018, 8, 16733. [Google Scholar]
- Li, J.; Lu, D.; Liu, H.; Williams, B.O.; Overbeek, P.A.; Lee, B.; Zheng, L.; Yang, T. SCLT1 deficiency causes cystic kidney by activating ERK and STAT3 signaling. Hum. Mol. Genet. 2017, 26, 2949–2960. [Google Scholar] [CrossRef]
- Slaats, G.G. Nephronophthisis-associated CEP164 regulates cell cycle progression, apoptosis and epithelial-to-mesenchymal transition. PLoS Genet. 2014, 10, e1004594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Airik, R.; Airik, M.; Schueler, M.; Bates, C.M.; Hildebrandt, F. Roscovitine blocks collecting duct cyst growth in CEP164-deficient kidneys. Kidney Int. 2019, 96, 320–326. [Google Scholar] [CrossRef]
- Veldman, B.C.; Kuper, W.F.; Lilien, M.; Schuurs-Hoeijmakers, J.H.; Marcelis, C.; Phan, M.; Hettinga, Y.; Talsma, H.E.; van Hasselt, P.M.; Haijes, H.A. Beyond nephronophthisis: Retinal dystrophy in the absence of kidney dysfunction in childhood expands the clinical spectrum of CEP83 deficiency. Am. J. Med. Genet. Part A 2021, 185, 2204–2210. [Google Scholar] [CrossRef]
- Kumar, D.; Rains, A.; Herranz-Perez, V.; Lu, Q.; Shi, X.; Swaney, D.L.; Stevenson, E.; Krogan, N.J.; Huang, B.; Westlake, C.; et al. A ciliopathy complex builds distal appendages to initiate ciliogenesis. J. Cell Biol. 2021, 220, e202011133. [Google Scholar] [CrossRef] [PubMed]
- Fliegauf, M.; Benzing, T.; Omran, H. When cilia go bad: Cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007, 8, 880–893. [Google Scholar] [CrossRef]
- Salomon, R.; Saunier, S.; Niaudet, P. Nephronophthisis. Pediatric Nephrol. 2009, 24, 2333–2344. [Google Scholar] [CrossRef] [Green Version]
- Stokman, M.; Lilien, M.; Knoers, N. Nephronophthisis. In GeneReviews® Internet; University of Washington: Seattle, WA, USA, 2016. [Google Scholar]
- Bisgrove, B.W.; Yost, H.J. The roles of cilia in developmental disorders and disease. Development 2006, 133, 4131–4143. [Google Scholar] [CrossRef] [Green Version]
- Hildebrandt, F.; Attanasio, M.; Otto, E. Nephronophthisis: Disease Mechanisms of a Ciliopathy. J. Am. Soc. Nephrol. 2009, 20, 23–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.; Molinari, E.; Raman, S.; Sayer, J.A. Many genes—one disease? Genetics of Nephronophthisis (NPHP) and NPHP-associated disorders. Front. Pediatrics 2018, 5, 287. [Google Scholar] [CrossRef]
- Luo, F.; Tao, Y.-H. Nephronophthisis: A review of genotype–phenotype correlation. Nephrology 2018, 23, 904–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snoek, R.; Van Setten, J.; Keating, B.J.; Israni, A.K.; Jacobson, P.A.; Oetting, W.S.; Matas, A.J.; Mannon, R.B.; Zhang, Z.; Zhang, W.; et al. NPHP1 (Nephrocystin-1) Gene Deletions Cause Adult-Onset ESRD. J. Am. Soc. Nephrol. 2018, 29, 1772–1779. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.T.; Hildebrandt, F. Nephronophthisis. Pediatric Nephrol. 2011, 26, 181–194. [Google Scholar] [CrossRef]
- Oud, M.M.; Van Bon, B.W.; Bongers, E.M.; Hoischen, A.; Marcelis, C.L.; De Leeuw, N.; Mol, S.J.J.; Mortier, G.; Knoers, N.V.A.M.; Brunner, H.G.; et al. Arts. Early presentation of cystic kidneys in a family with a homozygous INVS mutation. Am. J. Med. Genet. Part A 2014, 164, 1627–1634. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Zhou, W. Nephronophthisis-associated ciliopathies. J. Am. Soc. Nephrol. 2007, 18, 1855–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, E.M.; Conran, R.M.; Schroeder, J.W.; Rohena-Quinquilla, I.R.; Rooks, V.J. From the Radiologic Pathology Archives: Pediatric Polycystic Kidney Disease and Other Ciliopathies: Radiologic-Pathologic Correlation. RadioGraphics 2014, 34, 155–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, E.A.; Loeys, B.; Khanna, H.; Hellemans, J.; Sudbrak, R.; Fan, S.; Muerb, U.; O’Toole, J.F.; Helou, J.; Attanasio, M.; et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat. Genet. 2005, 37, 282–288. [Google Scholar] [CrossRef]
