Sequence-Dependent Nanofiber Structures of Phenylalanine and Isoleucine Tripeptides



Abstract
1. Introduction
2. Results
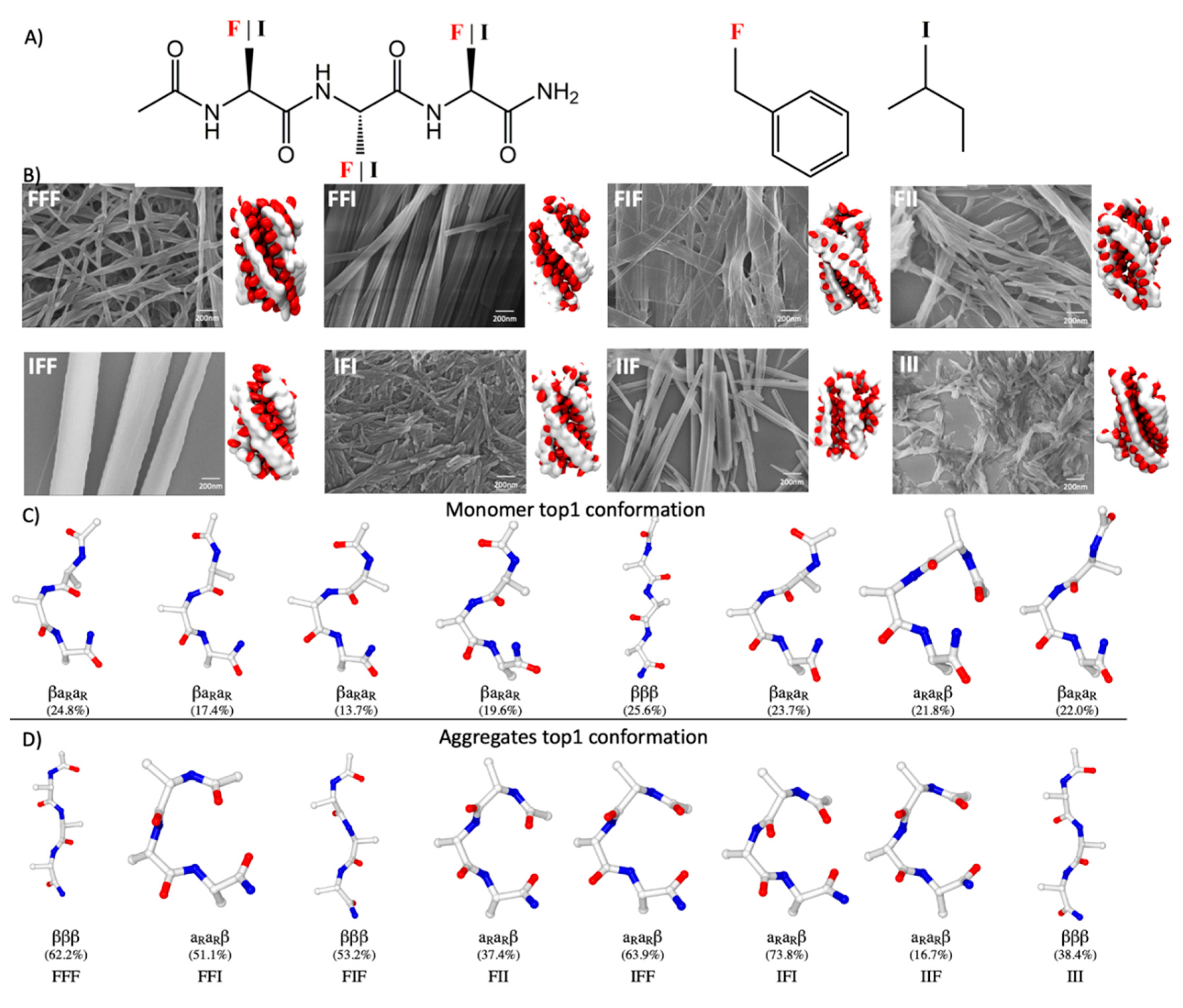
2.1. General Tendency of Model Tripeptides to Assemble into Fibrous Structures
2.2. Sequence Dependence of Peptide Conformations in Monomers and Assemblies
