
Defective Uteroplacental Vascular Remodeling in Preeclampsia: Key Molecular Factors Leading to Long Term Cardiovascular Disease


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Uteroplacental Vascular Development in Normal Pregnancy
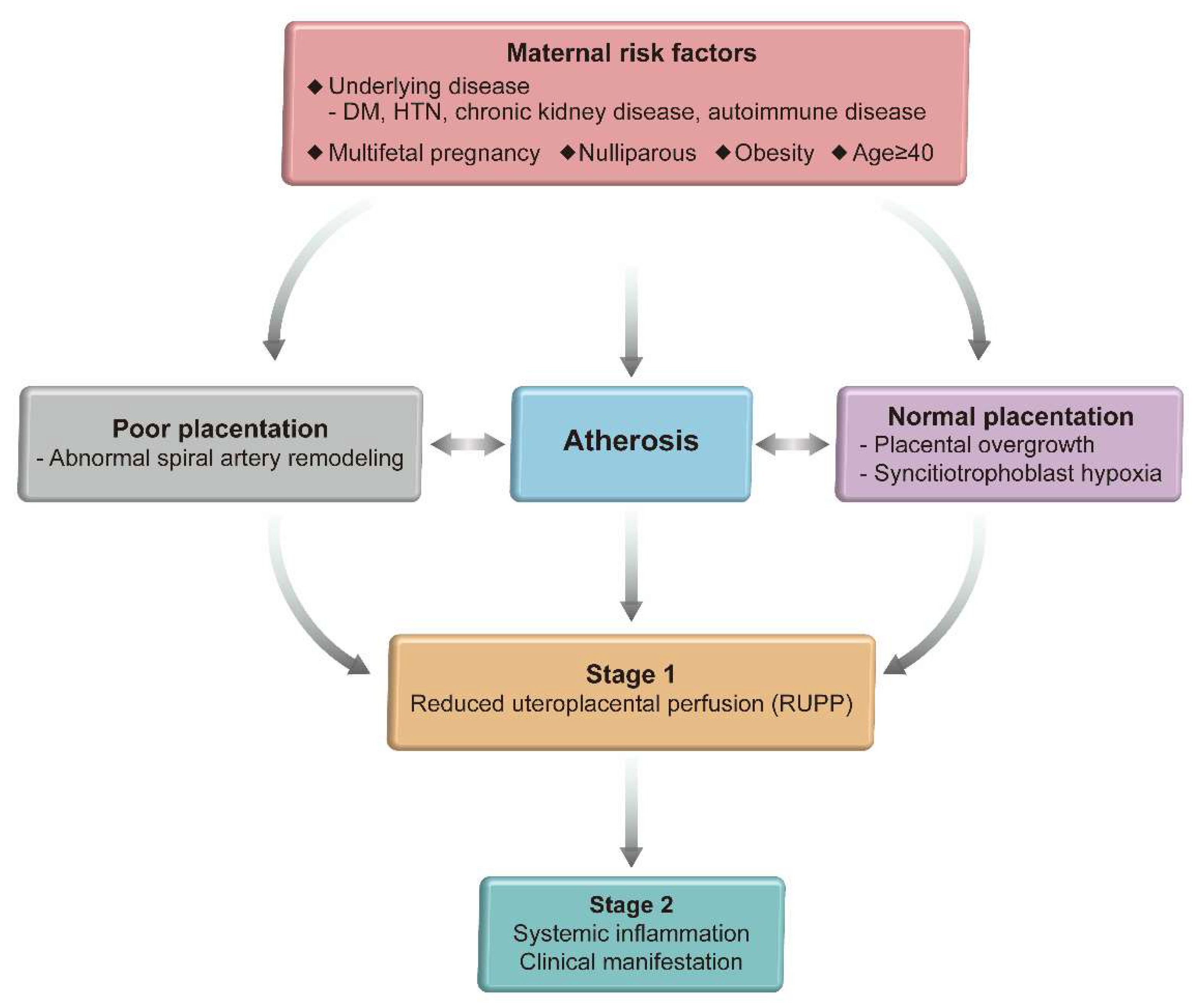
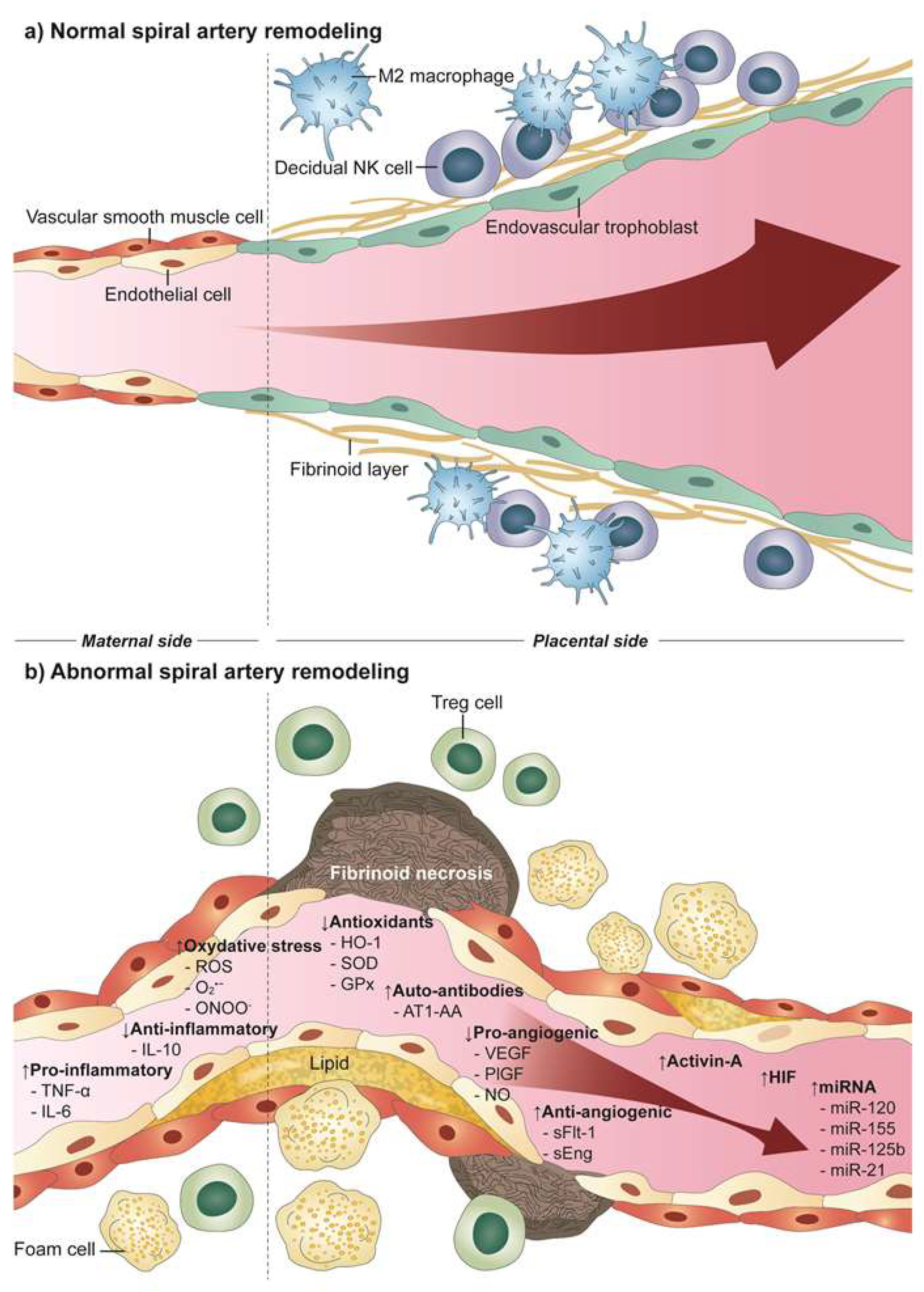
3. Defective Uteroplacental Vascular Remodeling in Preeclampsia
4. Two Step Model of Preeclampsia
5. Atherosis
6. Molecular Factors Resulting from Inadequate Uteroplacental Perfusion Leading to Preeclampsia
6.1. Inflammatory Factors
6.2. Reactive Oxygen Species (ROS)
6.3. Angiotensin II (AngII) and Angiotensin II Type 1 Receptor (AT1R) Autoantibodies (AT1-AA)
6.4. Angiogenic/Antiangiogenic Factors
6.4.1. Vascular Endothelial Growth Factors (VEGF)
6.4.2. Placental Growth Factor (PlGF)
6.4.3. Soluble FMS-Like Tyrosine Kinase I (sFlt-1)
6.4.4. Soluble Endoglin (sEng)
6.5. Activin A
6.6. Hypoxia Inducible Factor
6.7. MicroRNAs
7. Preeclampsia and Future Cardiovascular Health
7.1. Chronic Hypertension
7.2. Altered Vascular Structure
7.3. Coronary Artery Disease and Cerebrovascular Accident
7.4. Postpartum Management after HDP
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Brown, M.A.; Magee, L.A.; Kenny, L.C.; Karumanchi, S.A.; McCarthy, F.P.; Saito, S.; Hall, D.R.; Warren, C.E.; Adoyi, G.; Ishaku, S. The hypertensive disorders of pregnancy: ISSHP classification, diagnosis & management recommendations for international practice. Pregnancy Hypertens. 2018, 13, 291–310. [Google Scholar] [CrossRef]
- Brown, M.A.; Magee, L.A.; Kenny, L.C.; Karumanchi, S.A.; McCarthy, F.P.; Saito, S.; Hall, D.R.; Warren, C.E.; Adoyi, G.; Ishaku, S. Hypertensive Disorders of Pregnancy: ISSHP Classification, Diagnosis, and Management Recommendations for International Practice. Hypertension 2018, 72, 24–43. [Google Scholar] [CrossRef]
- Mosca, L.; Benjamin, E.J.; Berra, K.; Bezanson, J.L.; Dolor, R.J.; Lloyd-Jones, D.M.; Newby, L.K.; Piña, I.L.; Roger, V.L.; Shaw, L.J.; et al. Effectiveness-Based Guidelines for the Prevention of Cardiovascular Disease in Women—2011 Update. Circulation 2011, 123, 1243–1262. [Google Scholar] [CrossRef]
- Bellamy, L.; Casas, J.P.; Hingorani, A.D.; Williams, D.J. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: Systematic review and meta-analysis. BMJ 2007, 335, 974. [Google Scholar] [CrossRef]
- Hong, K.; Park, H.J.; Cha, D.H. Clinical implications of placenta-derived angiogenic/anti-angiogenic biomarkers in pre-eclampsia. Biomark. Med. 2021, 15, 523–536. [Google Scholar] [CrossRef]
- Lyall, F. The human placental bed revisited. Placenta 2002, 23, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Brosens, I.A.; Robertson, W.B.; Dixon, H.G. The role of the spiral arteries in the pathogenesis of preeclampsia. Obstet. Gynecol. Annu. 1972, 1, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Pijnenborg, R.; Vercruysse, L.; Hanssens, M. The uterine spiral arteries in human pregnancy: Facts and controversies. Placenta 2006, 27, 939–958. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y. Endovascular trophoblast and spiral artery remodeling. Mol. Cell. Endocrinol. 2020, 503, 110699. [Google Scholar] [CrossRef] [PubMed]
- Chaiworapongsa, T.; Chaemsaithong, P.; Yeo, L.; Romero, R. Pre-eclampsia part 1: Current understanding of its pathophysiology. Nat. Rev. Nephrol. 2014, 10, 466–480. [Google Scholar] [CrossRef]
- Sheppard, B.L.; Bonnar, J. An ultrastructural study of utero-placental spiral arteries in hypertensive and normotensive pregnancy and fetal growth retardation. Br. J. Obstet. Gynaecol. 1981, 88, 695–705. [Google Scholar] [CrossRef]
- Brosens, I.; Pijnenborg, R.; Vercruysse, L.; Romero, R. The “Great Obstetrical Syndromes” are associated with disorders of deep placentation. Am. J. Obstet. Gynecol. 2011, 204, 193–201. [Google Scholar] [CrossRef]
- Kim, Y.M.; Chaiworapongsa, T.; Gomez, R.; Bujold, E.; Yoon, B.H.; Rotmensch, S.; Thaler, H.T.; Romero, R. Failure of physiologic transformation of the spiral arteries in the placental bed in preterm premature rupture of membranes. Am. J. Obstet. Gynecol. 2002, 187, 1137–1142. [Google Scholar] [CrossRef]
- Romero, R.; Kusanovic, J.P.; Chaiworapongsa, T.; Hassan, S.S. Placental bed disorders in preterm labor, preterm PROM, spontaneous abortion and abruptio placentae. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Raymond, D.; Peterson, E. A critical review of early-onset and late-onset preeclampsia. Obstet. Gynecol. Surv. 2011, 66, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Staff, A.C. The two-stage placental model of preeclampsia: An update. J. Reprod. Immunol. 2019, 134-135, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Woods, A.W.; Jauniaux, E.; Kingdom, J.C. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta 2009, 30, 473–482. [Google Scholar] [CrossRef]
- Potter, J.M.; Nestel, P.J. The hyperlipidemia of pregnancy in normal and complicated pregnancies. Am. J. Obstet. Gynecol. 1979, 133, 165–170. [Google Scholar] [CrossRef]
- Belo, L.; Santos-Silva, A.; Quintanilha, A.; Rebelo, I. Similarities between pre-eclampsia and atherosclerosis: A protective effect of physical exercise? Curr. Med. Chem. 2008, 15, 2223–2229. [Google Scholar] [CrossRef]
- Labarrere, C.A. Acute atherosis. A histopathological hallmark of immune aggression? Placenta 1988, 9, 95–108. [Google Scholar] [CrossRef]
- Labarrere, C.A.; DiCarlo, H.L.; Bammerlin, E.; Hardin, J.W.; Kim, Y.M.; Chaemsaithong, P.; Haas, D.M.; Kassab, G.S.; Romero, R. Failure of physiologic transformation of spiral arteries, endothelial and trophoblast cell activation, and acute atherosis in the basal plate of the placenta. Am. J. Obstet. Gynecol. 2017, 216, 287.e1–287.e16. [Google Scholar] [CrossRef]
- Staff, A.C.; Dechend, R.; Pijnenborg, R. Learning from the placenta: Acute atherosis and vascular remodeling in preeclampsia-novel aspects for atherosclerosis and future cardiovascular health. Hypertension 2010, 56, 1026–1034. [Google Scholar] [CrossRef]
- Alnaes-Katjavivi, P.; Lyall, F.; Roald, B.; Redman, C.W.; Staff, A.C. Acute atherosis in vacuum suction biopsies of decidua basalis: An evidence based research definition. Placenta 2016, 37, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Chaemsaithong, P.; Romero, R.; Shaman, M.; Kim, C.J.; Kim, J.S.; Qureshi, F.; Jacques, S.M.; Ahmed, A.I.; Chaiworapongsa, T.; et al. The frequency of acute atherosis in normal pregnancy and preterm labor, preeclampsia, small-for-gestational age, fetal death and midtrimester spontaneous abortion. J. Matern. Fetal Neonatal. Med. 2015, 28, 2001–2009. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.U.; Al-Nasiry, S.; Bulten, J.; Spaanderman, M.E. Decidual vasculopathy and adverse perinatal outcome in preeclamptic pregnancy. Placenta 2012, 33, 630–633. [Google Scholar] [CrossRef]
- Harsem, N.K.; Roald, B.; Braekke, K.; Staff, A.C. Acute atherosis in decidual tissue: Not associated with systemic oxidative stress in preeclampsia. Placenta 2007, 28, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Staff, A.C.; Fjeldstad, H.E.; Fosheim, I.K.; Moe, K.; Turowski, G.; Johnsen, G.M.; Alnaes-Katjavivi, P.; Sugulle, M. Failure of physiological transformation and spiral artery atherosis: Their roles in preeclampsia. Am. J. Obstet. Gynecol. 2020. [Google Scholar] [CrossRef]
- Moreno-Eutimio, M.A.; Tovar-Rodríguez, J.M.; Vargas-Avila, K.; Nieto-Velázquez, N.G.; Frías-De-León, M.G.; Sierra-Martinez, M.; Acosta-Altamirano, G. Increased serum levels of inflammatory mediators and low frequency of regulatory T cells in the peripheral blood of preeclamptic Mexican women. Biomed. Res. Int. 2014, 2014, 413249. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Aranguren, L.C.; Prada, C.E.; Riaño-Medina, C.E.; Lopez, M. Endothelial dysfunction and preeclampsia: Role of oxidative stress. Front. Physiol. 2014, 5, 372. [Google Scholar] [CrossRef]
- Lockwood, C.J.; Yen, C.F.; Basar, M.; Kayisli, U.A.; Martel, M.; Buhimschi, I.; Buhimschi, C.; Huang, S.J.; Krikun, G.; Schatz, F. Preeclampsia-related inflammatory cytokines regulate interleukin-6 expression in human decidual cells. Am. J. Pathol. 2008, 172, 1571–1579. [Google Scholar] [CrossRef]
- Wallace, K.; Richards, S.; Dhillon, P.; Weimer, A.; Edholm, E.S.; Bengten, E.; Wilson, M.; Martin, J.N., Jr.; LaMarca, B. CD4+ T-helper cells stimulated in response to placental ischemia mediate hypertension during pregnancy. Hypertension 2011, 57, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, B.; Brewer, J.; Wallace, K. IL-6-induced pathophysiology during pre-eclampsia: Potential therapeutic role for magnesium sulfate? Int. J. Interferon. Cytokine Mediat. Res. 2011, 2011, 59–64. [Google Scholar] [CrossRef]
- Peixoto, A.B.; Araujo Júnior, E.; Ribeiro, J.U.; Rodrigues, D.B.; Castro, E.C.; Caldas, T.M.; Rodrigues Júnior, V. Evaluation of inflammatory mediators in the deciduas of pregnant women with pre-eclampsia/eclampsia. J. Matern. Fetal Neonatal. Med. 2016, 29, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, D.C.; Amaral, L.M.; Harmon, A.; Wallace, K.; Thomas, A.J.; Campbell, N.; Scott, J.; Herse, F.; Haase, N.; Moseley, J.; et al. An increased population of regulatory T cells improves the pathophysiology of placental ischemia in a rat model of preeclampsia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R884–R891. [Google Scholar] [CrossRef] [PubMed]
- Nath, M.C.; Cubro, H.; McCormick, D.J.; Milic, N.M.; Garovic, V.D. Preeclamptic Women Have Decreased Circulating IL-10 (Interleukin-10) Values at the Time of Preeclampsia Diagnosis: Systematic Review and Meta-Analysis. Hypertension 2020, 76, 1817–1827. [Google Scholar] [CrossRef]
- Shah, D.A.; Khalil, R.A. Bioactive factors in uteroplacental and systemic circulation link placental ischemia to generalized vascular dysfunction in hypertensive pregnancy and preeclampsia. Biochem. Pharmacol. 2015, 95, 211–226. [Google Scholar] [CrossRef]
- Reslan, O.M.; Khalil, R.A. Molecular and vascular targets in the pathogenesis and management of the hypertension associated with preeclampsia. Cardiovasc. Hematol. Agents Med. Chem. 2010, 8, 204–226. [Google Scholar] [CrossRef]
- Bainbridge, S.A.; Belkacemi, L.; Dickinson, M.; Graham, C.H.; Smith, G.N. Carbon monoxide inhibits hypoxia/reoxygenation-induced apoptosis and secondary necrosis in syncytiotrophoblast. Am. J. Pathol. 2006, 169, 774–783. [Google Scholar] [CrossRef][Green Version]
- Chambers, J.C.; Fusi, L.; Malik, I.S.; Haskard, D.O.; De Swiet, M.; Kooner, J.S. Association of maternal endothelial dysfunction with preeclampsia. JAMA 2001, 285, 1607–1612. [Google Scholar] [CrossRef]
- Sutton, E.F.; Gemmel, M.; Powers, R.W. Nitric oxide signaling in pregnancy and preeclampsia. Nitric Oxide 2020, 95, 55–62. [Google Scholar] [CrossRef]
- Guerby, P.; Tasta, O.; Swiader, A.; Pont, F.; Bujold, E.; Parant, O.; Vayssiere, C.; Salvayre, R.; Negre-Salvayre, A. Role of oxidative stress in the dysfunction of the placental endothelial nitric oxide synthase in preeclampsia. Redox Biol. 2021, 40, 101861. [Google Scholar] [CrossRef] [PubMed]
- Chassagne, C.; Eddahibi, S.; Adamy, C.; Rideau, D.; Marotte, F.; Dubois-Randé, J.L.; Adnot, S.; Samuel, J.L.; Teiger, E. Modulation of angiotensin II receptor expression during development and regression of hypoxic pulmonary hypertension. Am. J. Respir. Cell. Mol. Biol. 2000, 22, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Anton, L.; Merrill, D.C.; Neves, L.A.; Diz, D.I.; Corthorn, J.; Valdes, G.; Stovall, K.; Gallagher, P.E.; Moorefield, C.; Gruver, C.; et al. The uterine placental bed Renin-Angiotensin system in normal and preeclamptic pregnancy. Endocrinology 2009, 150, 4316–4325. [Google Scholar] [CrossRef]
- Quitterer, U.; Fu, X.; Pohl, A.; Bayoumy, K.M.; Langer, A.; AbdAlla, S. Beta-Arrestin1 Prevents Preeclampsia by Downregulation of Mechanosensitive AT1-B2 Receptor Heteromers. Cell 2019, 176, 318–333.e19. [Google Scholar] [CrossRef]
- Zanaty, M.; Seara, F.A.C.; Nakagawa, P.; Deng, G.; Mathieu, N.M.; Balapattabi, K.; Karnik, S.S.; Grobe, J.L.; Sigmund, C.D. β-Arrestin-Biased Agonist Targeting the Brain AT(1)R (Angiotensin II Type 1 Receptor) Increases Aversion to Saline and Lowers Blood Pressure in Deoxycorticosterone Acetate-Salt Hypertension. Hypertension 2021, 77, 420–431. [Google Scholar] [CrossRef]
- Siddiqui, A.H.; Irani, R.A.; Zhang, W.; Wang, W.; Blackwell, S.C.; Kellems, R.E.; Xia, Y. Angiotensin receptor agonistic autoantibody-mediated soluble fms-like tyrosine kinase-1 induction contributes to impaired adrenal vasculature and decreased aldosterone production in preeclampsia. Hypertension 2013, 61, 472–479. [Google Scholar] [CrossRef]
- George, E.M.; Granger, J.P. Endothelin: Key mediator of hypertension in preeclampsia. Am. J. Hypertens. 2011, 24, 964–969. [Google Scholar] [CrossRef]
- Novotny, S.R.; Wallace, K.; Heath, J.; Moseley, J.; Dhillon, P.; Weimer, A.; Wallukat, G.; Herse, F.; Wenzel, K.; Martin, J.N., Jr.; et al. Activating autoantibodies to the angiotensin II type I receptor play an important role in mediating hypertension in response to adoptive transfer of CD4+ T lymphocytes from placental ischemic rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R1197–R1201. [Google Scholar] [CrossRef]
- Hubel, C.A.; Wallukat, G.; Wolf, M.; Herse, F.; Rajakumar, A.; Roberts, J.M.; Markovic, N.; Thadhani, R.; Luft, F.C.; Dechend, R. Agonistic angiotensin II type 1 receptor autoantibodies in postpartum women with a history of preeclampsia. Hypertension 2007, 49, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Rieber-Mohn, A.B.; Sugulle, M.; Wallukat, G.; Alnæs-Katjavivi, P.; Leite Størvold, G.; Bolstad, N.; Redman, C.W.; Dechend, R.; Staff, A.C. Auto-antibodies against the angiotensin II type I receptor in women with uteroplacental acute atherosis and preeclampsia at delivery and several years postpartum. J. Reprod. Immunol. 2018, 128, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Bdolah, Y.; Sukhatme, V.P.; Karumanchi, S.A. Angiogenic imbalance in the pathophysiology of preeclampsia: Newer insights. Semin. Nephrol. 2004, 24, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Boeldt, D.S.; Bird, I.M. Vascular adaptation in pregnancy and endothelial dysfunction in preeclampsia. J. Endocrinol. 2017, 232, R27–R44. [Google Scholar] [CrossRef] [PubMed]
- Charnock-Jones, D.S.; Sharkey, A.M.; Boocock, C.A.; Ahmed, A.; Plevin, R.; Ferrara, N.; Smith, S.K. Vascular endothelial growth factor receptor localization and activation in human trophoblast and choriocarcinoma cells. Biol. Reprod. 1994, 51, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Athanassiades, A.; Hamilton, G.S.; Lala, P.K. Vascular endothelial growth factor stimulates proliferation but not migration or invasiveness in human extravillous trophoblast. Biol. Reprod. 1998, 59, 643–654. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.S.; Babcock, S.A.; Granger, J.P. Hypertension produced by reduced uterine perfusion in pregnant rats is associated with increased soluble fms-like tyrosine kinase-1 expression. Hypertension 2007, 50, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Albonici, L.; Giganti, M.G.; Modesti, A.; Manzari, V.; Bei, R. Multifaceted Role of the Placental Growth Factor (PlGF) in the Antitumor Immune Response and Cancer Progression. Int. J. Mol. Sci. 2019, 20, 2970. [Google Scholar] [CrossRef] [PubMed]
- March, M.I.; Geahchan, C.; Wenger, J.; Raghuraman, N.; Berg, A.; Haddow, H.; McKeon, B.A.; Narcisse, R.; David, J.L.; Scott, J.; et al. Circulating Angiogenic Factors and the Risk of Adverse Outcomes among Haitian Women with Preeclampsia. PLoS ONE 2015, 10, e0126815. [Google Scholar] [CrossRef]
- National Institute for Health and Care Excellence: Clinical Guidelines. Hypertension in Pregnancy: Diagnosis and Management; National Institute for Health and Care Excellence: London, UK, 2019. [Google Scholar]
- Wulff, C.; Wilson, H.; Dickson, S.E.; Wiegand, S.J.; Fraser, H.M. Hemochorial placentation in the primate: Expression of vascular endothelial growth factor, angiopoietins, and their receptors throughout pregnancy. Biol. Reprod. 2002, 66, 802–812. [Google Scholar] [CrossRef][Green Version]
- Geva, E.; Ginzinger, D.G.; Zaloudek, C.J.; Moore, D.H.; Byrne, A.; Jaffe, R.B. Human placental vascular development: Vasculogenic and angiogenic (branching and nonbranching) transformation is regulated by vascular endothelial growth factor-A, angiopoietin-1, and angiopoietin-2. J. Clin. Endocrinol. Metab. 2002, 87, 4213–4224. [Google Scholar] [CrossRef]
- Kendall, R.L.; Thomas, K.A. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 10705–10709. [Google Scholar] [CrossRef]
- Sircar, M.; Thadhani, R.; Karumanchi, S.A. Pathogenesis of preeclampsia. Curr. Opin. Nephrol. Hypertens. 2015, 24, 131–138. [Google Scholar] [CrossRef]
- Verlohren, S.; Herraiz, I.; Lapaire, O.; Schlembach, D.; Zeisler, H.; Calda, P.; Sabria, J.; Markfeld-Erol, F.; Galindo, A.; Schoofs, K.; et al. New gestational phase-specific cutoff values for the use of the soluble fms-like tyrosine kinase-1/placental growth factor ratio as a diagnostic test for preeclampsia. Hypertension 2014, 63, 346–352. [Google Scholar] [CrossRef]
- Lim, S.; Li, W.; Kemper, J.; Nguyen, A.; Mol, B.W.; Reddy, M. Biomarkers and the Prediction of Adverse Outcomes in Preeclampsia: A Systematic Review and Meta-analysis. Obstet. Gynecol. 2021, 137, 72–81. [Google Scholar] [CrossRef]
- Cheifetz, S.; Bellón, T.; Calés, C.; Vera, S.; Bernabeu, C.; Massagué, J.; Letarte, M. Endoglin is a component of the transforming growth factor-beta receptor system in human endothelial cells. J. Biol. Chem. 1992, 267, 19027–19030. [Google Scholar] [CrossRef]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; ten Dijke, P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Sorenson, C.M.; Sheibani, N. PECAM-1 isoforms, eNOS and endoglin axis in regulation of angiogenesis. Clin. Sci. 2015, 129, 217–234. [Google Scholar] [CrossRef]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.H.; Yuan, H.T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Rabinovici, J.; Goldsmith, P.C.; Librach, C.L.; Jaffe, R.B. Localization and regulation of the activin-A dimer in human placental cells. J. Clin. Endocrinol. Metab. 1992, 75, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Muttukrishna, S.; Knight, P.G.; Groome, N.P.; Redman, C.W.; Ledger, W.L. Activin A and inhibin A as possible endocrine markers for pre-eclampsia. Lancet 1997, 349, 1285–1288. [Google Scholar] [CrossRef]
- Manuelpillai, U.; Schneider-Kolsky, M.; Dole, A.; Wallace, E.M. Activin A and activin receptors in gestational tissue from preeclamptic pregnancies. J. Endocrinol. 2001, 171, 57–64. [Google Scholar] [CrossRef]
- Muttukrishna, S.; North, R.A.; Morris, J.; Schellenberg, J.C.; Taylor, R.S.; Asselin, J.; Ledger, W.; Groome, N.; Redman, C.W. Serum inhibin A and activin A are elevated prior to the onset of pre-eclampsia. Hum. Reprod. 2000, 15, 1640–1645. [Google Scholar] [CrossRef]
- Lim, R.; Acharya, R.; Delpachitra, P.; Hobson, S.; Sobey, C.G.; Drummond, G.R.; Wallace, E.M. Activin and NADPH-oxidase in preeclampsia: Insights from in vitro and murine studies. Am. J. Obstet. Gynecol. 2015, 212, 86.e1–86.e12. [Google Scholar] [CrossRef]
- Hobson, S.R.; Lim, R.; Mockler, J.C.; Gurusinghe, S.; Wallace, E.M. Role of Activin A in the Pathogenesis of Endothelial Cell Dysfunction in Preeclampsia. Methods Mol. Biol. 2018, 1710, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Shahul, S.; Ramadan, H.; Nizamuddin, J.; Mueller, A.; Patel, V.; Dreixler, J.; Tung, A.; Lang, R.M.; Weinert, L.; Nasim, R.; et al. Activin A and Late Postpartum Cardiac Dysfunction Among Women With Hypertensive Disorders of Pregnancy. Hypertension 2018, 72, 188–193. [Google Scholar] [CrossRef] [PubMed]
- de Martelly, V.A.; Dreixler, J.; Tung, A.; Mueller, A.; Heimberger, S.; Fazal, A.A.; Naseem, H.; Lang, R.; Kruse, E.; Yamat, M.; et al. Long-Term Postpartum Cardiac Function and Its Association With Preeclampsia. J. Am. Heart Assoc. 2021, 10, e018526. [Google Scholar] [CrossRef] [PubMed]
- Daikoku, T.; Matsumoto, H.; Gupta, R.A.; Das, S.K.; Gassmann, M.; DuBois, R.N.; Dey, S.K. Expression of hypoxia-inducible factors in the peri-implantation mouse uterus is regulated in a cell-specific and ovarian steroid hormone-dependent manner. Evidence for differential function of HIFs during early pregnancy. J. Biol. Chem. 2003, 278, 7683–7691. [Google Scholar] [CrossRef] [PubMed]
- Akhilesh, M.; Mahalingam, V.; Nalliah, S.; Ali, R.M.; Ganesalingam, M.; Haleagrahara, N. Hypoxia-inducible factor-1α as a predictive marker in pre-eclampsia. Biomed. Rep. 2013, 1, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Tal, R. The role of hypoxia and hypoxia-inducible factor-1alpha in preeclampsia pathogenesis. Biol. Reprod. 2012, 87, 134. [Google Scholar] [CrossRef]
- Iriyama, T.; Wang, W.; Parchim, N.F.; Song, A.; Blackwell, S.C.; Sibai, B.M.; Kellems, R.E.; Xia, Y. Hypoxia-independent upregulation of placental hypoxia inducible factor-1α gene expression contributes to the pathogenesis of preeclampsia. Hypertension 2015, 65, 1307–1315. [Google Scholar] [CrossRef]
- Tianthong, W.; Phupong, V. Serum hypoxia-inducible factor-1α and uterine artery Doppler ultrasound during the first trimester for prediction of preeclampsia. Sci. Rep. 2021, 11, 6674. [Google Scholar] [CrossRef] [PubMed]
- Krek, A.; Grün, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ge, Q.; Guo, L.; Lu, Z. Maternal plasma miRNAs expression in preeclamptic pregnancies. Biomed. Res. Int. 2013, 2013, 970265. [Google Scholar] [CrossRef] [PubMed]
- Pineles, B.L.; Romero, R.; Montenegro, D.; Tarca, A.L.; Han, Y.M.; Kim, Y.M.; Draghici, S.; Espinoza, J.; Kusanovic, J.P.; Mittal, P.; et al. Distinct subsets of microRNAs are expressed differentially in the human placentas of patients with preeclampsia. Am J. Obstet. Gynecol. 2007, 196, 261.e1–261.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fei, M.; Xue, G.; Zhou, Q.; Jia, Y.; Li, L.; Xin, H.; Sun, S. Elevated levels of hypoxia-inducible microRNA-210 in pre-eclampsia: New insights into molecular mechanisms for the disease. J. Cell. Mol. Med. 2012, 16, 249–259. [Google Scholar] [CrossRef]
- Bounds, K.R.; Chiasson, V.L.; Pan, L.J.; Gupta, S.; Chatterjee, P. MicroRNAs: New Players in the Pathobiology of Preeclampsia. Front. Cardiovasc. Med. 2017, 4, 60. [Google Scholar] [CrossRef]
- Lázár, L.; Rigó, J. PP088. The role of microRNA in pathogenesis of preeclampsia-miRNA network analysis. Pregnancy Hypertens. 2013, 3, 99. [Google Scholar] [CrossRef]
- Zhang, Y.; Diao, Z.; Su, L.; Sun, H.; Li, R.; Cui, H.; Hu, Y. MicroRNA-155 contributes to preeclampsia by down-regulating CYR61. Am. J. Obstet. Gynecol. 2010, 202, 466.e1–466.e7. [Google Scholar] [CrossRef]
- Gellhaus, A.; Schmidt, M.; Dunk, C.; Lye, S.J.; Kimmig, R.; Winterhager, E. Decreased expression of the angiogenic regulators CYR61 (CCN1) and NOV (CCN3) in human placenta is associated with pre-eclampsia. Mol. Hum. Reprod. 2006, 12, 389–399. [Google Scholar] [CrossRef]
- He, J.; Jing, Y.; Li, W.; Qian, X.; Xu, Q.; Li, F.S.; Liu, L.Z.; Jiang, B.H.; Jiang, Y. Roles and mechanism of miR-199a and miR-125b in tumor angiogenesis. PLoS ONE 2013, 8, e56647. [Google Scholar] [CrossRef]
- Licini, C.; Avellini, C.; Picchiassi, E.; Mensà, E.; Fantone, S.; Ramini, D.; Tersigni, C.; Tossetta, G.; Castellucci, C.; Tarquini, F.; et al. Pre-eclampsia predictive ability of maternal miR-125b: A clinical and experimental study. Transl. Res. 2021, 228, 13–27. [Google Scholar] [CrossRef]
- Gao, F.; Bian, F.; Ma, X.; Kalinichenko, V.V.; Das, S.K. Control of regional decidualization in implantation: Role of FoxM1 downstream of Hoxa10 and cyclin D3. Sci. Rep. 2015, 5, 13863. [Google Scholar] [CrossRef]
- Zhou, F.; Sun, Y.; Gao, Q.; Wang, H. microRNA-21 regulates the proliferation of placental cells via FOXM1 in preeclampsia. Exp. Ther. Med. 2020, 20, 1871–1878. [Google Scholar] [CrossRef]
- Wu, P.; Haththotuwa, R.; Kwok, C.S.; Babu, A.; Kotronias, R.A.; Rushton, C.; Zaman, A.; Fryer, A.A.; Kadam, U.; Chew-Graham, C.A.; et al. Preeclampsia and Future Cardiovascular Health: A Systematic Review and Meta-Analysis. Circ. Cardiovasc. Qual. Outcomes 2017, 10, e003497. [Google Scholar] [CrossRef]
- Brown, M.C.; Best, K.E.; Pearce, M.S.; Waugh, J.; Robson, S.C.; Bell, R. Cardiovascular disease risk in women with pre-eclampsia: Systematic review and meta-analysis. Eur. J. Epidemiol. 2013, 28, 1–19. [Google Scholar] [CrossRef]
- Lo, C.C.W.; Lo, A.C.Q.; Leow, S.H.; Fisher, G.; Corker, B.; Batho, O.; Morris, B.; Chowaniec, M.; Vladutiu, C.J.; Fraser, A.; et al. Future Cardiovascular Disease Risk for Women With Gestational Hypertension: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2020, 9, e013991. [Google Scholar] [CrossRef]
- Okoth, K.; Chandan, J.S.; Marshall, T.; Thangaratinam, S.; Thomas, G.N.; Nirantharakumar, K.; Adderley, N.J. Association between the reproductive health of young women and cardiovascular disease in later life: Umbrella review. BMJ 2020, 371, m3502. [Google Scholar] [CrossRef]
- McDonald, S.D.; Malinowski, A.; Zhou, Q.; Yusuf, S.; Devereaux, P.J. Cardiovascular sequelae of preeclampsia/eclampsia: A systematic review and meta-analyses. Am. Heart J. 2008, 156, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Jaffe, I.Z.; Karumanchi, S.A. Pre-eclampsia and cardiovascular disease. Cardiovasc. Res. 2014, 101, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Hypertension in Pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet. Gynecol. 2013, 122, 1122–1131. [Google Scholar] [CrossRef]
- Wallis, A.B.; Saftlas, A.F.; Hsia, J.; Atrash, H.K. Secular Trends in the Rates of Preeclampsia, Eclampsia, and Gestational Hypertension, United States, 1987–2004. Am. J. Hypertens. 2008, 21, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Leaños-Miranda, A.; Méndez-Aguilar, F.; Ramírez-Valenzuela, K.L.; Serrano-Rodríguez, M.; Berumen-Lechuga, G.; Molina-Pérez, C.J.; Isordia-Salas, I.; Campos-Galicia, I. Circulating angiogenic factors are related to the severity of gestational hypertension and preeclampsia, and their adverse outcomes. Medicine 2017, 96, e6005. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, C.; McCullough, L.D.; Awad, I.A.; Chireau, M.V.; Fedder, W.N.; Furie, K.L.; Howard, V.J.; Lichtman, J.H.; Lisabeth, L.D.; Piña, I.L.; et al. Guidelines for the Prevention of Stroke in Women. Stroke 2014, 45, 1545–1588. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.B.; Bushnell, C.D.; Adams, R.J.; Appel, L.J.; Braun, L.T.; Chaturvedi, S.; Creager, M.A.; Culebras, A.; Eckel, R.H.; Hart, R.G.; et al. Guidelines for the primary prevention of stroke: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2011, 42, 517–584. [Google Scholar] [CrossRef] [PubMed]
- Haug, E.B.; Horn, J.; Markovitz, A.R.; Fraser, A.; Vatten, L.J.; Macdonald-Wallis, C.; Tilling, K.; Romundstad, P.R.; Rich-Edwards, J.W.; Åsvold, B.O. Life Course Trajectories of Cardiovascular Risk Factors in Women With and Without Hypertensive Disorders in First Pregnancy: The HUNT Study in Norway. J. Am. Heart Assoc. 2018, 7, e009250. [Google Scholar] [CrossRef]
- Egeland, G.M.; Klungsøyr, K.; Øyen, N.; Tell, G.S.; Næss, Ø.; Skjærven, R. Preconception cardiovascular risk factor differences between gestational hypertension and preeclampsia: Cohort Norway Study. Hypertension 2016, 67, 1173–1180. [Google Scholar] [CrossRef]
- Berends, A.L.; de Groot, C.J.; Sijbrands, E.J.; Sie, M.P.; Benneheij, S.H.; Pal, R.; Heydanus, R.; Oostra, B.A.; van Duijn, C.M.; Steegers, E.A. Shared constitutional risks for maternal vascular-related pregnancy complications and future cardiovascular disease. Hypertension 2008, 51, 1034–1041. [Google Scholar] [CrossRef]
- Craici, I.; Wagner, S.; Garovic, V.D. Preeclampsia and future cardiovascular risk: Formal risk factor or failed stress test? Ther. Adv. Cardiovasc. Dis. 2008, 2, 249–259. [Google Scholar] [CrossRef]
- Rodie, V.A.; Freeman, D.J.; Sattar, N.; Greer, I.A. Pre-eclampsia and cardiovascular disease: Metabolic syndrome of pregnancy? Atherosclerosis 2004, 175, 189–202. [Google Scholar] [CrossRef]
- Staff, A.C.; Redman, C.W.G. IFPA Award in Placentology Lecture: Preeclampsia, the decidual battleground and future maternal cardiovascular disease. Placenta 2014, 35, S26–S31. [Google Scholar] [CrossRef]
- Melchiorre, K.; Thilaganathan, B.; Giorgione, V.; Ridder, A.; Memmo, A.; Khalil, A. Hypertensive Disorders of Pregnancy and Future Cardiovascular Health. Front. Cardiovasc. Med. 2020, 7, 59. [Google Scholar] [CrossRef]
- Barr, L.C.; Liblik, K.; Johri, A.M.; Smith, G.N. Maternal Cardiovascular Function following a Pregnancy Complicated by Preeclampsia. Am. J. Perinatol. 2020. [Google Scholar] [CrossRef]
- Wu, R.; Wang, T.; Gu, R.; Xing, D.; Ye, C.; Chen, Y.; Liu, X.; Chen, L. Hypertensive Disorders of Pregnancy and Risk of Cardiovascular Disease-Related Morbidity and Mortality: A Systematic Review and Meta-Analysis. Cardiology 2020, 145, 633–647. [Google Scholar] [CrossRef]
- Behrens, I.; Basit, S.; Melbye, M.; Lykke, J.A.; Wohlfahrt, J.; Bundgaard, H.; Thilaganathan, B.; Boyd, H.A. Risk of post-pregnancy hypertension in women with a history of hypertensive disorders of pregnancy: Nationwide cohort study. BMJ 2017, 358, j3078. [Google Scholar] [CrossRef] [PubMed]
- Grandi, S.M.; Filion, K.B.; Yoon, S.; Ayele, H.T.; Doyle, C.M.; Hutcheon, J.A.; Smith, G.N.; Gore, G.C.; Ray, J.G.; Nerenberg, K.; et al. Cardiovascular Disease-Related Morbidity and Mortality in Women With a History of Pregnancy Complications. Circulation 2019, 139, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Heida, K.Y.; Franx, A.; van Rijn, B.B.; Eijkemans, M.J.; Boer, J.M.; Verschuren, M.W.; Oudijk, M.A.; Bots, M.L.; van der Schouw, Y.T. Earlier Age of Onset of Chronic Hypertension and Type 2 Diabetes Mellitus After a Hypertensive Disorder of Pregnancy or Gestational Diabetes Mellitus. Hypertension 2015, 66, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Haas, D.M.; Parker, C.B.; Marsh, D.J.; Grobman, W.A.; Ehrenthal, D.B.; Greenland, P.; Bairey Merz, C.N.; Pemberton, V.L.; Silver, R.M.; Barnes, S.; et al. Association of Adverse Pregnancy Outcomes With Hypertension 2 to 7 Years Postpartum. J. Am. Heart Assoc. 2019, 8, e013092. [Google Scholar] [CrossRef]
- Foo, L.; Tay, J.; Lees, C.C.; McEniery, C.M.; Wilkinson, I.B. Hypertension in pregnancy: Natural history and treatment options. Curr. Hypertens. Rep. 2015, 17, 36. [Google Scholar] [CrossRef]
- Brouwers, L.; van der Meiden-van Roest, A.J.; Savelkoul, C.; Vogelvang, T.E.; Lely, A.T.; Franx, A.; van Rijn, B.B. Recurrence of pre-eclampsia and the risk of future hypertension and cardiovascular disease: A systematic review and meta-analysis. BJOG 2018, 125, 1642–1654. [Google Scholar] [CrossRef]
- Melchiorre, K.; Sutherland, G.R.; Liberati, M.; Thilaganathan, B. Preeclampsia is associated with persistent postpartum cardiovascular impairment. Hypertension 2011, 58, 709–715. [Google Scholar] [CrossRef]
- Kirollos, S.; Skilton, M.; Patel, S.; Arnott, C. A Systematic Review of Vascular Structure and Function in Pre-eclampsia: Non-invasive Assessment and Mechanistic Links. Front. Cardiovasc. Med. 2019, 6, 166. [Google Scholar] [CrossRef]
- Yuan, L.J.; Xue, D.; Duan, Y.Y.; Cao, T.S.; Yang, H.G.; Zhou, N. Carotid arterial intima–media thickness and arterial stiffness in pre-eclampsia: Analysis with a radiofrequency ultrasound technique. Ultrasound Obstet. Gynecol. 2013, 42, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Stergiotou, I.; Crispi, F.; Valenzuela-Alcaraz, B.; Bijnens, B.; Gratacos, E. Patterns of maternal vascular remodeling and responsiveness in early- versus late-onset preeclampsia. Am. J. Obstet. Gynecol. 2013, 209, 558.e1–558.e14. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Watanabe, K.; Iwasaki, A.; Kimura, C.; Matsushita, H.; Shinohara, K.; Wakatsuki, A. Differences in vascular reactivity between pregnant women with chronic hypertension and preeclampsia. Hypertens. Res. 2014, 37, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Zoet, G.A.; Meun, C.; Benschop, L.; Boersma, E.; Budde, R.P.J.; Fauser, B.; de Groot, C.J.M.; van der Lugt, A.; Maas, A.; Moons, K.G.M.; et al. Cardiovascular RiskprofilE—IMaging and gender-specific disOrders (CREw-IMAGO): Rationale and design of a multicenter cohort study. BMC Womens Health 2017, 17, 60. [Google Scholar] [CrossRef] [PubMed]
- White, W.M.; Mielke, M.M.; Araoz, P.A.; Lahr, B.D.; Bailey, K.R.; Jayachandran, M.; Miller, V.M.; Garovic, V.D. A history of preeclampsia is associated with a risk for coronary artery calcification 3 decades later. Am. J. Obstet. Gynecol. 2016, 214, 519. [Google Scholar] [CrossRef] [PubMed]
- Zoet, G.A.; Benschop, L.; Boersma, E.; Budde, R.P.J.; Fauser, B.; van der Graaf, Y.; de Groot, C.J.M.; Maas, A.; Roeters van Lennep, J.E.; Steegers, E.A.P.; et al. Prevalence of Subclinical Coronary Artery Disease Assessed by Coronary Computed Tomography Angiography in 45- to 55-Year-Old Women With a History of Preeclampsia. Circulation 2018, 137, 877–879. [Google Scholar] [CrossRef] [PubMed]