- Braun, D.A.; Hildebrandt, F. Ciliopathies. Cold Spring Harb. Perspect. Biol. 2017, 9, a028191. [Google Scholar] [CrossRef] [PubMed]
- Ronquillo, C.C.; Bernstein, P.S.; Baehr, W. Senior-Løken syndrome: A syndromic form of retinal dystrophy associated with nephronophthisis. Vis. Res. 2012, 75, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Sattar, S.; Gleeson, J.G. The ciliopathies in neuronal development: A clinical approach to investigation of Joubert syndrome and Joubert syndrome-related disorders. Dev. Med. Child Neurol. 2011, 53, 793–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louie, C.M.; Gleeson, J.G. Genetic basis of Joubert syndrome and related disorders of cerebellar development. Hum. Mol. Genet. 2005, 15, R235–R242. [Google Scholar] [CrossRef] [Green Version]
- Helou, J.; Otto, E.A.; Attanasio, M.; Allen, S.J.; Parisi, M.A.; Glass, I.; Utsch, B.; Hashmi, S.; Fazzi, E.; Omran, H.; et al. Mutation analysis of NPHP6/CEP290 in patients with Joubert syndrome and Senior–Løken syndrome. J. Med Genet. 2007, 44, 657–663. [Google Scholar] [CrossRef] [Green Version]
- Hartill, V.; Szymanska, K.; Sharif, S.M.; Wheway, G.; Johnson, C.A. Meckel-Gruber Syndrome: An Update on Diagnosis, Clinical Management, and Research Advances. Front. Pediatrics 2017, 5, 244. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, E.; Zaloszyc, A.; Lauer, J.; Durand, M.; Stutzmann, F.; Perdomo-Trujillo, Y.; Redin, C.; Bennouna Greene, V.A.; Toutain, L.; Perrin, M.; et al. Mutations in SDCCAG8/NPHP10 Cause Bardet-Biedl Syndrome and Are Associated with Penetrant Renal Disease and Absent Polydactyly. Mol. Syndr. 2011, 1, 273–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobin, J.L.; Beales, P.L. Bardet-Biedl syndrome: Beyond the cilium. Pediatric Nephrol. 2007, 22, 926–936. [Google Scholar] [CrossRef] [Green Version]
- Sang, L.; Miller, J.J.; Corbit, K.C.; Giles, R.H.; Brauer, M.J.; Otto, E.A.; Baye, L.M.; Wen, X.; Scales, S.J.; Kwong, M.; et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 2011, 145, 513–528. [Google Scholar] [CrossRef] [Green Version]
- Gupta, G.D.; Coyaud, É.; Gonçalves, J.; Mojarad, B.A.; Liu, Y.; Wu, Q.; Gheiratmand, L.; Comartin, D.; Tkach, J.M.; Cheung, S.W.; et al. A Dynamic Protein Interaction Landscape of the Human Centrosome-Cilium Interface. Cell 2015, 163, 1484–1499. [Google Scholar] [CrossRef] [Green Version]
- Habbig, S.; Bartram, M.P.; Müller, R.U.; Schwarz, R.; Andriopoulos, N.; Chen, S.; Sägmüller, J.G.; Hoehne, M.; Burst, V.; Liebau, M.C.; et al. NPHP4, a cilia-associated protein, negatively regulates the Hippo pathway. J. Cell Biol. 2011, 193, 633–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gonzalo, F.R.; Corbit, K.C.; Sirerol-Piquer, M.S.; Ramaswami, G.; Otto, E.A.; Noriega, T.R.; Reiter, J.F. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet. 2011, 43, 776–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, H.W.; Gustavsson, A.K.; Bayas, C.A.; Petrov, P.N.; Mooney, N.; Moerner, W.E.; Jackson, P.K. Novel fibrillar structure in the inversin compartment of primary cilia revealed by 3D single-molecule superresolution microscopy. Mol. Biol. Cell 2020, 31, 619–639. [Google Scholar] [CrossRef]
