
Sulforaphane Causes Cell Cycle Arrest and Apoptosis in Human Glioblastoma U87MG and U373MG Cell Lines under Hypoxic Conditions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. The Effects of SFN on Cell Viability
2.2. The Effects of SFN on Apoptotic Cell Death
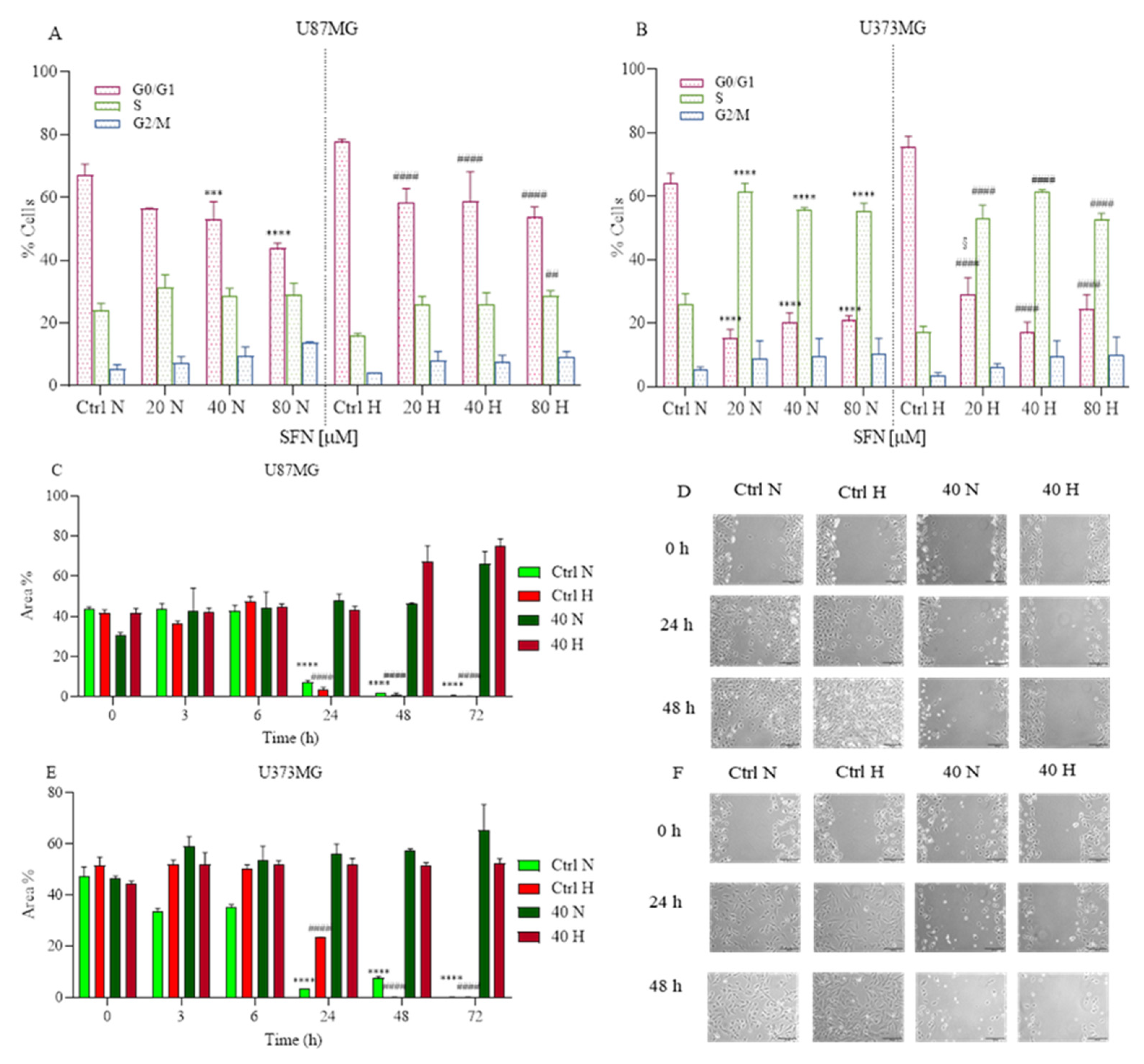
2.3. The Effects of SFN on Cell Cycle and Cell Motility
2.4. The Effects of SFN on Mitochondrial and Oxidative Status
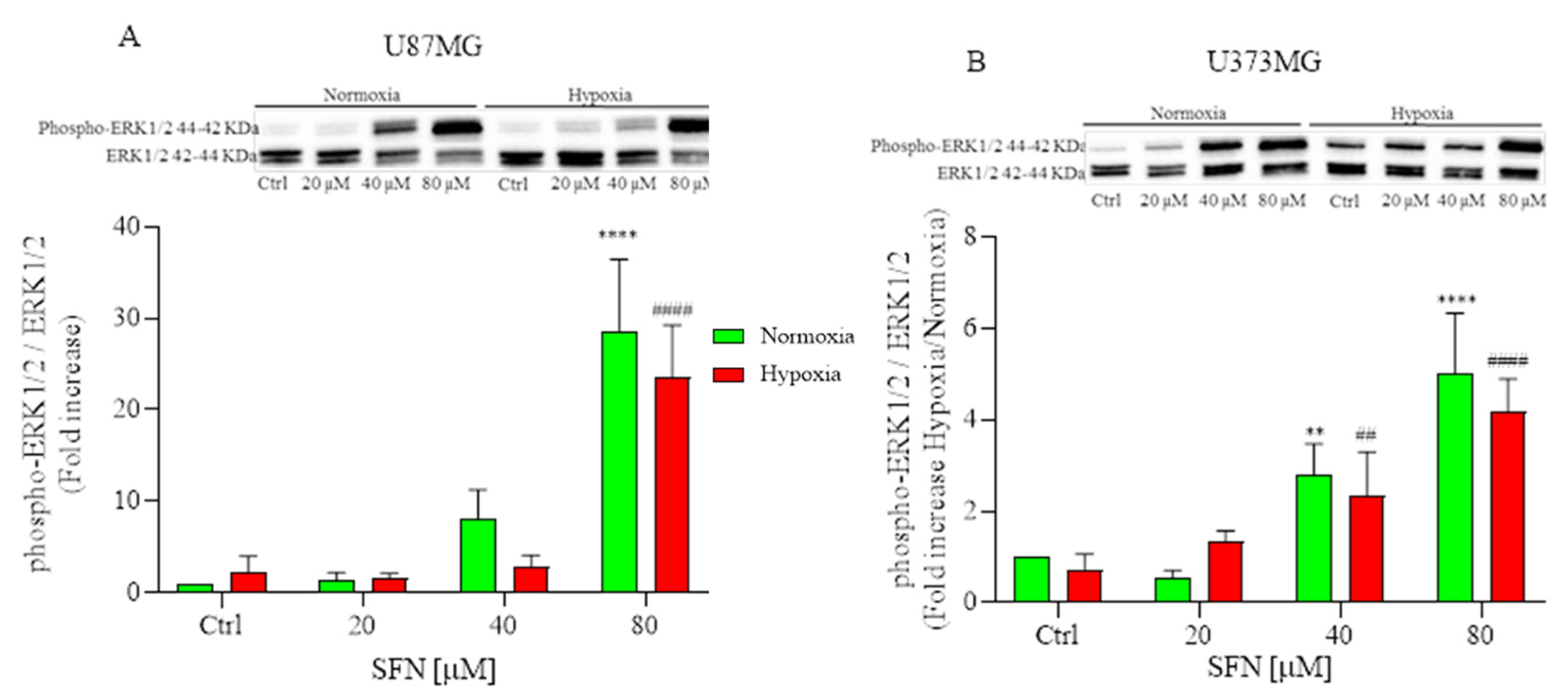
2.5. The Effect of SFN on MAPK Signaling Pathway
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Determination of Cell Viability
4.3. Determination of Apoptotic Cell Death
4.4. Analysis of Cell Cycle
4.5. Determination of Cell Motility
4.6. Determination of GSH Levels
4.7. Determination of Phospho-ERK1/2 and Cytochrome c
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McKinnon, C.; Nandhabalan, M.; Murray, S.A.; Plaha, P. Glioblastoma: Clinical presentation, diagnosis, and management. BMJ 2021, 374, n1560. [Google Scholar] [CrossRef]
- Wen, P.; Weller, M.; Lee, E.; Alexander, B.; Barnholtz-Sloan, J.; Barthel, F.; Batchelor, T.; Bindra, R.; Chang, S.; Chiocca, E.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro-Oncology 2020, 22, 1073–1113. [Google Scholar] [CrossRef]
- Ostrom, Q.; Fahmideh, M.A.; Cote, D.; Muskens, I.; Schraw, J.; Scheurer, M. ML Bondy Risk factors for childhood and adult primary brain tumors. Neuro-Oncology 2019, 21, 1357–1375. [Google Scholar] [CrossRef]
- Hutóczki, G.; Virga, J.; Birkó, Z.; Klekner, A. Novel Concepts of Glioblastoma Therapy Concerning Its Heterogeneity. Int. J. Mol. Sci. 2021, 22, 10005. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yan, Q.; Liao, B.; Zhao, L.; Xiong, S.; Wang, J.; Zou, D.; Pan, J.; Wu, L.; Deng, Y.; et al. The HIF1α/HIF2α-miR210-3p network regulates glioblastoma cell proliferation, dedifferentiation and chemoresistance through EGF under hypoxic conditions. Cell Death Dis. 2020, 11, 992. [Google Scholar] [CrossRef] [PubMed]
- Torrisi, F.; Vicario, N.; Spitale, F.M.; Cammarata, F.P.; Minafra, L.; Salvatorelli, L.; Russo, G.; Cuttone, G.; Valable, S.; Gulino, R.; et al. The role of hypoxia and src tyrosine kinase in glioblastoma invasiveness and radioresistance. Cancers 2020, 12, 2860. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Gupta, S.C.; Pandey, M.K.; Tyagi, A.K.; Deb, L. Oxidative stress and cancer: Advances and challenges. Oxidative Med. Cell. Longev. 2016, 2016, 5010423. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.R.; Danai, L.V.; Lewis, C.A.; Chan, S.H.; Gui, D.Y.; Kunchok, T.; Dennstedt, E.A.; Heiden, M.G.V.; Muir, A. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 2019, 8, e44235. [Google Scholar] [CrossRef] [PubMed]
- Fendt, S.-M.; Frezza, C.; Erez, A. Targeting Metabolic Plasticity and Flexibility Dynamics for Cancer Therapy. Cancer Discov. 2020, 10, 1797–1807. [Google Scholar] [CrossRef]
- Bensinger, S.; Christofk, H. New aspects of the Warburg effect in cancer cell biology. Semin. Cell Dev. Biol. 2012, 23, 352–361. [Google Scholar] [CrossRef]
- Sajadimajd, S.; Khazaei, M. Oxidative stress and cancer: The role of Nrf2. Curr. Cancer Drug Targets 2017, 18, 538–557. [Google Scholar] [CrossRef]
- Moindjie, H.; Rodrigues-Ferreira, S.; Nahmias, C. Mitochondrial Metabolism in Carcinogenesis and Cancer Therapy. Cancers 2021, 13, 3311. [Google Scholar] [CrossRef]
- George, S.; Abrahamse, H. Redox Potential of Antioxidants in Cancer Progression and Prevention. Antioxidants 2020, 9, 1156. [Google Scholar] [CrossRef]
- Luengo, A.; Abbott, K.L.; Davidson, S.M.; Hosios, A.M.; Faubert, B.; Chan, S.H.; Freinkman, E.; Zacharias, L.G.; Mathews, T.P.; Clish, C.B.; et al. Reactive metabolite production is a targetable liability of glycolytic metabolism in lung cancer. Nat. Commun. 2019, 10, 5604. [Google Scholar] [CrossRef]
- Bansal, A.; Celeste Simon, M. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef]
- Morroni, F.; Sita, G.; Djemil, A.; D’Amico, M.; Pruccoli, L.; Cantelli-Forti, G.; Hrelia, P.; Tarozzi, A. Comparison of Adaptive Neuroprotective Mechanisms of Sulforaphane and its Interconversion Product Erucin in in Vitro and in Vivo Models of Parkinson’s Disease. J. Agric. Food Chem. 2018, 66, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Yuanfeng, W.; Chengzhi, L.; Ligen, Z.; Juan, S.; Xinjie, S.; Yao, Z.; Jianwei, M. Approaches for enhancing the stability and formation of sulforaphane. Food Chem. 2021, 345, 128771. [Google Scholar] [CrossRef]
- Zhang, Y.; Talalay, P.; Cho, C.; Posner, G. A major inducer of anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, H.; Dong, N.; Su, X.; Duan, M.; Wei, Y.; Wei, J.; Liu, G.; Peng, Q.; Zhao, Y. Sulforaphane induces S-phase arrest and apoptosis via p53-dependent manner in gastric cancer cells. Sci. Rep. 2021, 11, 2504. [Google Scholar] [CrossRef]
- Fimognari, C.; Hrelia, P. Sulforaphane as a promising molecule for fighting cancer. Mutat. Res. Mutat. Res. 2007, 635, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, J.; Tang, L. Cancer-preventive isothiocyanates: Dichotomous modulators of oxidative stress. Free Radic. Biol. Med. 2005, 38, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Morroni, F.; Tarozzi, A.; Sita, G.; Bolondi, C.; Zolezzi Moraga, J.M.; Cantelli-Forti, G.; Hrelia, P. Neuroprotective effect of sulforaphane in 6-hydroxydopamine-lesioned mouse model of Parkinson’s disease. Neurotoxicology 2013, 36, 63–71. [Google Scholar] [CrossRef]
- Sita, G.; Hrelia, P.; Graziosi, A.; Morroni, F. Sulforaphane from cruciferous vegetables: Recent advances to improve glioblastoma treatment. Nutrients 2018, 10, 1755. [Google Scholar] [CrossRef]
- Schito, L.; Semenza, G.L. Hypoxia-inducible factors: Master regulators of cancer progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, C.; Shang, L.; Zhang, Y.; Zou, R.; Zhan, Y.; Bi, B. Sulforaphane induces apoptosis and inhibits invasion in U251MG glioblastoma cells. Springerplus 2016, 5, 235. [Google Scholar] [CrossRef]
- Karmakar, S.; Weinberg, M.S.; Banik, N.L.; Patel, S.J.; Ray, S.K. Activation of multiple molecular mechanisms for apoptosis in human malignant glioblastoma T98G and U87MG cells treated with sulforaphane. Neuroscience 2006, 141, 1265–1280. [Google Scholar] [CrossRef]
- Miao, Z.; Yu, F.; Ren, Y.; Yang, J. D,L-Sulforaphane induces ROS-dependent apoptosis in human gliomablastoma cells by inactivating STAT3 signaling pathway. Int. J. Mol. Sci. 2017, 18, 72. [Google Scholar] [CrossRef]
- Kerr, C.; Adhikary, G.; Grun, D.; George, N.; Eckert, R.L. Combination cisplatin and sulforaphane treatment reduces proliferation, invasion, and tumor formation in epidermal squamous cell carcinoma. Mol. Carcinog. 2018, 57, 3–11. [Google Scholar] [CrossRef]
- Hamilton, C.; Fox, J.P.; Longley, D.B.; Higgins, C.A. Therapeutics Targeting the Core Apoptotic Machinery. Cancers 2021, 13, 2618. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Marzetti, E. Cell death and inflammation: The role of mitochondria in health and disease. Cells 2021, 10, 537. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Yang, J.; Jones, D.P. Mitochondrial control of apoptosis: The role of cytochrome c. Biochim. Biophys. Acta-Bioenerg. 1998, 1366, 139–149. [Google Scholar] [CrossRef]
- Kohan, R.; Collin, A.; Guizzardi, S.; Tolosa de Talamoni, N.; Picotto, G. Reactive oxygen species in cancer: A paradox between pro- and anti-tumour activities. Cancer Chemother. Pharmacol. 2020, 86, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Zhou, Y.; Zhang, H.; Demizu, Y.; Chen, Z.; Pelicano, H.; Chiao, P.; Achanta, G.; Arlinghaus, R.; Liu, J.; et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Byun, S.; Shin, S.; Park, J.; Lim, S.; Lee, E.; Lee, C.; Sung, D.; Farrand, L.; Lee, S.; Kim, K.; et al. Sulforaphene suppresses growth of colon cancer-derived tumors via induction of glutathione depletion and microtubule depolymerization. Mol. Nutr. Food Res. 2016, 60, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhou, Q.-H.; Xu, K. Are isothiocyanates potential anti-cancer drugs? Acta Pharmacol. Sin. 2009, 30, 501–512. [Google Scholar] [CrossRef]
- Wu, S.; Zhou, Y.; Yang, G.; Tian, H.; Geng, Y.; Hu, Y.; Lin, K.; Wu, W. Sulforaphane-cysteine induces apoptosis by sustained activation of ERK1/2 and caspase 3 in human glioblastoma U373MG and U87MG cells. Oncol. Rep. 2017, 37, 2829–2838. [Google Scholar] [CrossRef]
- Park, J.H.; de Lomana, A.L.G.; Marzese, D.M.; Juarez, T.; Feroze, A.; Hothi, P.; Cobbs, C.; Patel, A.P.; Kesari, S.; Huang, S.; et al. A systems approach to brain tumor treatment. Cancers 2021, 13, 3152. [Google Scholar] [CrossRef]
- Cavazos, D.A.; Brenner, A.J. Hypoxia in astrocytic tumors and implications for therapy. Neurobiol. Dis. 2016, 85, 227–233. [Google Scholar] [CrossRef][Green Version]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef]
- Brahimi-Horn, M.C.; Chiche, J.; Pouysségur, J. Hypoxia and cancer. J. Mol. Med. 2007, 85, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298. [Google Scholar] [CrossRef]
- Virtuoso, A.; Giovannoni, R.; De Luca, C.; Gargano, F.; Cerasuolo, M.; Maggio, N.; Lavitrano, M.; Papa, M. The glioblastoma microenvironment: Morphology, metabolism, and molecular signature of glial dynamics to discover metabolic rewiring sequence. Int. J. Mol. Sci. 2021, 22, 3301. [Google Scholar] [CrossRef]
- Stadlbauer, A.; Kinfe, T.M.; Eyüpoglu, I.; Zimmermann, M.; Kitzwögerer, M.; Podar, K.; Buchfelder, M.; Heinz, G.; Oberndorfer, S.; Marhold, F. Tissue hypoxia and alterations in microvascular architecture predict glioblastoma recurrence in humans. Clin. Cancer Res. 2021, 27, 1641–1649. [Google Scholar] [CrossRef]
- Mahase, S.; Rattenni, R.N.; Wesseling, P.; Leenders, W.; Baldotto, C.; Jain, R.; Zagzag, D. Hypoxia-mediated mechanisms associated with antiangiogenic treatment resistance in glioblastomas. Am. J. Pathol. 2017, 187, 940–953. [Google Scholar] [CrossRef]
- Dapash, M.; Castro, B.; Hou, D.; Lee-Chang, C. Current Immunotherapeutic Strategies for the Treatment of Glioblastoma. Cancers 2021, 13, 4548. [Google Scholar] [CrossRef]
- Gong, T.T.; Guo, Q.; Li, X.; Zhang, T.N.; Liu, F.H.; He, X.H.; Lin, B.; Wu, Q.J. Isothiocyanate Iberin inhibits cell proliferation and induces cell apoptosis in the progression of ovarian cancer by mediating ROS accumulation and GPX1 expression. Biomed. Pharmacother. 2021, 142, 111533. [Google Scholar] [CrossRef]
- Xie, H.; Chun, F.K.; Rutz, J.; Blaheta, R.A. Sulforaphane impact on reactive oxygen species (ROS) in bladder carcinoma. Int. J. Mol. Sci. 2021, 22, 5938. [Google Scholar] [CrossRef] [PubMed]
- Dinh, T.N.; Parat, M.O.; Ong, Y.S.; Khaw, K.Y. Anticancer activities of dietary benzyl isothiocyanate: A comprehensive review. Pharmacol. Res. 2021, 169, 105666. [Google Scholar] [CrossRef]
- Rabben, H.L.; Kodama, Y.; Nakamura, M.; Bones, A.M.; Wang, T.C.; Chen, D.; Zhao, C.M.; Øverby, A. Chemopreventive effects of dietary isothiocyanates in animal models of gastric cancer and synergistic anticancer effects with cisplatin in human gastric cancer cells. Front. Pharmacol. 2021, 12, 613458. [Google Scholar] [CrossRef] [PubMed]
- Kamal, M.M.; Akter, S.; Lin, C.N.; Nazzal, S. Sulforaphane as an anticancer molecule: Mechanisms of action, synergistic effects, enhancement of drug safety, and delivery systems. Arch. Pharm. Res. 2020, 43, 371–384. [Google Scholar] [CrossRef]
- Bijangi-Vishehsaraei, K.; Saadatzadeh, M.R.; Wang, H.; Nguyen, A.; Kamocka, M.M.; Cai, W.; Cohen-Gadol, A.A.; Halum, S.L.; Sarkaria, J.N.; Pollok, K.E.; et al. Sulforaphane suppresses the growth of glioblastoma cells, glioblastoma stem cell–like spheroids, and tumor xenografts through multiple cell signaling pathways. J. Neurosurg. 2017, 127, 1219. [Google Scholar] [CrossRef]
- Li, C.; Zhou, Y.; Peng, X.; Du, L.; Tian, H.; Yang, G.; Niu, J.; Wu, W. Sulforaphane inhibits invasion via activating ERK1/2 signaling in human glioblastoma U87MG and U373MG cells. PLoS ONE 2014, 9, e90520. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; de Mooij, T.; Peterson, T.E.; Kaptzan, T.; Johnson, A.J.; Daniels, D.J.; Parney, I.F. Modulating glioma-mediated myeloid-derived suppressor cell development with sulforaphane. PLoS ONE 2017, 12, e0179012. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.; Bjerke, M.; Edlund, H.; Nelander, S.; Westermark, B. Origin of the U87MG glioma cell line: Good news and bad news. Sci. Transl. Med. 2016, 8, 354re3. [Google Scholar] [CrossRef] [PubMed]
- Zhai, K.; Siddiqui, M.; Abdellatif, B.; Liskova, A.; Kubatka, P.; Büsselberg, D. Natural compounds in glioblastoma therapy: Preclinical insights, mechanistic pathways, and outlook. Cancers 2021, 13, 2317. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, A.S.; Mandave, P.C.; Deshpande, M.; Ranjekar, P.; Prakash, O. Phytochemicals in cancer treatment: From preclinical studies to clinical practice. Front. Pharmacol. 2020, 10, 1614. [Google Scholar] [CrossRef]
- Gomez-Manzano, C.; Fueyo, J.; Kyritsis, A.P.; Steck, P.A.; Levin, V.A.; Alfred Yung, W.K.; McDonnell, T.J. Characterization of p53 and p21 functional interactions in glioma cells en route to apoptosis. JNCI J. Natl. Cancer Inst. 1997, 89, 1036–1044. [Google Scholar] [CrossRef]
- Cordani, M.; Butera, G.; Pacchiana, R.; Masetto, F.; Mullappilly, N.; Riganti, C.; Donadelli, M. Mutant p53-associated molecular mechanisms of ROS regulation in cancer cells. Biomolecules 2020, 10, 361. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.G.; Ding, X.Z.; Talamonti, M.S.; Bell, R.H.; Adrian, T.E. LTB4 stimulates growth of human pancreatic cancer cells via MAPK and PI-3 kinase pathways. Biochem. Biophys. Res. Commun. 2005, 335, 949–956. [Google Scholar] [CrossRef]
- Lee, W.J.; Hsiao, M.; Chang, J.L.; Yang, S.F.; Tseng, T.H.; Cheng, C.W.; Chow, J.M.; Lin, K.H.; Lin, Y.W.; Liu, C.C.; et al. Quercetin induces mitochondrial-derived apoptosis via reactive oxygen species-mediated ERK activation in HL-60 leukemia cells and xenograft. Arch. Toxicol. 2015, 89, 1103–1117. [Google Scholar] [CrossRef]
- Lan, H.; Yuan, H.; Lin, C. Sulforaphane induces p53-deficient SW480 cell apoptosis via the ROS-MAPK signaling pathway. Mol. Med. Rep. 2017, 16, 7796–7804. [Google Scholar] [CrossRef]
- Liu, H.-J.; Wang, L.; Kang, L.; Du, J.; Li, S.; Cui, H.-X. Sulforaphane-N-acetyl-cysteine induces autophagy through activation of ERK1/2 in U87MG and U373MG cells. Cell. Physiol. Biochem. 2018, 51, 528–542. [Google Scholar] [CrossRef]
- Lu, N.; Malemud, C.J. Extracellular signal-regulated kinase: A regulator of cell growth, inflammation, chondrocyte and bone cell receptor-mediated gene expression. Int. J. Mol. Sci. 2019, 20, 3792. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of cell viability by the alamarblue assay. Cold Spring Harb. Protoc. 2018, 2018, 462–464. [Google Scholar] [CrossRef]
- Tarozzi, A.; Morroni, F.; Merlicco, A.; Hrelia, S.; Angeloni, C.; Cantelli-Forti, G.; Hrelia, P. Sulforaphane as an inducer of glutathione prevents oxidative stress-induced cell death in a dopaminergic-like neuroblastoma cell line. J. Neurochem. 2009, 111, 1161–1171. [Google Scholar] [CrossRef]
- Morroni, F.; Sita, G.; Graziosi, A.; Turrini, E.; Fimognari, C.; Tarozzi, A.; Hrelia, P. Protective effects of 6-(methylsulfinyl)hexyl isothiocyanate on Aβ1-42-induced cognitive deficit, oxidative stress, inflammation, and apoptosis in mice. Int. J. Mol. Sci. 2018, 19, 2083. [Google Scholar] [CrossRef] [PubMed]
- Traka, M.; Gasper, A.V.; Smith, J.A.; Hawkey, C.J.; Bao, Y.; Mithen, R.F. Transcriptome analysis of human colon Caco-2 cells exposed to sulforaphane. J. Nutr. 2005, 135, 1865–1872. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sita, G.; Graziosi, A.; Hrelia, P.; Morroni, F. Sulforaphane Causes Cell Cycle Arrest and Apoptosis in Human Glioblastoma U87MG and U373MG Cell Lines under Hypoxic Conditions. Int. J. Mol. Sci. 2021, 22, 11201. https://doi.org/10.3390/ijms222011201
Sita G, Graziosi A, Hrelia P, Morroni F. Sulforaphane Causes Cell Cycle Arrest and Apoptosis in Human Glioblastoma U87MG and U373MG Cell Lines under Hypoxic Conditions. International Journal of Molecular Sciences. 2021; 22(20):11201. https://doi.org/10.3390/ijms222011201
Chicago/Turabian StyleSita, Giulia, Agnese Graziosi, Patrizia Hrelia, and Fabiana Morroni. 2021. "Sulforaphane Causes Cell Cycle Arrest and Apoptosis in Human Glioblastoma U87MG and U373MG Cell Lines under Hypoxic Conditions" International Journal of Molecular Sciences 22, no. 20: 11201. https://doi.org/10.3390/ijms222011201
APA StyleSita, G., Graziosi, A., Hrelia, P., & Morroni, F. (2021). Sulforaphane Causes Cell Cycle Arrest and Apoptosis in Human Glioblastoma U87MG and U373MG Cell Lines under Hypoxic Conditions. International Journal of Molecular Sciences, 22(20), 11201. https://doi.org/10.3390/ijms222011201