Computational Evolution of Beta-2-Microglobulin Binding Peptides for Nanopatterned Surface Sensors

 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Results and Discussion
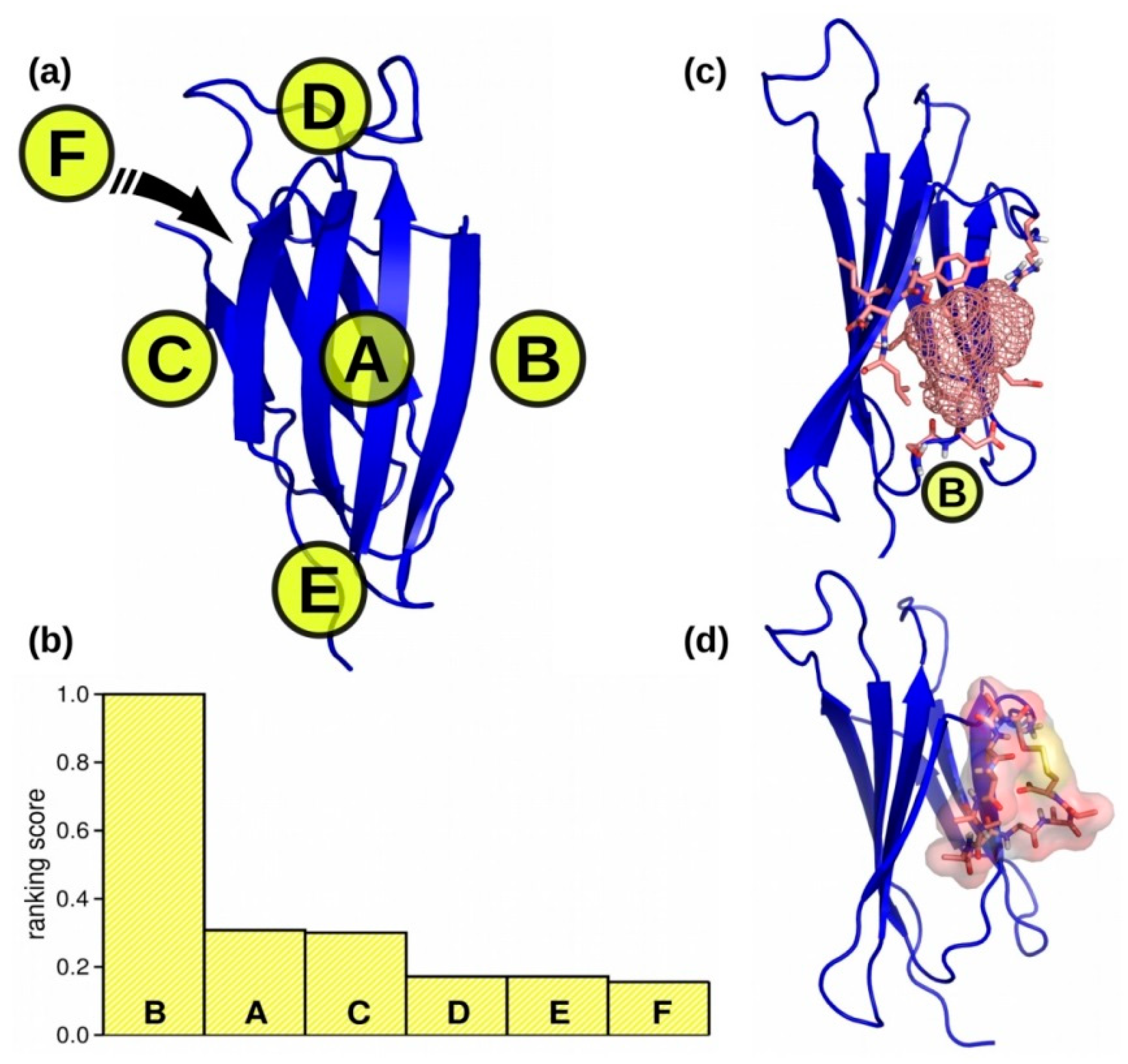
2.1. Biding Site Selection
2.2. Peptides Generation
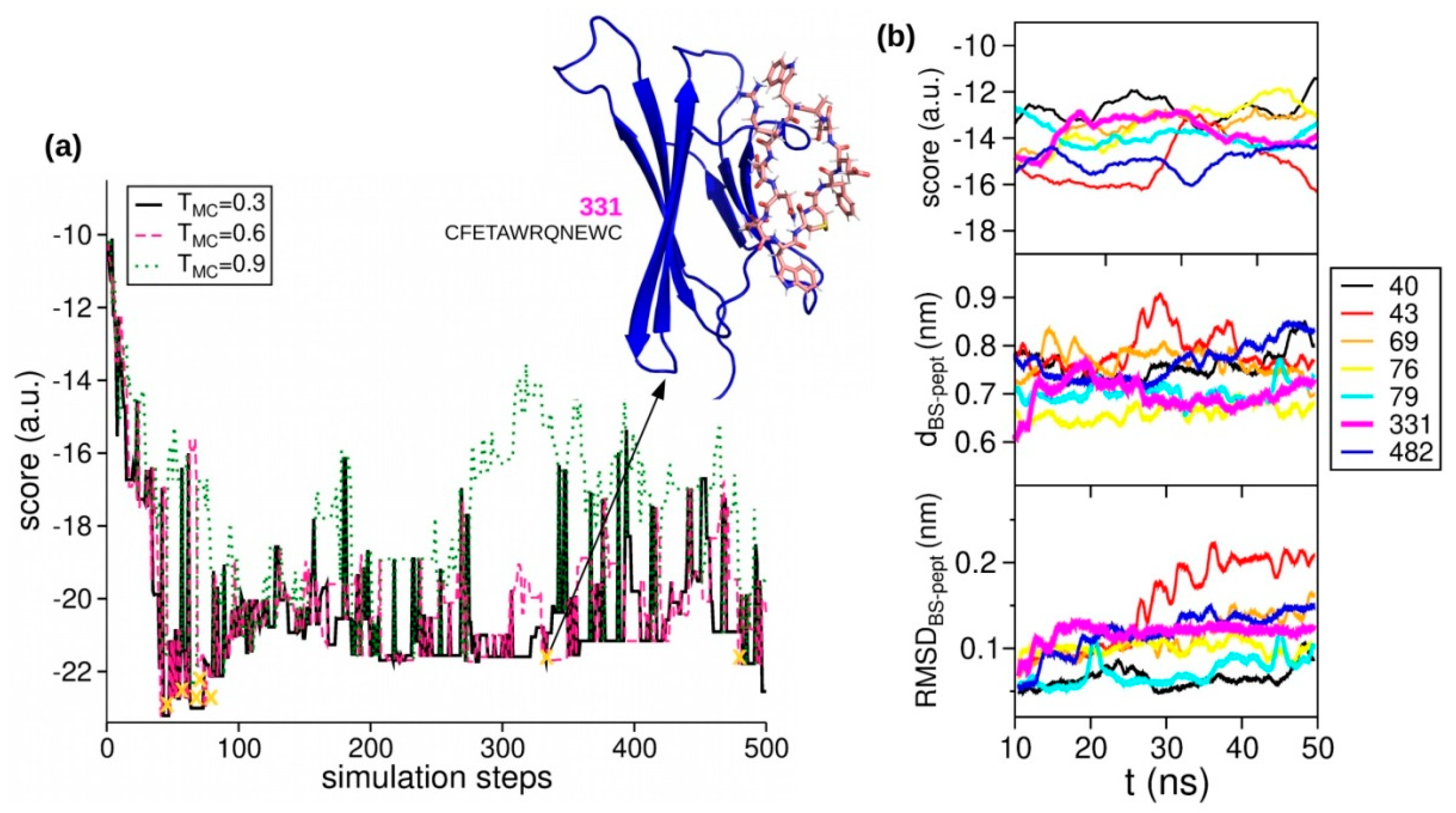
2.3. Peptides Screening
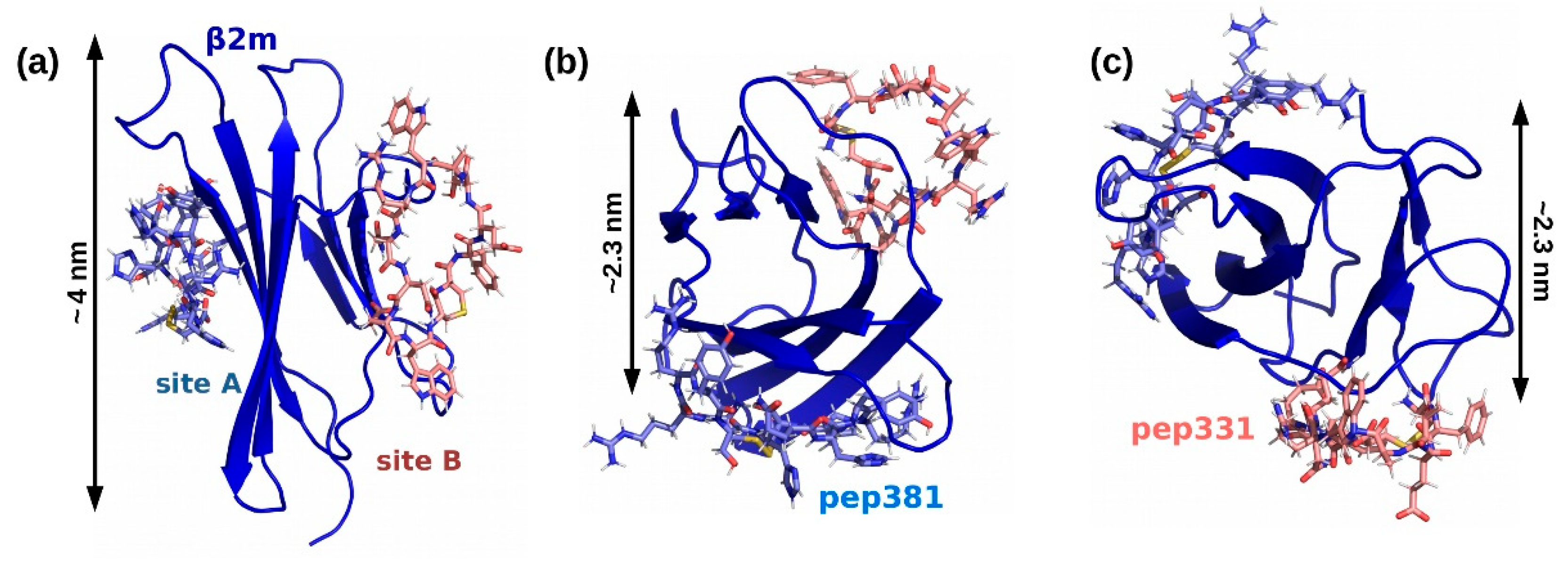
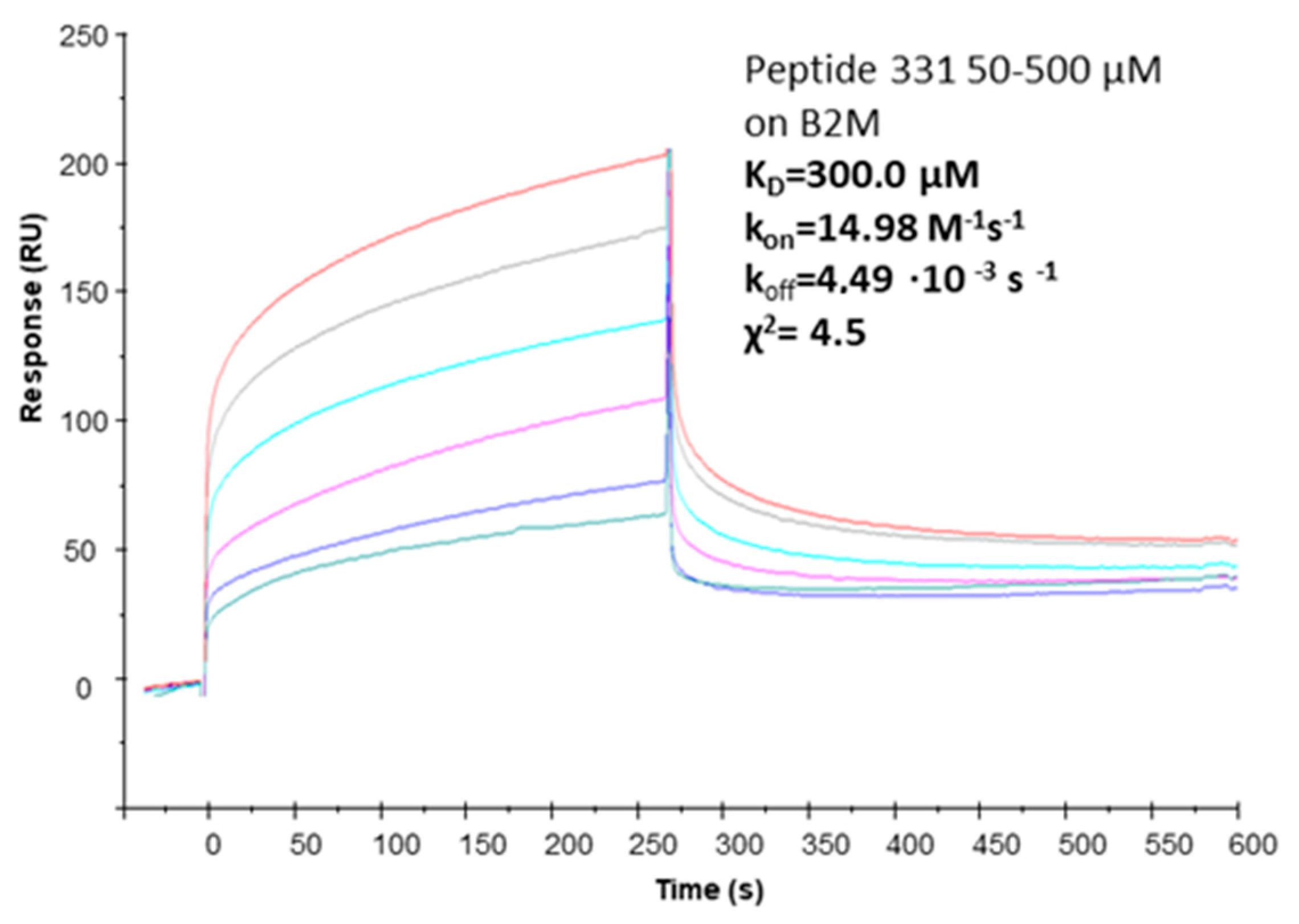
2.4. Affinity Measures
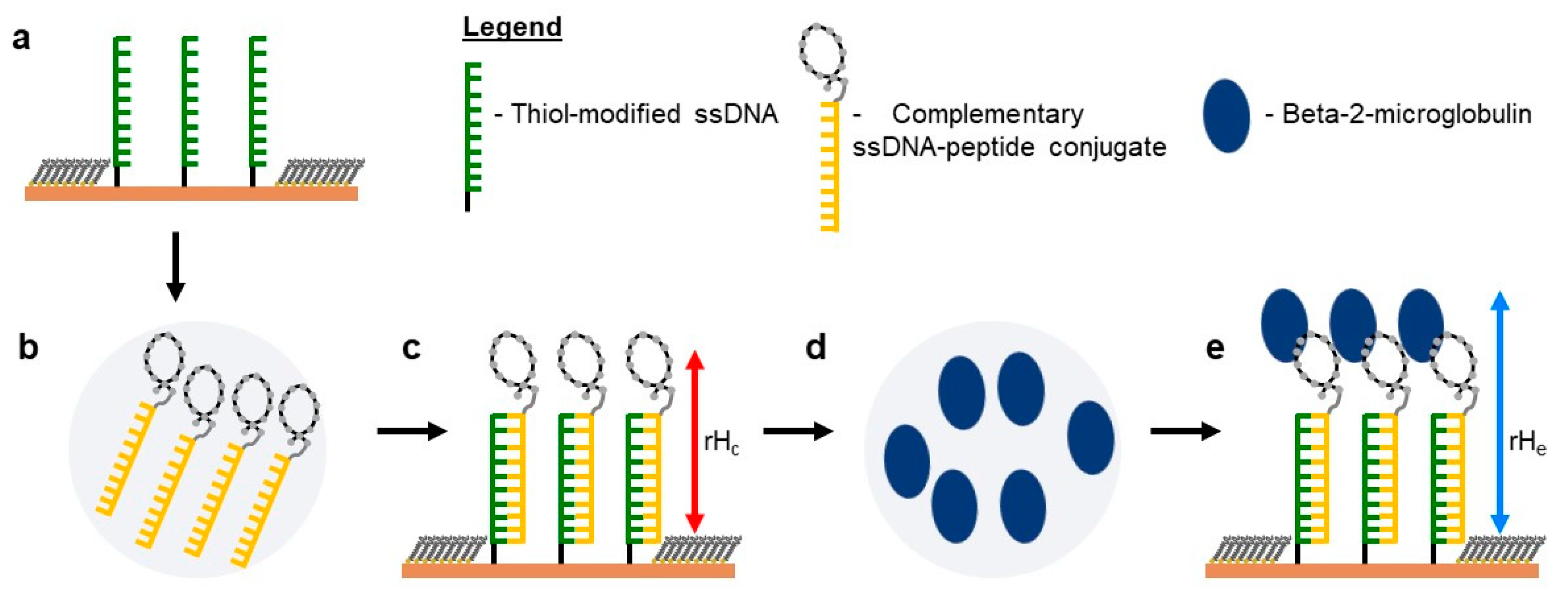
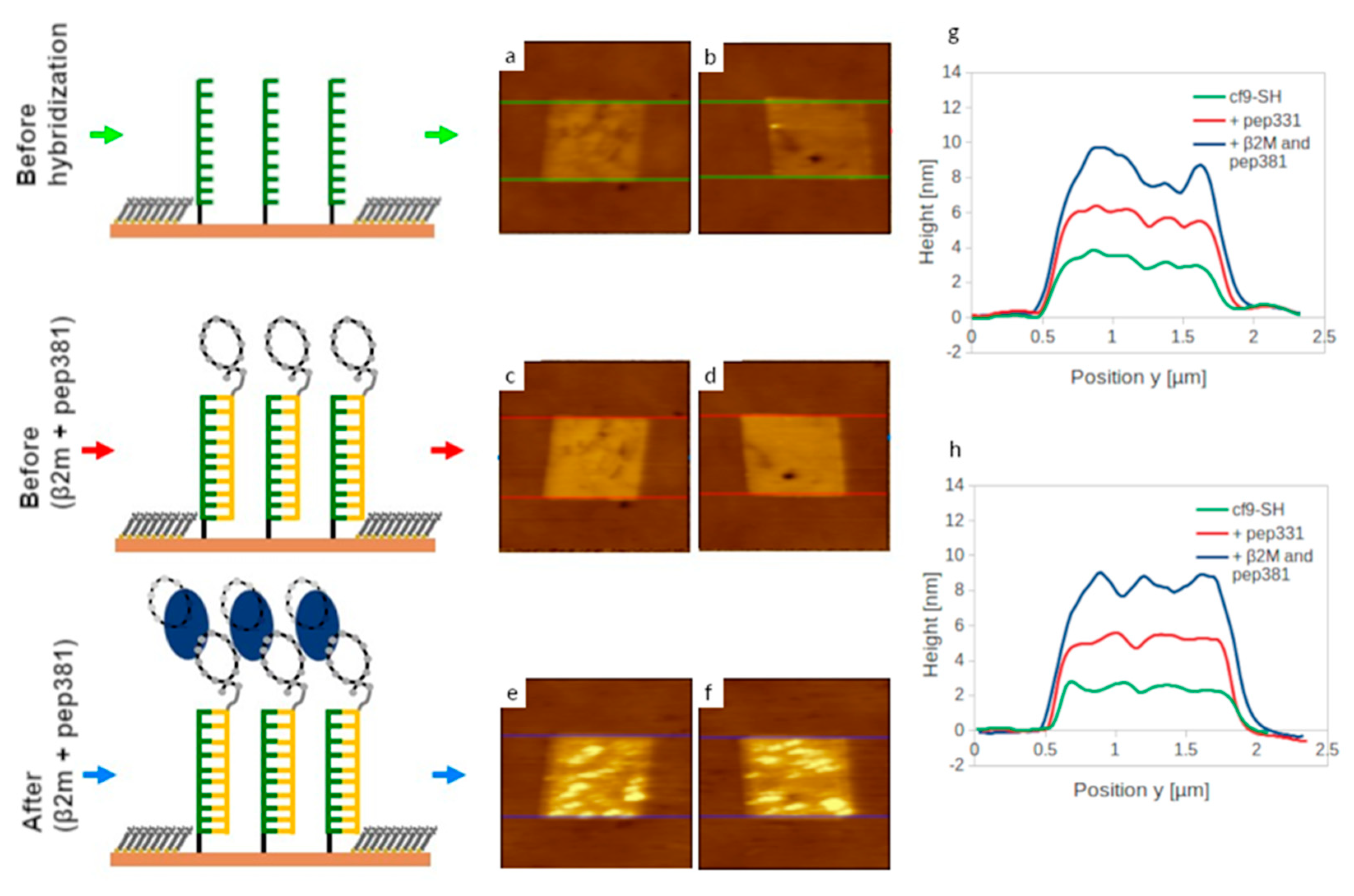
2.5. Self-Assembly of Functional dsDNA–peptide Nanopatch
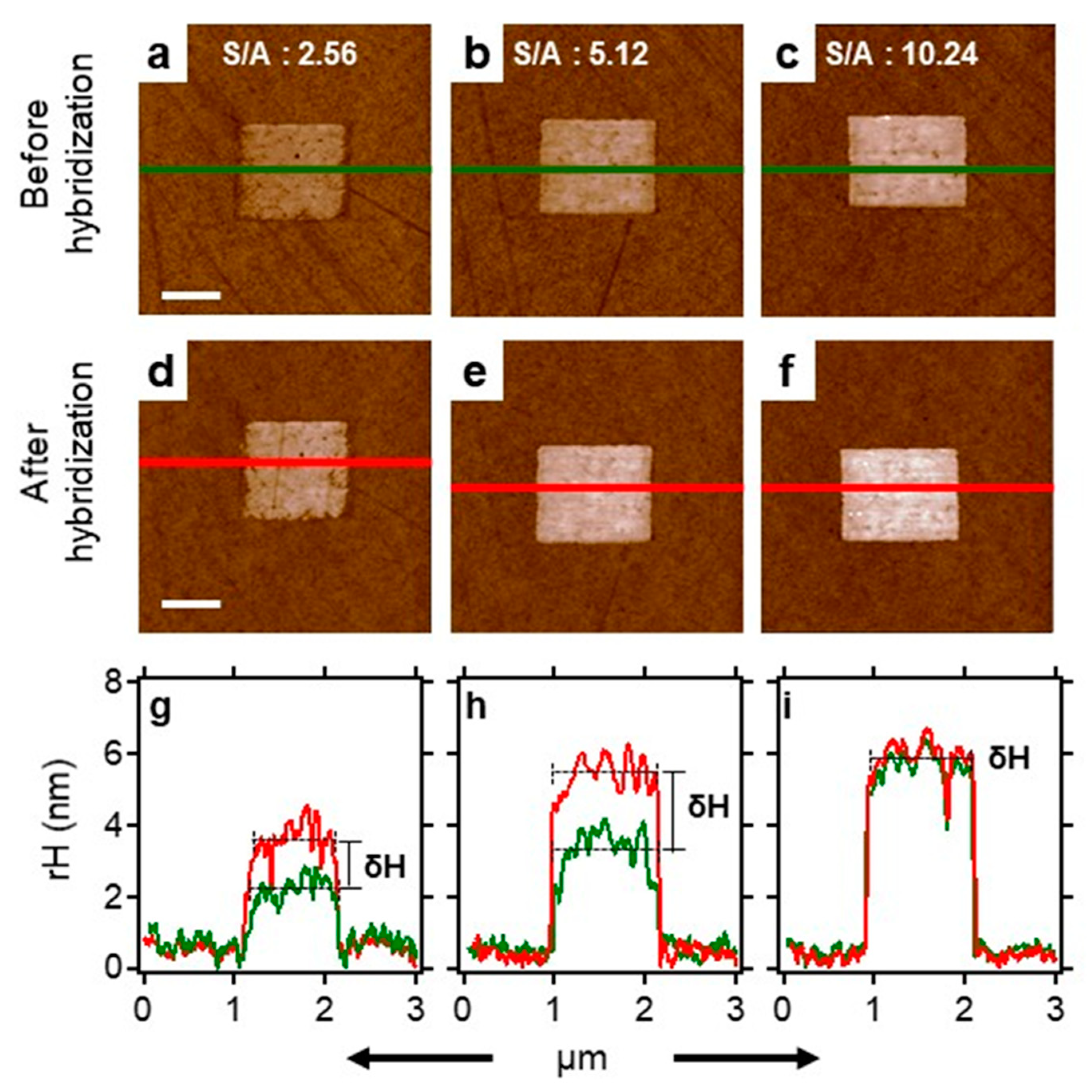
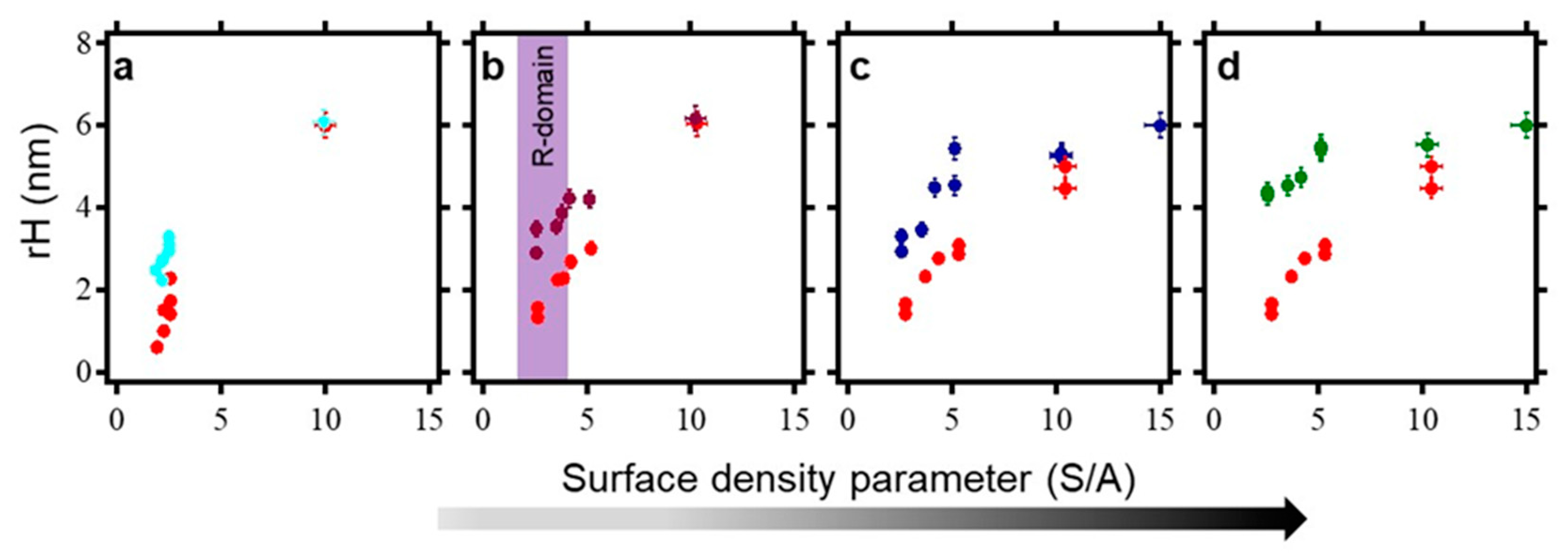
2.6. Surface Density Controls the Hybridization Efficiency of ssDNA Nanopatch
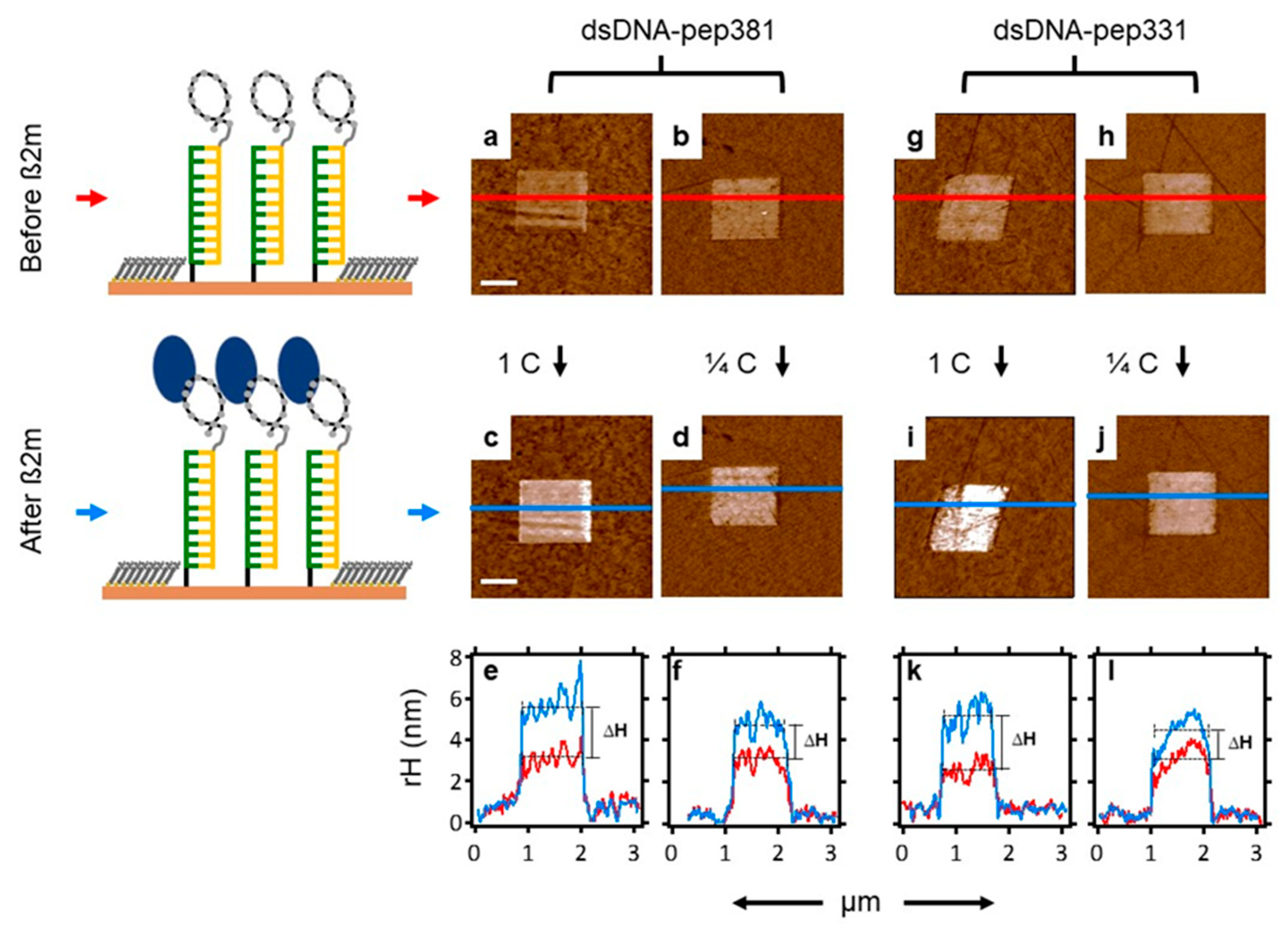
2.7. Recognition of β2m by dsDNA–peptide Conjugate Nanopatches
2.8. Competition Assay
3. Materials and Methods
3.1. Binding Site Selection
3.2. Peptide Design
3.3. Molecular Dynamics Simulations
3.4. Materials
3.5. Surface Plasmon Resonance
3.6. ssDNA–peptide Conjugates Preparation
3.7. Preparation of Ultra-Flat Gold Substrate
3.8. Preparation of Top-Oligo-Ethylene-Glycol SAM (TOEGSAM) on the Ultra-Flat Gold Substrate
3.9. Nanografting of Thiol-Modified ssDNA in Contact Mode
3.10. Hybridization with Complementary ssDNA–Peptide Conjugates
3.11. Beta-2-Microglobulin Binding Assay
3.12. AFM Imaging of dsDNA–peptide Assemblages before and after the β2m Recognition Assay
3.13. Competition Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ß2m | Beta-2-microglobulin |
| DNA | Deoxyribonucleic acid |
| ss | Single stranded |
| ds | Double stranded |
| AFM | Atomic force microscopy |
| SPR | Surface plasmon resonance |
| DDI | DNA-directed immobilization |
| MD | Molecular dynamics |
| REMD | Replica-exchange molecular dynamics |
| RMSD | Root mean square deviation |
| KD | Dissociation constant |
| S/A | Surface density parameter |
| BS | Binding site |
References
- Liu, Y.; Miyoshi, H.; Nakamura, M. Nanomedicine for drug delivery and imaging: A promising avenue for cancer therapy and diagnosis using targeted functional nanoparticles. Int. J. Cancer 2007, 120, 2527–2537. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, R.; Yang, L.; Tan, W. Nucleic acid conjugated nanomaterials for enhanced molecular recognition. ACS Nano 2009, 3, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Wink, T.; Van Zuilen, S.; Bult, A.; Van Bennekom, W. Self-assembled monolayers for biosensors. Analyst 1997, 122, 43R–50R. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Kaur, N.; Comini, E. The role of self-assembled monolayers in electronic devices. J. Mater. Chem. C 2020, 8, 3938–3955. [Google Scholar] [CrossRef]
- Bolivar, J.M.; Gallego, F.L. Characterization and evaluation of immobilized enzymes for applications in flow-reactors. Curr. Opin. Green Sustain. Chem. 2020. [Google Scholar] [CrossRef]
- Szymczak, L.C.; Kuo, H.-Y.; Mrksich, M. Peptide arrays: Development and application. Anal. Chem. 2018, 90, 266–282. [Google Scholar] [CrossRef]
- Niemeyer, C.M. Functional devices from DNA and proteins. Nano Today 2007, 2, 42–52. [Google Scholar] [CrossRef]
- Niemeyer, C.M.; Sano, T.; Smith, C.L.; Cantor, C.R. Oligonucleotide-directed self-assembly of proteins: Semisynthetic DNA—streptavidin hybrid molecules as connectors for the generation of macroscopic arrays and the construction of supramolecular bioconjugates. Nucleic Acids Res. 1994, 22, 5530–5539. [Google Scholar] [CrossRef]
- Ganau, M.; Bosco, A.; Palma, A.; Corvaglia, S.; Parisse, P.; Fruk, L.; Beltrami, A.; Cesselli, D.; Casalis, L.; Scoles, G. A DNA-based nano-immunoassay for the label-free detection of glial fibrillary acidic protein in multicell lysates. Nanomed.: Nanotechnol. Biol. Med. 2015, 11, 293–300. [Google Scholar] [CrossRef]
- Bano, F.; Fruk, L.; Sanavio, B.; Glettenberg, M.; Casalis, L.; Niemeyer, C.M.; Scoles, G. Toward multiprotein nanoarrays using nanografting and DNA directed immobilization of proteins. Nano Lett. 2009, 9, 2614–2618. [Google Scholar] [CrossRef]
- Bosco, A.; Ambrosetti, E.; Mavri, J.; Capaldo, P.; Casalis, L. Miniaturized aptamer-based assays for protein detection. Chemosensors 2016, 4, 18. [Google Scholar] [CrossRef]
- Deutscher, S. Phage display to detect and identify autoantibodies in disease. N. Engl. J. Med. 2019, 381, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P.; Petrenko, V.A. Phage Display. Chem. Rev. 1997, 97, 391–410. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- La Manna, S.; Lopez-Sanz, L.; Mercurio, F.A.; Fortuna, S.; Leone, M.; Gomez-Guerrero, C.; Marasco, D. Chimeric peptidomimetics of SOCS 3 able to interact with JAK2 as anti-inflammatory compounds. ACS Med. Chem. Lett. 2020, 11, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Aiello, C.; Grieco, P.; Marasco, D. Targeting “undruggable” proteins: Design of synthetic cyclopeptides. Curr. Med. Chem. 2016, 23, 748–762. [Google Scholar] [CrossRef]
- Soler, M.A.; Fortuna, S.; Scoles, G. Computational design of peptides as probes for the recognition of protein biomarkers. Eur. Biophys. J. 2015, 44, S149. [Google Scholar]
- Soler, M.A.; Rodriguez, A.; Russo, A.; Adedeji, A.F.; Foumthuim, C.J.D.; Cantarutti, C.; Ambrosetti, E.; Casalis, L.; Corazza, A.; Scoles, G.; et al. Computational design of cyclic peptides for the customized oriented immobilization of globular proteins. Phys. Chem. Chem. Phys. 2017, 19, 2740–2748. [Google Scholar] [CrossRef]
- Russo, A.; Scognamiglio, P.L.; Enriquez, R.P.H.; Santambrogio, C.; Grandori, R.; Marasco, D.; Giordano, A.; Scoles, G.; Fortuna, S. In silico generation of peptides by replica exchange Monte Carlo: Docking-based optimization of maltose-binding-protein ligands. PLoS ONE 2015, 10, e0133571. [Google Scholar] [CrossRef]
- Lathbridge, A.; Mason, J.M. Computational competitive and negative design to derive a specific cJun antagonist. Biochemistry 2018, 57, 6108–6118. [Google Scholar] [CrossRef]
- Soler, M.A.; Medagli, B.; Semrau, M.S.; Storici, P.; Bajc, G.; De Marco, A.; Laio, A.; Fortuna, S. A consensus protocol for the in silico optimisation of antibody fragments. Chem. Commun. 2019, 55, 14043–14046. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, R.; Soler, M.A.; Laio, A.; Cossio, P. PARCE: Protocol for amino acid refinement through computational evolution. Comput. Phys. Commun. 2021, 260, 107716. [Google Scholar] [CrossRef]
- Enriquez, R.P.H.; Pavan, S.; Benedetti, F.; Tossi, A.; Savoini, A.; Berti, F.; Laio, A. Designing short peptides with high affinity for organic molecules: A combined docking, molecular dynamics, and Monte Carlo approach. J. Chem. Theory Comput. 2012, 8, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Guida, F.; Battisti, A.; Gladich, I.; Buzzo, M.; Marangon, E.; Giodini, L.; Toffoli, G.; Laio, A.; Berti, F. Peptide biosensors for anticancer drugs: Design in silico to work in denaturizing environment. Biosens. Bioelectron. 2018, 100, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Chi, L.A.; Vargas, M.C. In silico design of peptides as potential ligands to resistin. J. Mol. Model 2020, 26, 1–14. [Google Scholar] [CrossRef]
- Verdone, G.; Corazza, A.; Viglino, P.; Pettirossi, F.; Giorgetti, S.; Mangione, P.; Andreola, A.; Stoppini, M.; Bellotti, V.; Esposito, G. The solution structure of human β2-microglobulin reveals the prodromes of its amyloid transition. Protein Sci. 2009, 11, 487–499. [Google Scholar] [CrossRef]
- Saper, M.; Bjorkman, P.; Wiley, D. Refined structure of the human histocompatibility antigen HLA-A2 at 2.6 Å resolution. J. Mol. Biol. 1991, 219, 277–319. [Google Scholar] [CrossRef]
- Fung, E.T. A recipe for proteomics diagnostic test development: The OVA1 test, from biomarker discovery to FDA clearance. Clin. Chem. 2010, 56, 327–329. [Google Scholar] [CrossRef]
- Locatelli, F.; Mastrangelo, F.; Redaelli, B.; Ronco, C.; Marcelli, D.; La Greca, G.; Orlandini, G. The Italian Cooperative Dialysis Study Group. Effects of different membranes and dialysis technologies on patient treatment tolerance and nutritional parameters. Kidney Int. 1996, 50, 1293–1302. [Google Scholar] [CrossRef]
- Xu, S.; Liu, G.-Y. Nanometer-scale fabrication by simultaneous nanoshaving and molecular self-assembly. Langmuir 1997, 13, 127–129. [Google Scholar] [CrossRef]
- Mirmomtaz, E.; Castronovo, M.; Grunwald, C.; Bano, F.; Scaini, D.; Ensafi, A.A.; Scoles, G.; Casalis, L. Quantitative Study of the Effect of Coverage on the Hybridization Efficiency of Surface-Bound DNA Nanostructures. Nano Lett. 2008, 8, 4134–4139. [Google Scholar] [CrossRef] [PubMed]
- Lavi, A.; Ngan, C.H.; Movshovitz-Attias, D.; Bohnuud, T.; Yueh, C.; Beglov, D.; Schueler-Furman, O.; Dima, K. Detection of peptide-binding sites on protein surfaces: The first step toward the modeling and targeting of peptide-mediated interactions. Proteins 2013, 81, 2096–2105. [Google Scholar] [CrossRef] [PubMed]
- Brenke, R.; Kozakov, D.; Chuang, G.-Y.; Beglov, D.; Hall, D.; Landon, M.R.; Mattos, C.; Vajda, S. Fragment-based identification of druggable ‘hot spots’ of proteins using Fourier domain correlation techniques. Bioinformatics 2009, 25, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide folding: When simulation meets experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Iba, Y. Extended ensemble Monte Carlo. Int. J. Mod. Phys. C 2001, 12, 623–656. [Google Scholar] [CrossRef]
- Salmaso, V.; Sturlese, M.; Cuzzolin, A.; Moro, S. Exploring protein-peptide recognition pathways using a supervised molecular dynamics approach. Structure 2017, 25, 655–662. [Google Scholar] [CrossRef]
- Castronovo, M.; Lucesoli, A.; Parisse, P.; Kurnikova, A.; Malhotra, A.; Grassi, M.; Grassi, G.; Scaggiante, B.; Casalis, L.; Scoles, G. Two-dimensional enzyme diffusion in laterally confined DNA monolayers. Nat. Commun. 2011, 2, 297. [Google Scholar] [CrossRef]
- Castronovo, M.; Radovic, S.; Grunwald, C.; Casalis, L.; Morgante, M.; Scoles, G. Control of Steric Hindrance on Restriction Enzyme Reactions with Surface-Bound DNA Nanostructures. Nano Lett. 2008, 8, 4140–4145. [Google Scholar] [CrossRef]
- Di Natale, C.; Natale, C.F.; Florio, D.; Netti, P.A.; Morelli, G.; Ventre, M.; Marasco, D. Effects of surface nanopatterning on internalization and amyloid aggregation of the fragment 264-277 of Nucleophosmin 1. Colloids Surf. B Biointerfaces 2020, 197, 111439. [Google Scholar] [CrossRef]
- Bosco, A.; Bano, F.; Parisse, P.; Casalis, L.; DeSimone, A.; Micheletti, C. Hybridization in nanostructured DNA monolayers probed by AFM: Theory versus experiment. Nanoscale 2011, 4, 1734. [Google Scholar] [CrossRef]
- Adedeji, A.F.; Ambrosetti, E.; Casalis, L.; Castronovo, M. Spatially Resolved Peptide-DNA Nanoassemblages for Biomarker Detection: A Synergy of DNA-Directed Immobilization and Nanografting. In Advanced Structural Safety Studies; Springer: Berlin/Heidelberg, Germany, 2018; pp. 151–162. [Google Scholar]
- Trinh, C.H.; Smith, D.P.; Kalverda, A.P.; Phillips, S.E.; Radford, S.E. Crystal structure of monomeric human β-2-microglobulin reveals clues to its amyloidogenic properties. Proc. Natl. Acad. Sci. USA 2002, 99, 9771–9776. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Schafmeister, C.; Ross, W.; Romanovski, W. Leap; University of California: San Francisco, CA, USA, 1995. [Google Scholar]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Sreejit, G.; Ahmed, A.; Parveen, N.; Jha, V.; Valluri, V.L.; Ghosh, S.; Mukhopadhyay, S. The ESAT-6 Protein of Mycobacterium tuberculosis Interacts with Beta-2-Microglobulin (β2M) Affecting Antigen Presentation Function of Macrophage. PLOS Pathog. 2014, 10, e1004446. [Google Scholar] [CrossRef]
- Redhu, S.K.; Castronovo, M.; Nicholson, A.W. Digital Imprinting of RNA Recognition and Processing on a Self-Assembled Nucleic Acid Matrix. Sci. Rep. 2013, 3, srep02550. [Google Scholar] [CrossRef]
- Soler, M.A.; Fortuna, S.; De Marco, A.; Laio, A. Binding affinity prediction of nanobody–protein complexes by scoring of molecular dynamics trajectories. Phys. Chem. Chem. Phys. 2018, 20, 3438–3444. [Google Scholar] [CrossRef]
- Ochoa, R.; Soler, M.A.; Laio, A.; Cossio, P. Assessing the capability of in silico mutation protocols for predicting the finite temperature conformation of amino acids. Phys. Chem. Chem. Phys. 2018, 20, 25901–25909. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, R.; Laio, A.; Cossio, P. Predicting the Affinity of Peptides to Major Histocompatibility Complex Class II by Scoring Molecular Dynamics Simulations. J. Chem. Inf. Model. 2019, 59, 3464–3473. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, S.; Fogolari, F.; Scoles, G. Chelating effect in short polymers for the design of bidentate binders of increased affinity and selectivity. Sci. Rep. 2015, 5, 15633. [Google Scholar] [CrossRef] [PubMed]
- Soler, M.A.; Fortuna, S. Influence of Linker Flexibility on the Binding Affinity of Bidentate Binders. J. Phys. Chem. B 2017, 121, 3918–3924. [Google Scholar] [CrossRef]
- Liese, S.; Netz, R.R. Quantitative Prediction of Multivalent Ligand–Receptor Binding Affinities for Influenza, Cholera, and Anthrax Inhibition. ACS Nano 2018, 12, 4140–4147. [Google Scholar] [CrossRef] [PubMed]
- Zumbro, E.; Alexander-Katz, A. Influence of Binding Site Affinity Patterns on Binding of Multivalent Polymers. ACS Omega 2020, 5, 10774–10781. [Google Scholar] [CrossRef] [PubMed]
- Soler, M.A.; de Marco, A.; Fortuna, S. Molecular dynamics simulations and docking enable to explore the biophysical factors controlling the yields of engineered nanobodies. Sci. Rep. 2016, 6, 34869. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Sequence | Remark | KD (SPR) |
|---|---|---|---|
| pep381 | [CRRYSHQHYRHC] | cyclic with S-S bridge | 38 ± 9 µM (Ref. [18]) |
| pep331 | [CFETAWRQNEWC] | cyclic with S-S bridge | 300.0 µM (χ2 = 4.5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adedeji Olulana, A.F.; Soler, M.A.; Lotteri, M.; Vondracek, H.; Casalis, L.; Marasco, D.; Castronovo, M.; Fortuna, S. Computational Evolution of Beta-2-Microglobulin Binding Peptides for Nanopatterned Surface Sensors. Int. J. Mol. Sci. 2021, 22, 812. https://doi.org/10.3390/ijms22020812
Adedeji Olulana AF, Soler MA, Lotteri M, Vondracek H, Casalis L, Marasco D, Castronovo M, Fortuna S. Computational Evolution of Beta-2-Microglobulin Binding Peptides for Nanopatterned Surface Sensors. International Journal of Molecular Sciences. 2021; 22(2):812. https://doi.org/10.3390/ijms22020812
Chicago/Turabian StyleAdedeji Olulana, Abimbola Feyisara, Miguel A. Soler, Martina Lotteri, Hendrik Vondracek, Loredana Casalis, Daniela Marasco, Matteo Castronovo, and Sara Fortuna. 2021. "Computational Evolution of Beta-2-Microglobulin Binding Peptides for Nanopatterned Surface Sensors" International Journal of Molecular Sciences 22, no. 2: 812. https://doi.org/10.3390/ijms22020812
APA StyleAdedeji Olulana, A. F., Soler, M. A., Lotteri, M., Vondracek, H., Casalis, L., Marasco, D., Castronovo, M., & Fortuna, S. (2021). Computational Evolution of Beta-2-Microglobulin Binding Peptides for Nanopatterned Surface Sensors. International Journal of Molecular Sciences, 22(2), 812. https://doi.org/10.3390/ijms22020812