- Otto, E.A.; Schermer, B.; Obara, T.; O’Toole, J.F.; Hiller, K.S.; Mueller, A.M.; Ruf, R.G.; Hoefele, J.; Beekmann, F.; Landau, D.; et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat. Genet. 2003, 34, 413–420. [Google Scholar] [CrossRef]
- Okada, M.; Sugimoto, K.; Shimada, Y.; Fujita, S.; Yanagida, H.; Yagi, K.; Takemura, T. Association of INVS (NPHP2) mutation in an adolescent exhibiting nephronophthisis (NPH) and complete situs inversus. Clin. Nephrol. 2008, 69, 135–141. [Google Scholar] [CrossRef]
- Tory, K.; Rousset-Rouviere, C.; Gubler, M.C.; Moriniere, V.; Pawtowski, A.; Becker, C.; Guyot, C.; Gié, S.; Frishberg, Y.; Nivet, H.; et al. Mutations of NPHP2 and NPHP3 in infantile nephronophthisis. Kidney Int. 2009, 75, 839–847. [Google Scholar] [CrossRef] [Green Version]
- Bellavia, S.; Dahan, K.; Terryn, S.; Cosyns, J.P.; Devuyst, O.; Pirson, Y. A homozygous mutation in INVS causing juvenile nephronophthisis with abnormal reactivity of the Wnt/beta-catenin pathway. Nephrol. Dial. Transplant. 2010, 25, 4097–4102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabriel, G.C.; Pazour, G.J.; Lo, C.W. Congenital Heart Defects and Ciliopathies Associated With Renal Phenotypes. Front. Pediatrics 2018, 6, 175. [Google Scholar] [CrossRef] [PubMed]
- Czarnecki, P.G.; Gabriel, G.C.; Manning, D.K.; Sergeev, M.; Lemke, K.; Klena, N.T.; Liu, X.; Chen, Y.; Li, Y.; San Agustin, J.T.; et al. ANKS6 is the critical activator of NEK8 kinase in embryonic situs determination and organ patterning. Nat. Commun. 2015, 6, 6023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, V.L.; Li, C.; Bowie, R.V.; Clarke, L.; Mohan, S.; Blacque, O.E.; Leroux, M.R. Formation of the transition zone by Mks5/Rpgrip1L establishes a ciliary zone of exclusion (CIZE) that compartmentalises ciliary signalling proteins and controls PIP2 ciliary abundance. EMBO J. 2015, 34, 2537–2556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegering, A.; Dildrop, R.; Vesque, C.; Khanna, H.; Schneider-Maunoury, S.; Gerhardt, C. Rpgrip1l controls ciliary gating by ensuring the proper amount of CEP290 at the vertebrate transition zone. Mol. Biol. Cell 2021, 32, 675–689. [Google Scholar] [CrossRef]
- Gerhardt, C.; Lier, J.M.; Burmühl, S.; Struchtrup, A.; Deutschmann, K.; Vetter, M.; Leu, T.; Reeg, S.; Grune, T.; Rüther, U. The transition zone protein Rpgrip1l regulates proteasomal activity at the primary cilium. J. Cell Biol. 2015, 210, 115–133. [Google Scholar] [CrossRef] [Green Version]
- Vierkotten, J.; Dildrop, R.; Peters, T.; Wang, B.; Rüther, U. Ftm is a novel basal body protein of cilia involved in Shh signalling. Development 2007, 134, 2569–2577. [Google Scholar] [CrossRef] [Green Version]
- Struchtrup, A.; Wiegering, A.; Stork, B.; Rüther, U.; Gerhardt, C. The ciliary protein RPGRIP1L governs autophagy independently of its proteasome-regulating function at the ciliary base in mouse embryonic fibroblasts. Autophagy 2018, 14, 567–583. [Google Scholar] [CrossRef] [Green Version]
- Delous, M.; Baala, L.; Salomon, R.; Laclef, C.; Vierkotten, J.; Tory, K.; Golzio, C.; Lacoste, T.; Besse, L.; Ozilou, C.; et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 2007, 39, 875–881. [Google Scholar] [CrossRef]
- Coppieters, F.; Lefever, S.; Leroy, B.P.; De Baere, E. CEP290, a gene with many faces: Mutation overview and presentation of CEP290base. Hum. Mutat. 2010, 31, 1097–1108. [Google Scholar] [CrossRef] [Green Version]
- Drivas, T.G.; Bennett, J. CEP290 and the primary cilium. Adv. Exp. Med. Biol. 2014, 801, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Weatherbee, S.D.; Niswander, L.A.; Anderson, K.V. A mouse model for Meckel syndrome reveals Mks1 is required for ciliogenesis and Hedgehog signaling. Hum. Mol. Genet. 2009, 18, 4565–4575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyttälä, M.; Tallila, J.; Salonen, R.; Kopra, O.; Kohlschmidt, N.; Paavola-Sakki, P.; Peltonen, L.; Kestilä, M. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat. Genet. 2006, 38, 155–157. [Google Scholar] [CrossRef]
- Plotnikova, O.V.; Golemis, E.A.; Pugacheva, E.N. Cell cycle-dependent ciliogenesis and cancer. Cancer Res. 2008, 68, 2058–2061. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, I.; Dynlacht, B.D. Cilium assembly and disassembly. Nat. Cell Biol. 2016, 18, 711–717. [Google Scholar] [CrossRef] [Green Version]
- Mirvis, M.; Stearns, T.; Nelson, W.J. Cilium structure, assembly, and disassembly regulated by the cytoskeleton. Biochem. J. 2018, 475, 2329–2353. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Insinna, C.; Ott, C.; Stauffer, J.; Pintado, P.A.; Rahajeng, J.; Baxa, U.; Walia, V.; Cuenca, A.; Hwang, Y.S.; et al. Early steps in primary cilium assembly require EHD1/EHD3-dependent ciliary vesicle formation. Nat. Cell Biol. 2015, 17, 228–240. [Google Scholar] [CrossRef] [Green Version]
- Yee, L.E.; Reiter, J.F. Ciliary vesicle formation: A prelude to ciliogenesis. Dev. Cell 2015, 32, 665–666. [Google Scholar] [CrossRef] [Green Version]
- Knödler, A.; Feng, S.; Zhang, J.; Zhang, X.; Das, A.; Peränen, J.; Guo, W. Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 6346–6351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westlake, C.J.; Baye, L.M.; Nachury, M.V.; Wright, K.J.; Ervin, K.E.; Phu, L.; Chalouni, C.; Beck, J.S.; Kirkpatrick, D.S.; Slusarski, D.C.; et al. Primary cilia membrane assembly is initiated by Rab11 and transport protein particle II (TRAPPII) complex-dependent trafficking of Rabin8 to the centrosome. Proc. Natl. Acad. Sci. USA 2011, 108, 2759–2764. [Google Scholar] [CrossRef] [Green Version]
- Reilly, M.L.; Benmerah, A. Ciliary kinesins beyond IFT: Cilium length, disassembly, cargo transport and signalling. Biol. Cell 2019, 111, 79–94. [Google Scholar] [CrossRef]
- Lopes, C.A.; Prosser, S.L.; Romio, L.; Hirst, R.A.; O’Callaghan, C.; Woolf, A.S.; Fry, A.M. Centriolar satellites are assembly points for proteins implicated in human ciliopathies, including oral-facial-digital syndrome 1. J. Cell Sci. 2011, 124, 600–612. [Google Scholar] [CrossRef] [Green Version]
- Klinger, M.; Wang, W.; Kuhns, S.; Bärenz, F.; Dräger-Meurer, S.; Pereira, G.; Gruss, O.J. The novel centriolar satellite protein SSX2IP targets Cep290 to the ciliary transition zone. Mol. Biol. Cell 2014, 25, 495–507. [Google Scholar] [CrossRef]
- Kurtulmus, B.; Wang, W.; Ruppert, T.; Neuner, A.; Cerikan, B.; Viol, L.; Dueñas-Sánchez, R.; Gruss, O.J.; Pereira, G. WDR8 is a centriolar satellite and centriole-associated protein that promotes ciliary vesicle docking during ciliogenesis. J. Cell Sci. 2016, 129, 621–636. [Google Scholar]
- Shao, W.; Yang, J.; He, M.; Yu, X.Y.; Lee, C.H.; Yang, Z.; Joyner, A.L.; Anderson, K.V.; Zhang, J.; Tsou, M.-F.B. Centrosome anchoring regulates progenitor properties and cortical formation. Nature 2020, 580, 106–112. [Google Scholar] [CrossRef]
- Lo, C.H.; Lin, I.; Yang, T.T.; Huang, Y.C.; Tanos, B.E.; Chou, P.C.; Chang, C.W.; Tsay, Y.G.; Liao, J.C.; Wang, W.J. Phosphorylation of CEP83 by TTBK2 is necessary for cilia initiation. J. Cell Biol. 2019, 218, 3489–3505. [Google Scholar] [CrossRef] [Green Version]