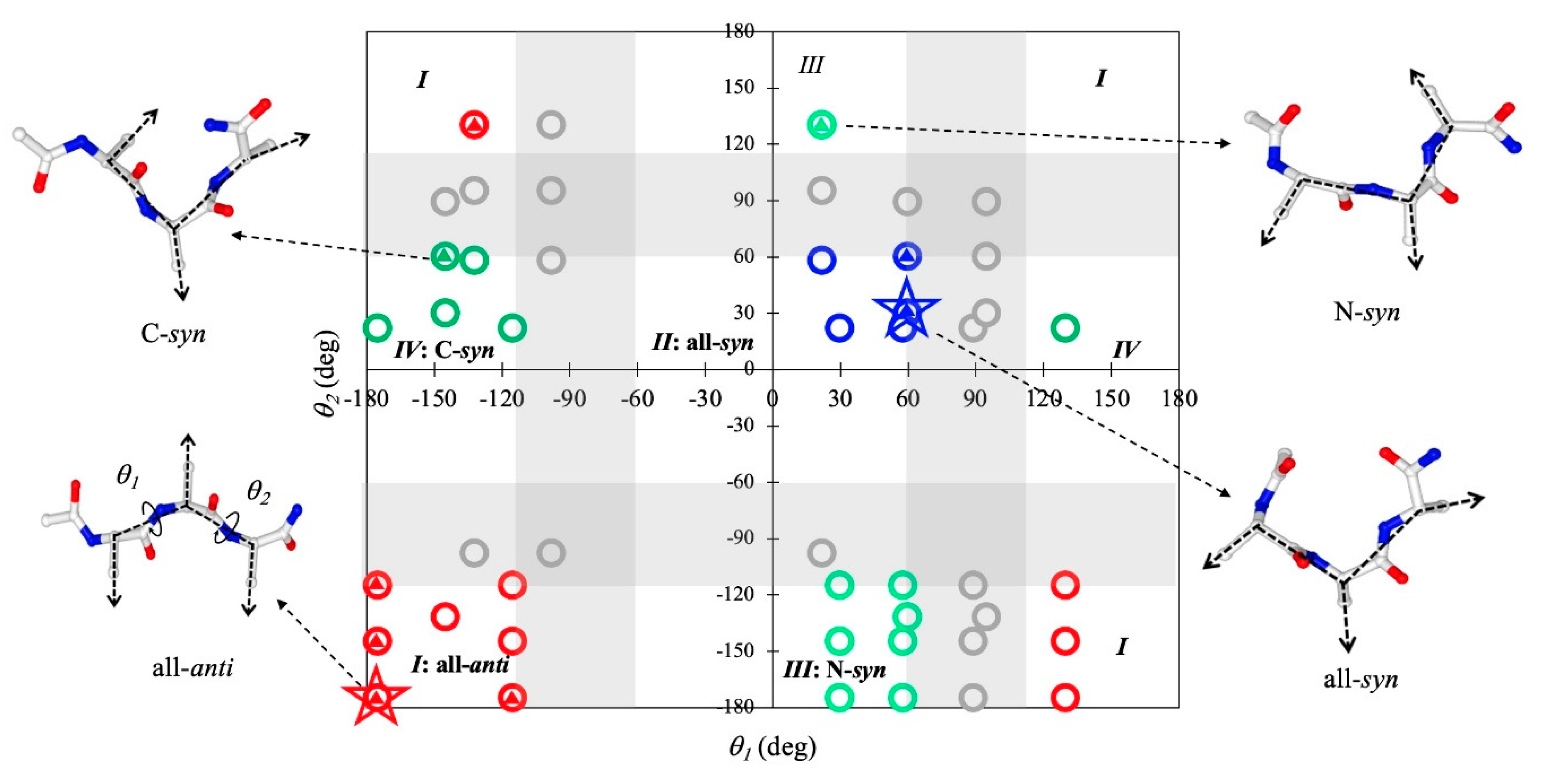
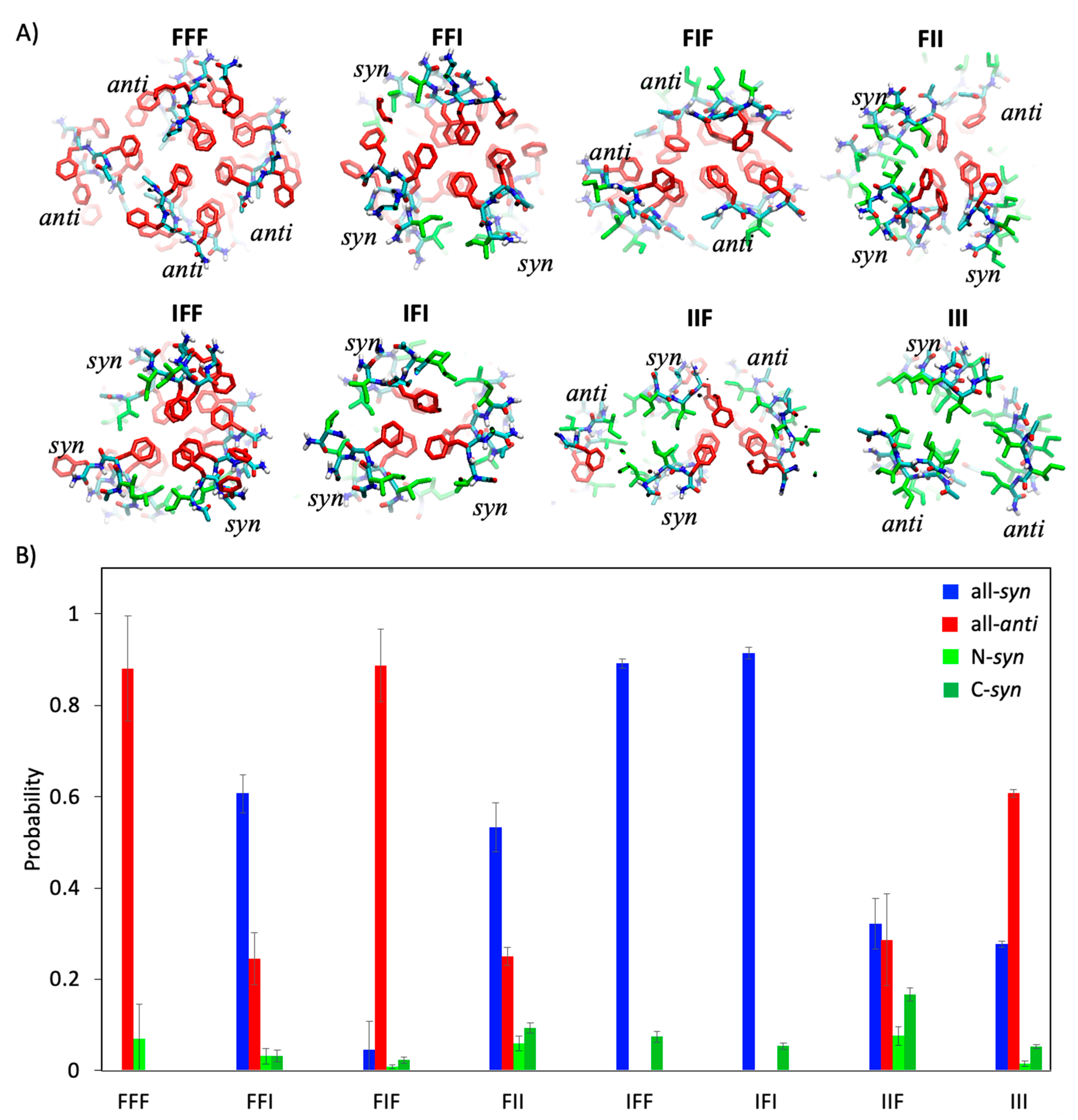
2.3. Geometric Features of Assembled Conformations of Tripeptides
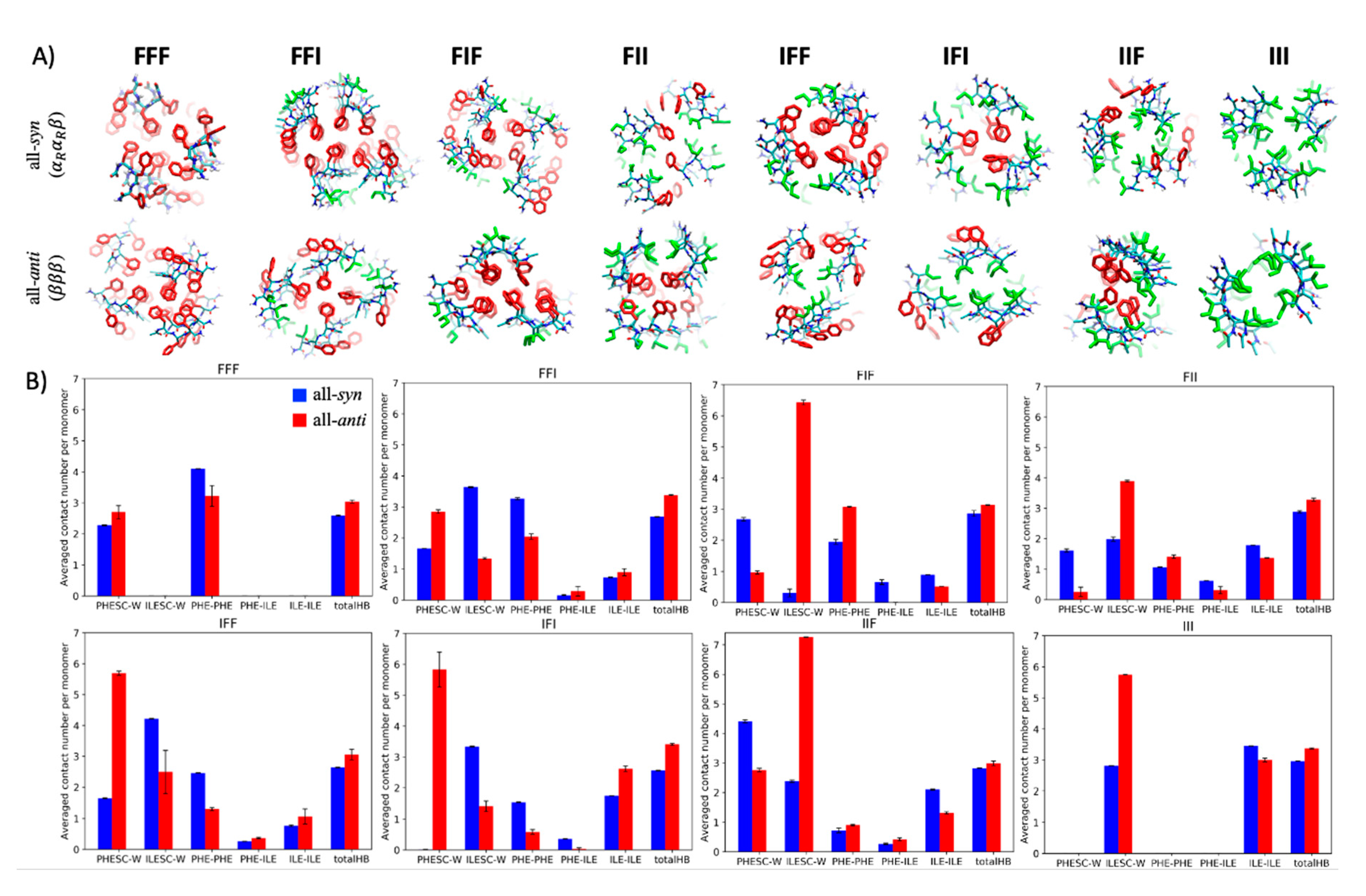
2.4. Sequence Dependence of Molecular Packing of Tripeptides in Fibrous Assemblies
2.5. Molecular Basis of Conformational Preference of Tripeptides in Self-Assembled Structures
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Preparation
4.3. Scanning Electron Microscopy
4.4. Models and Simulation Setup
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Amabilino, D.B.; Smith, D.K.; Steed, J.W. Supramolecular materials. Chem. Soc. Rev. 2017, 46, 2404–2420. [Google Scholar] [CrossRef]
- Makam, P.; Gazit, E. Minimalistic peptide supramolecular co-assembly: Expanding the conformational space for nanotechnology. Chem. Soc. Rev. 2018, 47, 3406–3420. [Google Scholar] [CrossRef]
- Wei, G.; Su, Z.; Reynolds, N.P.; Arosio, P.; Hamley, I.W.; