
High Resolution Structure of the Mature Capsid of Ralstonia solanacearum Bacteriophage ϕRSA1 by Cryo-Electron Microscopy


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
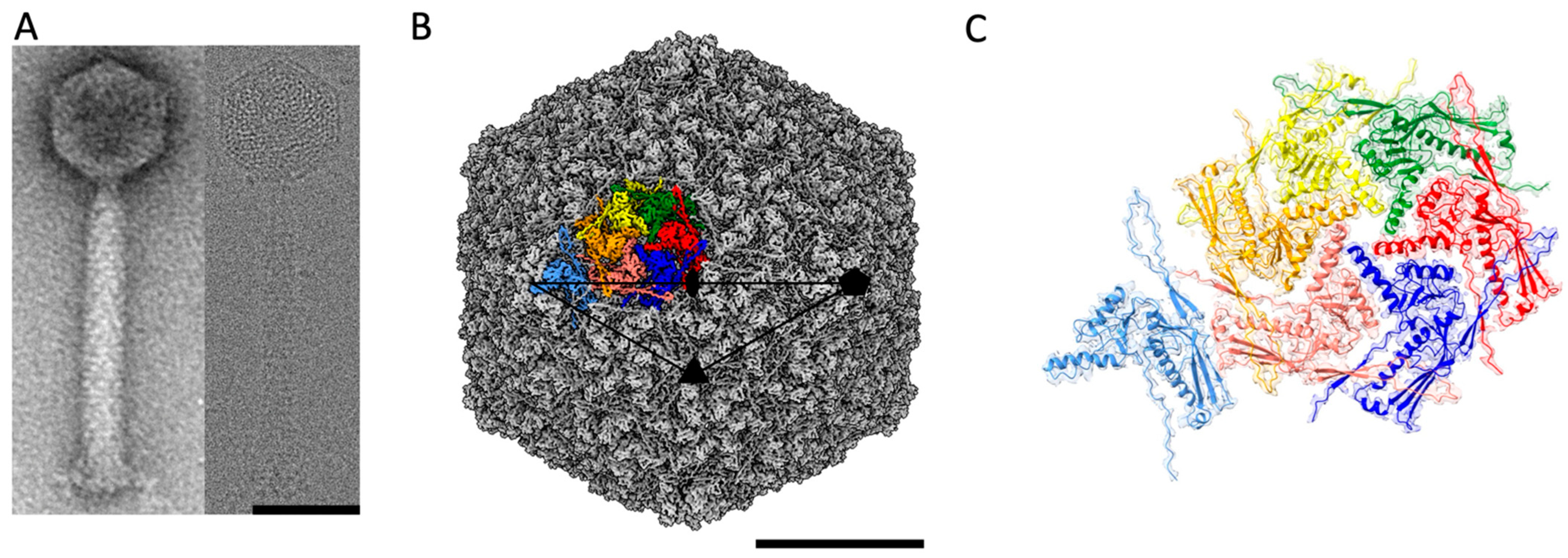
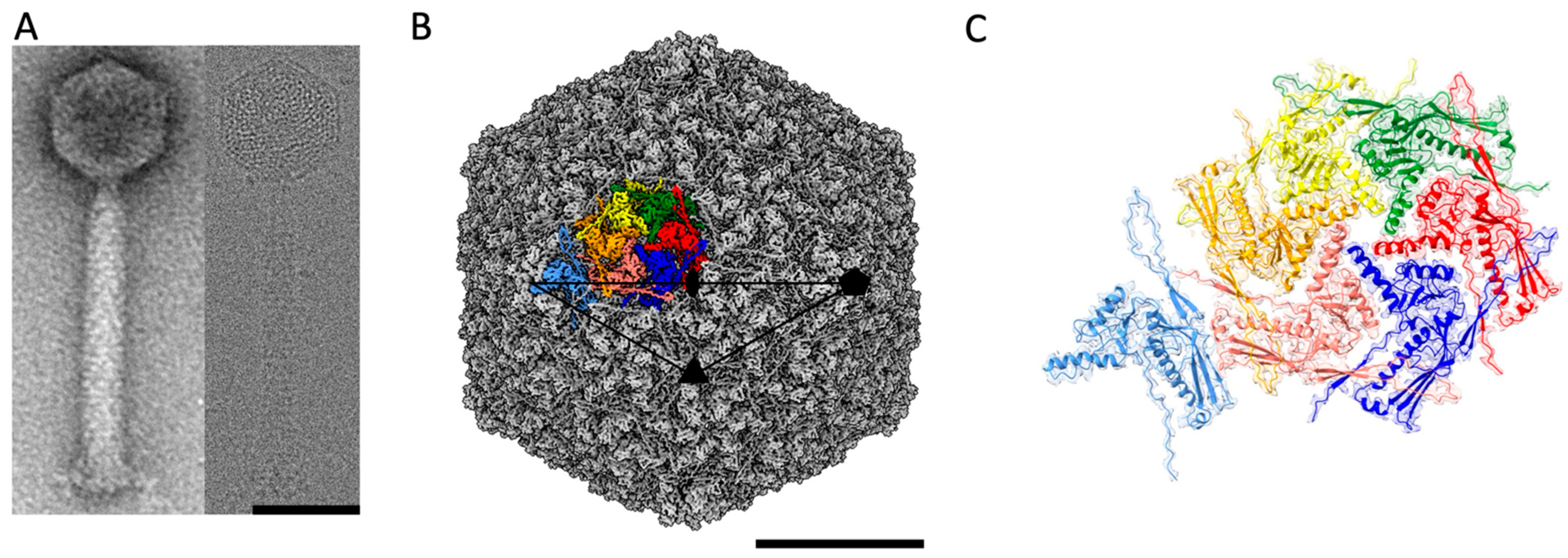
2.1. Structure of the ϕRSA1 Capsid by Cryo-EM
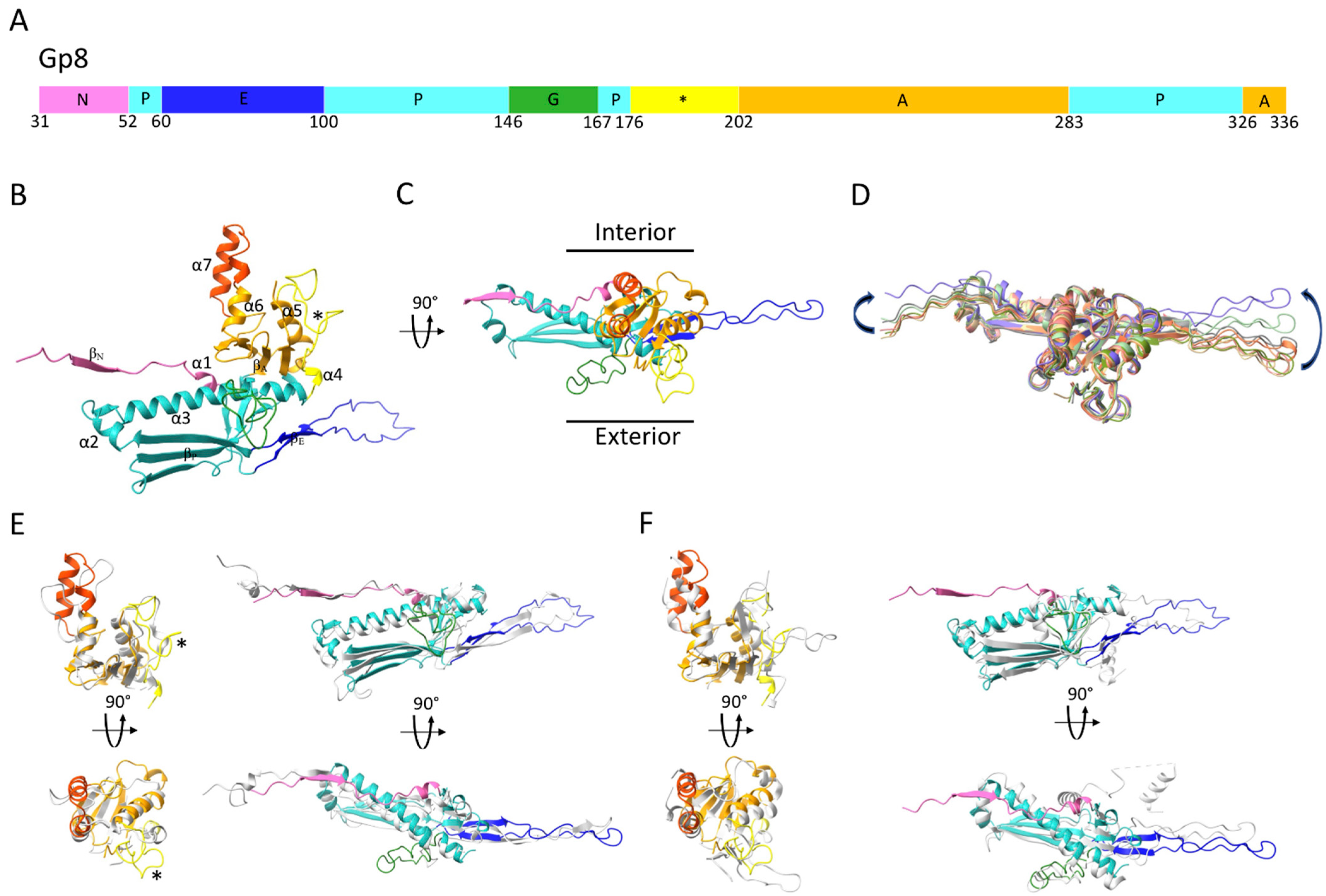
2.2. Structure of the ϕRSA1’s MCP
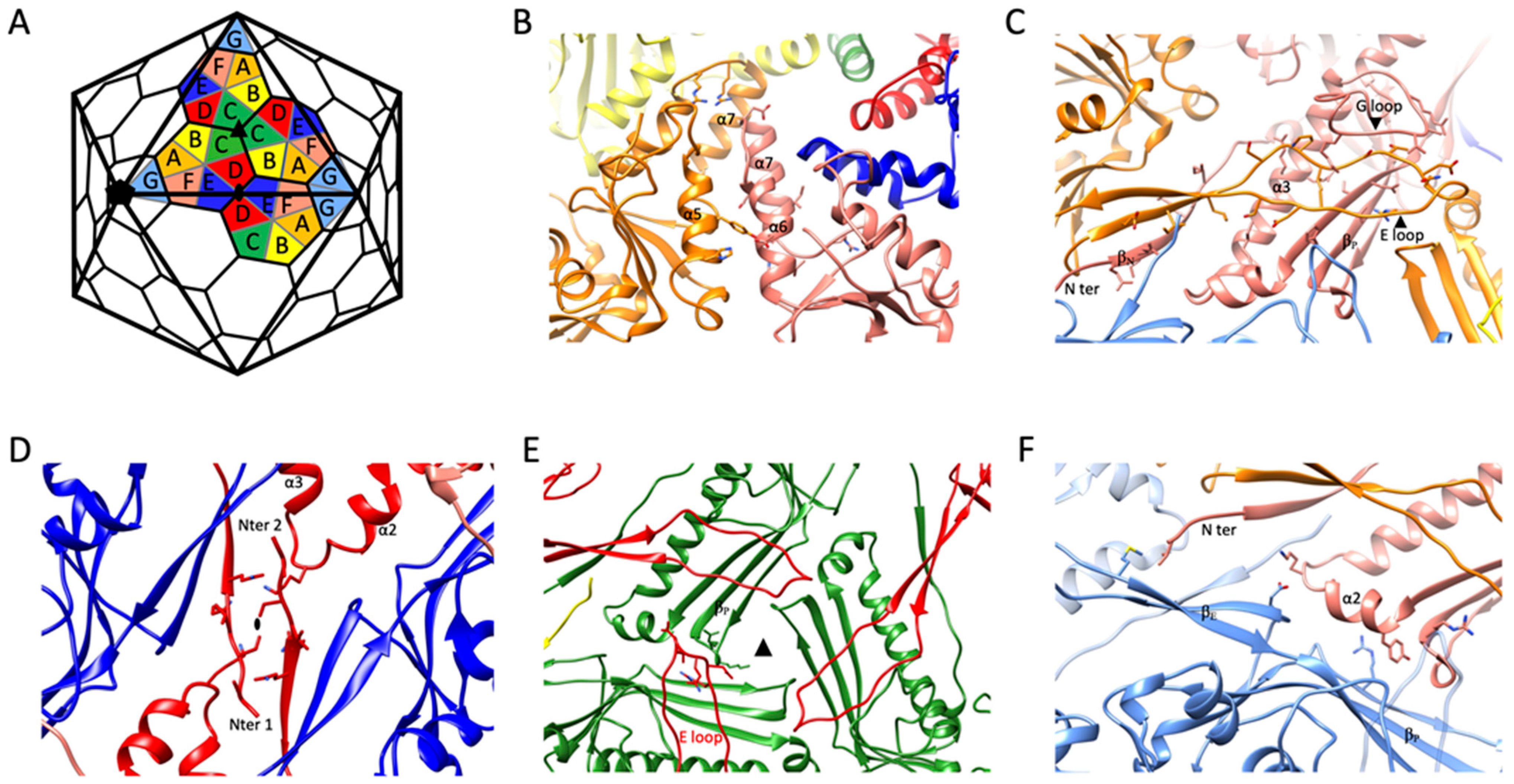
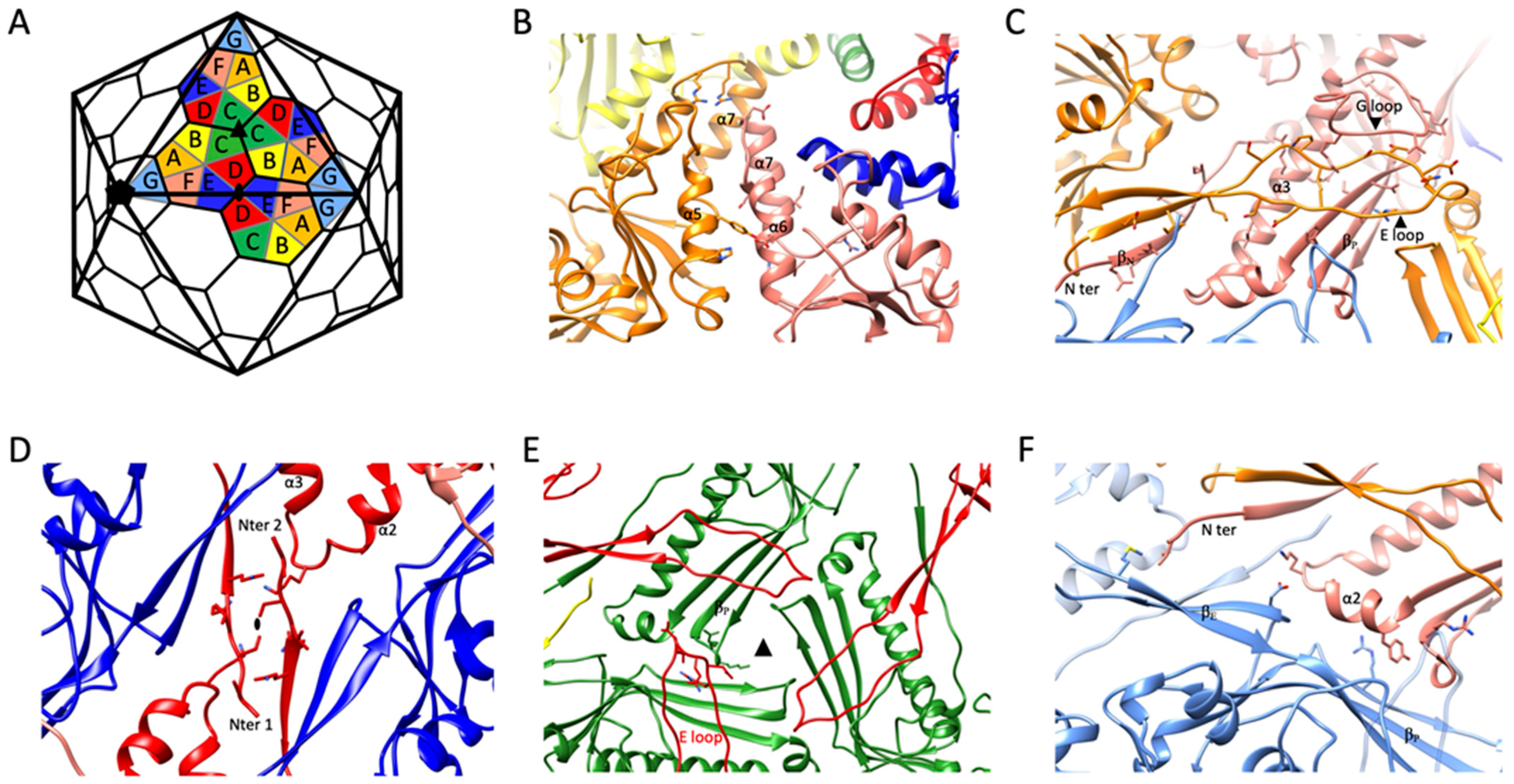
2.3. Intra-Capsomeric Molecular Interactions between the Capsid’s MCPs
2.4. Inter-Capsomeric Molecular Interactions between the Capsid’s MCPs
2.5. Comparison between the Right-(Dextro) and Left-(Laevo) Handed T = 7 Lattices
3. Conclusions
4. Materials and Methods
4.1. Bacteriophage Production and Purification
4.2. Negative Staining
4.3. Cryo-Electron Microscopy and Preprocessing
4.4. Image Analysis of the Capsid
4.5. Model Building of the ϕRSA1 Capsid
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3D | Tri-dimensional |
| Cryo-EM | Cryo Electron Microscopy |
| DALI | Distance mAtrix aLIgnment |
| dsDNA | Double Stranded DeoxyriboNucleic Acid |
| MCP | Major Capsid Protein |
| RMSD | Root Mean Square Deviation |
References
- Ackermann, H.-W. 5500 Phages Examined in the Electron Microscope. Arch. Virol. 2007, 152, 227–243. [Google Scholar] [CrossRef]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Davison, A.J.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; et al. Changes to Virus Taxonomy and to the International Code of Virus Classification and Nomenclature Ratified by the International Committee on Taxonomy of Viruses (2021). Arch. Virol. 2021, 166, 2633–2648. [Google Scholar] [CrossRef]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage Diversity, Genomics and Phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar] [CrossRef]
- Helgstrand, C.; Wikoff, W.R.; Duda, R.L.; Hendrix, R.W.; Johnson, J.E.; Liljas, L. The Refined Structure of a Protein Catenane: The HK97 Bacteriophage Capsid at 3.44 A Resolution. J. Mol. Biol. 2003, 334, 885–899. [Google Scholar] [CrossRef]
- Pietilä, M.K.; Laurinmäki, P.; Russell, D.A.; Ko, C.-C.; Jacobs-Sera, D.; Hendrix, R.W.; Bamford, D.H.; Butcher, S.J. Structure of the Archaeal Head-Tailed Virus HSTV-1 Completes the HK97 Fold Story. PNAS 2013, 110, 10604–10609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wikoff, W.R.; Liljas, L.; Duda, R.L.; Tsuruta, H.; Hendrix, R.W.; Johnson, J.E. Topologically Linked Protein Rings in the Bacteriophage HK97 Capsid. Science 2000, 289, 2129–2133. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Xiao, H.; Wang, X.; Song, S.; Han, Z.; Li, X.; Yang, F.; Wang, L.; Song, J.; Liu, H.; et al. Structural Changes of a Bacteriophage upon DNA Packaging and Maturation. Protein Cell 2020, 11, 374–379. [Google Scholar] [CrossRef] [PubMed]
- McNulty, R.; Cardone, G.; Gilcrease, E.B.; Baker, T.S.; Casjens, S.R.; Johnson, J.E. Cryo-EM Elucidation of the Structure of Bacteriophage P22 Virions after Genome Release. Biophys. J. 2018, 114, 1295–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Sun, L.; Zhang, Z.; Fokine, A.; Padilla-Sanchez, V.; Hanein, D.; Jiang, W.; Rossmann, M.G.; Rao, V.B. Cryo-EM Structure of the Bacteriophage T4 Isometric Head at 3.3-Å Resolution and Its Relevance to the Assembly of Icosahedral Viruses. Proc. Natl. Acad. Sci. USA 2017, 114, E8184–E8193. [Google Scholar] [CrossRef] [Green Version]
- Huet, A.; Duda, R.L.; Boulanger, P.; Conway, J.F. Capsid Expansion of Bacteriophage T5 Revealed by High Resolution Cryoelectron Microscopy. Proc. Natl. Acad. Sci. USA 2019, 116, 21037–21046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Effantin, G.; Hamasaki, R.; Kawasaki, T.; Bacia, M.; Moriscot, C.; Weissenhorn, W.; Yamada, T.; Schoehn, G. Cryo-Electron Microscopy Three-Dimensional Structure of the Jumbo Phage ΦRSL1 Infecting the Phytopathogen Ralstonia solanacearum. Structure 2013, 21, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Fokine, A.; Battisti, A.J.; Bowman, V.D.; Efimov, A.V.; Kurochkina, L.P.; Chipman, P.R.; Mesyanzhinov, V.V.; Rossmann, M.G. Cryo-EM Study of the Pseudomonas Bacteriophage PhiKZ. Structure 2007, 15, 1099–1104. [Google Scholar] [CrossRef] [Green Version]
- Neumann, E.; Kawasaki, T.; Effantin, G.; Estrozi, L.F.; Chatchawankanphanich, O.; Yamada, T.; Schoehn, G. 3D Structure of Three Jumbo Phage Heads. J. Gen. Virol. 2020, 101, 1219–1226. [Google Scholar] [CrossRef]
- Hua, J.; Huet, A.; Lopez, C.A.; Toropova, K.; Pope, W.H.; Duda, R.L.; Hendrix, R.W.; Conway, J.F. Capsids and Genomes of Jumbo-Sized Bacteriophages Reveal the Evolutionary Reach of the HK97 Fold. mBio 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Molineux, I.J.; Panja, D. Popping the Cork: Mechanisms of Phage Genome Ejection. Nat. Rev. Microbiol. 2013, 11, 194–204. [Google Scholar] [CrossRef]
- São-José, C.; de Frutos, M.; Raspaud, E.; Santos, M.A.; Tavares, P. Pressure Built by DNA Packing inside Virions: Enough to Drive DNA Ejection in Vitro, Largely Insufficient for Delivery into the Bacterial Cytoplasm. J. Mol. Biol. 2007, 374, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Evilevitch, A.; Lavelle, L.; Knobler, C.M.; Raspaud, E.; Gelbart, W.M. Osmotic Pressure Inhibition of DNA Ejection from Phage. Proc. Natl. Acad. Sci. USA 2003, 100, 9292–9295. [Google Scholar] [CrossRef] [Green Version]
- Duda, R.L. Protein Chainmail: Catenated Protein in Viral Capsids. Cell 1998, 94, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Lander, G.C.; Evilevitch, A.; Jeembaeva, M.; Potter, C.S.; Carragher, B.; Johnson, J.E. Bacteriophage Lambda Stabilization by Auxiliary Protein GpD: Timing, Location, and Mechanism of Attachment Determined by Cryo-EM. Structure 2008, 16, 1399–1406. [Google Scholar] [CrossRef] [Green Version]
- Effantin, G.; Figueroa-Bossi, N.; Schoehn, G.; Bossi, L.; Conway, J.F. The Tripartite Capsid Gene of Salmonella Phage Gifsy-2 Yields a Capsid Assembly Pathway Engaging Features from HK97 and Lambda. Virology 2010, 402, 355–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Hardies, S.C.; Fokine, A.; Klose, T.; Jiang, W.; Cho, B.C.; Rossmann, M.G. Structure of the Marine Siphovirus TW1: Evolution of Capsid-Stabilizing Proteins and Tail Spikes. Structure 2018, 26, 238–248.e3. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.M.; Dunstan, R.A.; Grinter, R.; Belousoff, M.J.; Wang, J.; Pickard, D.; Venugopal, H.; Dougan, G.; Lithgow, T.; Coulibaly, F. The Architecture and Stabilisation of Flagellotropic Tailed Bacteriophages. Nat. Commun. 2020, 11, 3748. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, K.; Lynn, A.Y.; Aron, K.E.; Yu, G.; Jiang, W.; Tang, L. Structure of a Headful DNA-Packaging Bacterial Virus at 2.9 Å Resolution by Electron Cryo-Microscopy. Proc. Natl. Acad. Sci. USA 2017, 114, 3601–3606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, G.; Askora, A.; Blanc-Mathieu, R.; Kawasaki, T.; Li, Y.; Nakano, M.; Ogata, H.; Yamada, T. Xanthomonas Citri Jumbo Phage XacN1 Exhibits a Wide Host Range and High Complement of TRNA Genes. Sci. Rep. 2018, 8, 4486. [Google Scholar] [CrossRef]
- Fokine, A.; Islam, M.Z.; Zhang, Z.; Bowman, V.D.; Rao, V.B.; Rossmann, M.G. Structure of the Three N-Terminal Immunoglobulin Domains of the Highly Immunogenic Outer Capsid Protein from a T4-like Bacteriophage. J. Virol. 2011, 85, 8141–8148. [Google Scholar] [CrossRef] [Green Version]
- Hayward, A.C. Biology and Epidemiology of Bacterial Wilt Caused by Pseudomonas Solanacearum. Annu. Rev. Phytopathol. 1991, 29, 65–87. [Google Scholar] [CrossRef]
- Yamada, T.; Kawasaki, T.; Nagata, S.; Fujiwara, A.; Usami, S.; Fujie, M. New Bacteriophages That Infect the Ralstonia solanacearum Solanacearum. Microbiology 2007, 153, 2630–2639. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Satoh, S.; Ishikawa, H.; Fujiwara, A.; Kawasaki, T.; Fujie, M.; Ogata, H. A Jumbo Phage Infecting the Phytopathogen Ralstonia solanacearum Defines a New Lineage of the Myoviridae Family. Virology 2010, 398, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhunchoth, A.; Blanc-Mathieu, R.; Mihara, T.; Nishimura, Y.; Askora, A.; Phironrit, N.; Leksomboon, C.; Chatchawankanphanich, O.; Kawasaki, T.; Nakano, M.; et al. Two Asian Jumbo Phages, ΦRSL2 and ΦRSF1, Infect Ralstonia solanacearum and Show Common Features of ΦKZ-Related Phages. Virology 2016, 494, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, A.; Kawasaki, T.; Usami, S.; Fujie, M.; Yamada, T. Genomic Characterization of Ralstonia solanacearum Phage PhiRSA1 and Its Related Prophage (PhiRSX) in Strain GMI1000. J. Bacteriol. 2008, 190, 143–156. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, S.; Bebeacua, C.; Orlov, I.; Tremblay, D.; Klaholz, B.P.; Moineau, S.; Cambillau, C. Cryo-Electron Microscopy Structure of Lactococcal Siphophage 1358 Virion. J. Virol. 2014, 88, 8900–8910. [Google Scholar] [CrossRef] [Green Version]
- Dearborn, A.D.; Laurinmaki, P.; Chandramouli, P.; Rodenburg, C.M.; Wang, S.; Butcher, S.J.; Dokland, T. Structure and Size Determination of Bacteriophage P2 and P4 Procapsids: Function of Size Responsiveness Mutations. J. Struct. Biol. 2012, 178, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Suhanovsky, M.M.; Teschke, C.M. Nature’s Favorite Building Block: Deciphering Folding and Capsid Assembly of Proteins with the HK97-Fold. Virology 2015, 479–480, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Hryc, C.F.; Chen, D.-H.; Afonine, P.V.; Jakana, J.; Wang, Z.; Haase-Pettingell, C.; Jiang, W.; Adams, P.D.; King, J.A.; Schmid, M.F.; et al. Accurate Model Annotation of a Near-Atomic Resolution Cryo-EM Map. Proc. Natl. Acad. Sci. USA 2017, 114, 3103–3108. [Google Scholar] [CrossRef] [Green Version]
- Ignatiou, A.; Brasilès, S.; El Sadek Fadel, M.; Bürger, J.; Mielke, T.; Topf, M.; Tavares, P.; Orlova, E.V. Structural Transitions during the Scaffolding-Driven Assembly of a Viral Capsid. Nat. Commun. 2019, 10, 4840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, M.L.; Hryc, C.F.; Zhang, Q.; Wu, W.; Jakana, J.; Haase-Pettingell, C.; Afonine, P.V.; Adams, P.D.; King, J.A.; Jiang, W.; et al. Validated Near-Atomic Resolution Structure of Bacteriophage Epsilon15 Derived from Cryo-EM and Modeling. Proc. Natl. Acad. Sci. USA 2013, 110, 12301–12306. [Google Scholar] [CrossRef] [Green Version]
- Baker, M.L.; Jiang, W.; Rixon, F.J.; Chiu, W. Common Ancestry of Herpesviruses and Tailed DNA Bacteriophages. J. Virol. 2005, 79, 14967–14970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayfield, O.W.; Klimuk, E.; Winkler, D.C.; Hesketh, E.L.; Chechik, M.; Cheng, N.; Dykeman, E.C.; Minakhin, L.; Ranson, N.A.; Severinov, K.; et al. Cryo-EM Structure and in Vitro DNA Packaging of a Thermophilic Virus with Supersized T=7 Capsids. Proc. Natl. Acad. Sci. USA 2019, 116, 3556–3561. [Google Scholar] [CrossRef] [Green Version]
- Stone, N.P.; Demo, G.; Agnello, E.; Kelch, B.A. Principles for Enhancing Virus Capsid Capacity and Stability from a Thermophilic Virus Capsid Structure. Nat. Commun. 2019, 10, 4471. [Google Scholar] [CrossRef] [Green Version]
- Kizziah, J.L.; Rodenburg, C.M.; Dokland, T. Structure of the Capsid Size-Determining Scaffold of “Satellite” Bacteriophage P4. Viruses 2020, 12, 953. [Google Scholar] [CrossRef]
- Lata, R.; Conway, J.F.; Cheng, N.; Duda, R.L.; Hendrix, R.W.; Wikoff, W.R.; Johnson, J.E.; Tsuruta, H.; Steven, A.C. Maturation Dynamics of a Viral Capsid: Visualization of Transitional Intermediate States. Cell 2000, 100, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Forrer, P.; Dauter, Z.; Conway, J.F.; Cheng, N.; Cerritelli, M.E.; Steven, A.C.; Plückthun, A.; Wlodawer, A. Novel Fold and Capsid-Binding Properties of the Lambda-Phage Display Platform Protein GpD. Nat. Struct. Biol. 2000, 7, 230–237. [Google Scholar] [CrossRef]
- Baker, T.S.; Olson, N.H.; Fuller, S.D. Adding the Third Dimension to Virus Life Cycles: Three-Dimensional Reconstruction of Icosahedral Viruses from Cryo-Electron Micrographs. Microbiol. Mol. Biol. Rev. 1999, 63, 862–922. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.Q.; Palovcak, E.; Armache, J.-P.; Verba, K.A.; Cheng, Y.; Agard, D.A. MotionCor2: Anisotropic Correction of Beam-Induced Motion for Improved Cryo-Electron Microscopy. Nat. Methods 2017, 14, 331–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K. Gctf: Real-Time CTF Determination and Correction. J. Struct. Biol. 2016, 193, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuller, S.D.; Butcher, S.J.; Cheng, R.H.; Baker, T.S. Three-Dimensional Reconstruction of Icosahedral Particles—The Uncommon Line. J. Struct. Biol. 1996, 116, 48–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, J.F.; Steven, A.C. Methods for Reconstructing Density Maps of “Single” Particles from Cryoelectron Micrographs to Subnanometer Resolution. J. Struct. Biol. 1999, 128, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Zivanov, J.; Nakane, T.; Forsberg, B.O.; Kimanius, D.; Hagen, W.J.; Lindahl, E.; Scheres, S.H. New Tools for Automated High-Resolution Cryo-EM Structure Determination in RELION-3. eLife 2018, 7, e42166. [Google Scholar] [CrossRef] [PubMed]