- Huang, H. Matrix metalloproteinase-9 (MMP-9) as a cancer biomarker and MMP-9 biosensors: Recent advances. Sensors 2018, 18, 3249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, N.; Zhang, D.; Li, F.; Chai, P.; Wang, S.; Teng, J.; Chen, J. M-Phase Phosphoprotein 9 regulates ciliogenesis by modulating CP110-CEP97 complex localization at the mother centriole. Nat. Commun. 2018, 9, 4511. [Google Scholar] [CrossRef] [PubMed]
- Joo, K.; Kim, C.G.; Lee, M.S.; Moon, H.Y.; Lee, S.H.; Kim, M.J.; Kweon, H.S.; Park, W.Y.; Kim, C.H.; Gleeson, J.G.; et al. CCDC41 is required for ciliary vesicle docking to the mother centriole. Proc. Natl. Acad. Sci. USA 2013, 110, 5987–5992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, M.C.; Weihbrecht, K.; Searby, C.C.; Li, Y.; Pope, R.M.; Sheffield, V.C.; Seo, S. ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc. Natl. Acad. Sci. USA 2012, 109, 19691–19696. [Google Scholar] [CrossRef] [Green Version]
- Chaki, M.; Airik, R.; Ghosh, A.K.; Giles, R.H.; Chen, R.; Slaats, G.G.; Wang, H.; Hurd, T.W.; Zhou, W.; Cluckey, A.; et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 2012, 150, 533–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimaru, T.; Kawanishi, K.; Mori, T.; Mishima, E.; Sekine, A.; Chiga, M.; Mizui, M.; Sato, N.; Yanagita, M.; Ooki, Y.; et al. Genetic Background and Clinicopathologic Features of Adult-onset Nephronophthisis. Kidney Int. Rep. 2021, 6, 1346–1354. [Google Scholar] [CrossRef] [PubMed]
- Maria, M.; Lamers, I.J.; Schmidts, M.; Ajmal, M.; Jaffar, S.; Ullah, E.; Mustafa, B.; Ahmad, S.; Nazmutdinova, K.; Hoskins, B.; et al. Genetic and clinical characterization of Pakistani families with Bardet-Biedl syndrome extends the genetic and phenotypic spectrum. Sci. Rep. 2016, 6, 34764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamseldin, H.E.; Al Mogarri, I.; Alqwaiee, M.M.; Alharbi, A.S.; Baqais, K.; AlSaadi, M.; AlAnzi, T.; Alhashem, A.; Saghier, A.; Ameen, W.; et al. An exome-first approach to aid in the diagnosis of primary ciliary dyskinesia. Hum. Genet. 2020, 139, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Siller, S.S.; Sharma, H.; Li, S.; Yang, J.; Zhang, Y.; Holtzman, M.J.; Winuthayanon, W.; Colognato, H.; Holdener, B.C.; Li, F.Q.; et al. Conditional knockout mice for the distal appendage protein CEP164 reveal its essential roles in airway multiciliated cell differentiation. PLoS Genet. 2017, 13, e1007128. [Google Scholar] [CrossRef]
- Adly, N.; Alhashem, A.; Ammari, A.; Alkuraya, F.S. Ciliary Genes TBC 1 D 32/C6orf170 and SCLT 1 are Mutated in Patients with OFD Type IX. Hum. Mutat. 2014, 35, 36–40. [Google Scholar] [CrossRef]
- Morisada, N.; Hamada, R.; Miura, K.; Ye, M.J.; Nozu, K.; Hattori, M.; Iijima, K. Bardet-Biedl syndrome in two unrelated patients with identical compound heterozygous SCLT1 mutations. CEN Case Rep. 2020, 9, 260–265. [Google Scholar] [CrossRef]
- Graser, S.; Stierhof, Y.D.; Lavoie, S.B.; Gassner, O.S.; Lamla, S.; Le Clech, M.; Nigg, E.A. CEP164, a novel centriole appendage protein required for primary cilium formation. J. Cell Biol. 2007, 179, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, K.N.; Kuhns, S.; Neuner, A.; Hub, B.; Zentgraf, H.; Pereira, G. Cep164 mediates vesicular docking to the mother centriole during early steps of ciliogenesis. J. Cell Biol. 2012, 199, 1083–1101. [Google Scholar] [CrossRef] [Green Version]
- Burke, M.C.; Li, F.-Q.; Cyge, B.; Arashiro, T.; Brechbuhl, H.M.; Chen, X.; Siller, S.S.; Weiss, M.A.; O’Connell, C.B.; Love, D.; et al. Chibby promotes ciliary vesicle formation and basal body docking during airway cell differentiation. J. Cell Biol. 2014, 207, 123–137. [Google Scholar] [CrossRef] [Green Version]