- Benschop, L.; Brouwers, L.; Zoet, G.A.; Meun, C.; Boersma, E.; Budde, R.P.J.; Fauser, B.; de Groot, C.M.J.; van der Schouw, Y.T.; Maas, A.; et al. Early Onset of Coronary Artery Calcification in Women With Previous Preeclampsia. Circ. Cardiovasc. Imaging 2020, 13, e010340. [Google Scholar] [CrossRef]
- Toth, P.P. Subclinical atherosclerosis: What it is, what it means and what we can do about it. Int. J. Clin. Pract. 2008, 62, 1246–1254. [Google Scholar] [CrossRef]
- Benschop, L.; Duvekot, J.J.; Roeters van Lennep, J.E. Future risk of cardiovascular disease risk factors and events in women after a hypertensive disorder of pregnancy. Heart 2019, 105, 1273–1278. [Google Scholar] [CrossRef]
- Riise, H.K.R.; Sulo, G.; Tell, G.S.; Igland, J.; Egeland, G.; Nygard, O.; Selmer, R.; Iversen, A.C.; Daltveit, A.K. Hypertensive pregnancy disorders increase the risk of maternal cardiovascular disease after adjustment for cardiovascular risk factors. Int. J. Cardiol. 2019, 282, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Haug, E.B.; Horn, J.; Markovitz, A.R.; Fraser, A.; Klykken, B.; Dalen, H.; Vatten, L.J.; Romundstad, P.R.; Rich-Edwards, J.W.; Åsvold, B.O. Association of Conventional Cardiovascular Risk Factors With Cardiovascular Disease After Hypertensive Disorders of Pregnancy: Analysis of the Nord-Trøndelag Health Study. JAMA Cardiol. 2019, 4, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Behrens, I.; Basit, S.; Lykke, J.A.; Ranthe, M.F.; Wohlfahrt, J.; Bundgaard, H.; Melbye, M.; Boyd, H.A. Association Between Hypertensive Disorders of Pregnancy and Later Risk of Cardiomyopathy. JAMA 2016, 315, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.J.; Tanz, L.J.; Missmer, S.A.; Rimm, E.B.; Spiegelman, D.; James-Todd, T.M.; Rich-Edwards, J.W. Hypertensive Disorders of Pregnancy and Maternal Cardiovascular Disease Risk Factor Development: An Observational Cohort Study. Ann. Intern. Med. 2018, 169, 224–232. [Google Scholar] [CrossRef]
- Auger, N.; Fraser, W.D.; Schnitzer, M.; Leduc, L.; Healy-Profitós, J.; Paradis, G. Recurrent pre-eclampsia and subsequent cardiovascular risk. Heart 2017, 103, 235–243. [Google Scholar] [CrossRef]
- Tooher, J.; Thornton, C.; Makris, A.; Ogle, R.; Korda, A.; Horvath, J.; Hennessy, A. Hypertension in pregnancy and long-term cardiovascular mortality: A retrospective cohort study. Am. J. Obstet. Gynecol. 2016, 214, 722.e1–722.e6. [Google Scholar] [CrossRef]
- Hiersch, L.; Ray, J.G.; Barrett, J.; Berger, H.; Geary, M.; McDonald, S.D.; Diong, C.; Gandhi, S.; Guan, J.; Murray-Davis, B.; et al. Maternal cardiovascular disease after twin pregnancies complicated by hypertensive disorders of pregnancy: A population-based cohort study. Cmaj 2021, 193, E1448–E1458. [Google Scholar] [CrossRef]
- McDonald, S.D.; Han, Z.; Walsh, M.W.; Gerstein, H.C.; Devereaux, P.J. Kidney disease after preeclampsia: A systematic review and meta-analysis. Am. J. Kidney Dis. 2010, 55, 1026–1039. [Google Scholar] [CrossRef]
- Riise, H.K.R.; Sulo, G.; Tell, G.S.; Igland, J.; Nygård, O.; Iversen, A.C.; Daltveit, A.K. Association Between Gestational Hypertension and Risk of Cardiovascular Disease Among 617,589 Norwegian Women. J. Am. Heart Assoc. 2018, 7, e008337. [Google Scholar] [CrossRef]
- ACOG Committee Opinion No. 736: Optimizing Postpartum Care. Obstet. Gynecol. 2018, 131, e140–e150. [CrossRef]
- Khosla, K.; Heimberger, S.; Nieman, K.M.; Tung, A.; Shahul, S.; Staff, A.C.; Rana, S. Long-Term Cardiovascular Disease Risk in Women After Hypertensive Disorders of Pregnancy: Recent Advances in Hypertension. Hypertension 2021, 78, 927–935. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, K.; Kim, S.H.; Cha, D.H.; Park, H.J. Defective Uteroplacental Vascular Remodeling in Preeclampsia: Key Molecular Factors Leading to Long Term Cardiovascular Disease. Int. J. Mol. Sci. 2021, 22, 11202. https://doi.org/10.3390/ijms222011202
Hong K, Kim SH, Cha DH, Park HJ. Defective Uteroplacental Vascular Remodeling in Preeclampsia: Key Molecular Factors Leading to Long Term Cardiovascular Disease. International Journal of Molecular Sciences. 2021; 22(20):11202. https://doi.org/10.3390/ijms222011202
Chicago/Turabian StyleHong, Kirim, Soo Hyun Kim, Dong Hyun Cha, and Hee Jin Park. 2021. "Defective Uteroplacental Vascular Remodeling in Preeclampsia: Key Molecular Factors Leading to Long Term Cardiovascular Disease" International Journal of Molecular Sciences 22, no. 20: 11202. https://doi.org/10.3390/ijms222011202
APA StyleHong, K., Kim, S. H., Cha, D. H., & Park, H. J. (2021). Defective Uteroplacental Vascular Remodeling in Preeclampsia: Key Molecular Factors Leading to Long Term Cardiovascular Disease. International Journal of Molecular Sciences, 22(20), 11202. https://doi.org/10.3390/ijms222011202