- Morin, A.; Eisenbraun, B.; Key, J.; Sanschagrin, P.C.; Timony, M.A.; Ottaviano, M.; Sliz, P. Collaboration Gets the Most out of Software. eLife 2013, 2, e01456. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein Structure and Function Prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.Y.-R.; Song, Y.; Barad, B.A.; Cheng, Y.; Fraser, J.S.; DiMaio, F. Automated Structure Refinement of Macromolecular Assemblies from Cryo-EM Maps Using Rosetta. eLife 2016, 5, e17219. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Effantin, G.; Fujiwara, A.; Kawasaki, T.; Yamada, T.; Schoehn, G. High Resolution Structure of the Mature Capsid of Ralstonia solanacearum Bacteriophage ϕRSA1 by Cryo-Electron Microscopy. Int. J. Mol. Sci. 2021, 22, 11053. https://doi.org/10.3390/ijms222011053
Effantin G, Fujiwara A, Kawasaki T, Yamada T, Schoehn G. High Resolution Structure of the Mature Capsid of Ralstonia solanacearum Bacteriophage ϕRSA1 by Cryo-Electron Microscopy. International Journal of Molecular Sciences. 2021; 22(20):11053. https://doi.org/10.3390/ijms222011053
Chicago/Turabian StyleEffantin, Grégory, Akiko Fujiwara, Takeru Kawasaki, Takashi Yamada, and Guy Schoehn. 2021. "High Resolution Structure of the Mature Capsid of Ralstonia solanacearum Bacteriophage ϕRSA1 by Cryo-Electron Microscopy" International Journal of Molecular Sciences 22, no. 20: 11053. https://doi.org/10.3390/ijms222011053
APA StyleEffantin, G., Fujiwara, A., Kawasaki, T., Yamada, T., & Schoehn, G. (2021). High Resolution Structure of the Mature Capsid of Ralstonia solanacearum Bacteriophage ϕRSA1 by Cryo-Electron Microscopy. International Journal of Molecular Sciences, 22(20), 11053. https://doi.org/10.3390/ijms222011053