- Goetz, S.C.; Liem, K.F., Jr.; Anderson, K.V. The spinocerebellar ataxia-associated gene Tau tubulin kinase 2 controls the initiation of ciliogenesis. Cell 2012, 151, 847–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, I.R.; Binó, L.; Johnson, C.M.; Rutherford, T.J.; Neuhaus, D.; Andreeva, A.; Čajánek, L.; van Breugel, M. Molecular mechanisms underlying the role of the centriolar CEP164-TTBK2 complex in ciliopathies. Structure 2021, 6, 1–5. [Google Scholar] [CrossRef]
- Hua, K.; Ferland, R.J. Primary Cilia Reconsidered in the Context of Ciliopathies: Extraciliary and Ciliary Functions of Cilia Proteins Converge on a Polarity theme? BioEssays News Rev. Mol. Cell. Dev. Biol. 2018, 40, e1700132. [Google Scholar] [CrossRef]
- Hua, K.; Ferland, R.J. Primary cilia proteins: Ciliary and extraciliary sites and functions. Cell. Mol. Life Sci. CMLS 2018, 75, 1521–1540. [Google Scholar] [CrossRef] [PubMed]
- Sillibourne, J.E.; Hurbain, I.; Grand-Perret, T.; Goud, B.; Tran, P.; Bornens, M. Primary ciliogenesis requires the distal appendage component Cep123. Biol. Open 2013, 2, 535–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.; Guo, W. Rabs and the exocyst in ciliogenesis, tubulogenesis and beyond. Trends Cell Biol. 2011, 21, 383–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nachury, M.V.; Loktev, A.V.; Zhang, Q.; Westlake, C.J.; Peränen, J.; Merdes, A.; Slusarski, D.C.; Scheller, R.H.; Bazan, J.F.; Sheffield, V.C. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 2007, 129, 1201–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, W.Y.; Bossard, C.; Khanna, H.; Peränen, J.; Swaroop, A.; Malhotra, V.; Dynlacht, B.D. CP110 suppresses primary cilia formation through its interaction with CEP290, a protein deficient in human ciliary disease. Dev. Cell 2008, 15, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Hernandez, V.; Pravincumar, P.; Diaz-Font, A.; May-Simera, H.; Jenkins, D.; Knight, M.; Beales, P.L. Bardet-Biedl syndrome proteins control the cilia length through regulation of actin polymerization. Hum. Mol. Genet. 2013, 22, 3858–3868. [Google Scholar] [CrossRef] [Green Version]
- Singla, V.; Romaguera-Ros, M.; Garcia-Verdugo, J.M.; Reiter, J.F. Ofd1, a human disease gene, regulates the length and distal structure of centrioles. Dev. Cell 2010, 18, 410–424. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.C.; Badano, J.L.; Sibold, S.; Esmail, M.A.; Hill, J.; Hoskins, B.E.; Leitch, C.C.; Venner, K.; Ansley, S.J.; Ross, A.J. The Bardet-Biedl protein BBS4 targets cargo to the pericentriolar region and is required for microtubule anchoring and cell cycle progression. Nat. Genet. 2004, 36, 462–470. [Google Scholar] [CrossRef] [Green Version]
- He, R.; Huang, N.; Bao, Y.; Zhou, H.; Teng, J.; Chen, J. LRRC45 is a centrosome linker component required for centrosome cohesion. Cell Rep. 2013, 4, 1100–1107. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Xu, Q.; Zhang, Y.; Li, Y.; Zhang, Q.; Hu, Z.; Harris, P.C.; Torres, V.E.; Ling, K.; Hu, J. Transition fibre protein FBF1 is required for the ciliary entry of assembled intraflagellar transport complexes. Nat. Commun. 2013, 4, 2750. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Ling, K.; Hu, J. The essential roles of transition fibers in the context of cilia. Curr. Opin. Cell Biol. 2015, 35, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Lee, K.; Rhee, K. CEP90 is required for the assembly and centrosomal accumulation of centriolar satellites, which is essential for primary cilia formation. PLoS ONE 2012, 7, e48196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodani, A.; Timothy, W.Y.; Johnson, J.R.; Jayaraman, D.; Johnson, T.L.; Al-Gazali, L.; Sztriha, L.; Partlow, J.N.; Kim, H.; Krup, A.L. Centriolar satellites assemble centrosomal microcephaly proteins to recruit CDK2 and promote centriole duplication. eLife 2015, 4, e07519. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Krishnaswami, S.R.; Gleeson, J.G. CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium. Hum. Mol. Genet. 2008, 17, 3796–3805. [Google Scholar] [CrossRef] [Green Version]
- Hinchcliffe, E.H.; Li, C.; Thompson, E.A.; Maller, J.L.; Sluder, G. Requirement of Cdk2-cyclin E activity for repeated centrosome reproduction in Xenopus egg extracts. Science 1999, 283, 851–854. [Google Scholar] [CrossRef] [PubMed]
- Löffler, H.; Fechter, A.; Matuszewska, M.; Saffrich, R.; Mistrik, M.; Marhold, J.; Hornung, C.; Westermann, F.; Bartek, J.; Krämer, A. Cep63 recruits Cdk1 to the centrosome: Implications for regulation of mitotic entry, centrosome amplification, and genome maintenance. Cancer Res. 2011, 71, 2129–2139. [Google Scholar] [CrossRef] [Green Version]
- Ott, T.; Kaufmann, L.; Granzow, M.; Hinderhofer, K.; Bartram, C.R.; Theiß, S.; Paramasivam, N.; Schulz, A.; Moog, U.; Blum, M. The Frog Xenopus as a Model to Study Joubert Syndrome: The Case of a Human Patient with Compound Heterozygous Variants in PIBF1. Front. Physiol. 2019, 10, 134. [Google Scholar] [CrossRef]
- Wheway, G.; Schmidts, M.; Mans, D.A.; Szymanska, K.; Nguyen, T.M.T.; Racher, H.; Phelps, I.G.; Toedt, G.; Kennedy, J.; Wunderlich, K.A.; et al. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat. Cell Biol. 2015, 17, 1074–1087. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Mutations | Renal Phenotype | Extra-Renal Manifestations | Cilia Phenotype | Ref. |
|---|---|---|---|---|---|
| CEP83 | Hs In 9 individuals diagnosed with NPHP-RC homozygous or compound heterozygous CEP83 mutations has been identified. 1 individual has been reported with a homozygous missense, 1 with a homozygous protein-truncating mutation. 7 individuals carry at least one loss of function allele. | Nephronophthisis, Tubulointerstitial nephritis, Corticomedullary cysts, Tubular atrophy, and End-stage renal disease. | Eye (in some patients): Retinitis Strabismus Liver (in some patients): Cholestasis, Hepatic cytolysis, Portal fibrosis Central Nervous System (in some patients): Intellectual disability, Hydrocephalus. | (a) primary fibroblasts:impaired ciliation (b) renal biopsy sample: increased ciliary length (c) overexpression of disease construct in RPE-1/ IMCD3: abolished centrosomal localization of CEP83 abrogated protein interaction with CEP164 and IFT20 nuclear accumulation of CEP83 (d) depletion in RPE-1: abolished cilia formation. | [36] |
| Cep83 | Mm Selective deletion of Cep83 in cortical radial glial progenitors (RGPs). | Enlarged brain with abnormal folding. | RGPs lacking Cep83 display a lack of primary cilia. | [96] | |
| cep83 | Dr Morpholino-mediated knockdown of ccdc41 (CEP83 ortholog in Zebrafish). | No defect in left/right body asymmetry was observed. | Olfactory placodes showed reduction in cilium formation. | [100] | |
| CEP164 | Hs Homozygous and compound heterozygous has been identified in 4 individuals with NPHP-RC. 2 individuals carried loss of function mutations. Compound heterozygous missense mutations lead to Bardet–Biedl syndrome in 1 individual and a homozygous loss mutation to primary ciliary dyskinesia (PCD) in 1 individual. | Nephronophthisis | Eyes: Retinal degeneration, Leber congenital amaurosis, Nystagmus (in 2 patients) Liver (in some patients): Liver failure Central Nervous System: Developmental delay (in 1 patient), Seizures (in 1 patient), Cerebellar vermis hypoplasia (in 1 patient) Skeletal: Polydactyly (in 2 patients) Obesity (in 2 patients) Short stature (in 1 patient) Bilateral bronchiectasis (in 1 patient). | (a) Overexpression of disease construct in IMCD3 cells: abolished centrosomal localization (b) Overexpression of disease construct in hTERT RPE: compromise interaction with TTBK2 (c) Depletion in RPE-1: abolish cilia formation. | [101,102,103,104,105] |
| cep164 | Dr | Pronephric tubule cysts. | Abnormal heart looping, hydrocephalus, and retinal dysplasia. | n/a | [102] |
| Cep164 | Mm | Only in the collecting duct-specific deletion of CEP164 mice: Cystic kidneys [40]. | (a) Global CEP164 deficiency mice: early embryonic lethality, holoprosencephaly, cardiac looping defects, and a truncated posterior trunk [106]. (b) collecting duct-specific deletion of CEP164 mice: only renal cyst growth. (c) CEP164 loss in FOXJ1-positive tissues in mice: results in hydrocephalus. | (a) Global deficiency: abolish cilia formation in neuronal tube (b) Collecting duct-specific deletion: abolishes primary cilia formation in epithelial cells (c) FOXJ1-specific deletion:reduction number of l multiciliated cells. | [40,106] |
| SCLT1 | Hs Compound heterozygous missense mutations has been reported in 1 individual with oro-facio-digital syndrome. Biallelic loss of function mutations have been identified in 1 individual withSenior-Løken syndrom, and in 1 individual with Bardet–Biedl syndrome. | Nephronophthisis (1 patient) bilateral hyperechogenicity, cortico-medullary renal cysts (1 Patient) ESRD at 11 years of age (1 Patient). | (a) Orofaciodigital syndrome type IX (OFD type IX) (2 patients): midline cleft, microcephaly, colobomatous microphthalmia/ anophthalmia, polydactyly, absent pituitary, and congenital heart disease. (b) Senior–Løken syndrome (1 patient): Nystagmus, hepatic dysfunction, megacystis, mild learning disability, autism, obesity (c) Bardet–Biedl syndrome (2 patients): intellectual disability, autism, and motor developmental delay, hepatic fibrosis, short stature, truncal obesity, retinitis pigmentosa. | (a) Depletion in RPE-1: abolish cilia formation. | [107,108] |
| Sclt1 | Mm | Cystic kidneys | Cleft palate and polydactyly. | Global deficiency: disrupted cilia assembly. | [37,38,108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mansour, F.; Boivin, F.J.; Shaheed, I.B.; Schueler, M.; Schmidt-Ott, K.M. The Role of Centrosome Distal Appendage Proteins (DAPs) in Nephronophthisis and Ciliogenesis. Int. J. Mol. Sci. 2021, 22, 12253. https://doi.org/10.3390/ijms222212253
Mansour F, Boivin FJ, Shaheed IB, Schueler M, Schmidt-Ott KM. The Role of Centrosome Distal Appendage Proteins (DAPs) in Nephronophthisis and Ciliogenesis. International Journal of Molecular Sciences. 2021; 22(22):12253. https://doi.org/10.3390/ijms222212253
Chicago/Turabian StyleMansour, Fatma, Felix J. Boivin, Iman B. Shaheed, Markus Schueler, and Kai M. Schmidt-Ott. 2021. "The Role of Centrosome Distal Appendage Proteins (DAPs) in Nephronophthisis and Ciliogenesis" International Journal of Molecular Sciences 22, no. 22: 12253. https://doi.org/10.3390/ijms222212253
APA StyleMansour, F., Boivin, F. J., Shaheed, I. B., Schueler, M., & Schmidt-Ott, K. M. (2021). The Role of Centrosome Distal Appendage Proteins (DAPs) in Nephronophthisis and Ciliogenesis. International Journal of Molecular Sciences, 22(22), 12253. https://doi.org/10.3390/ijms222212253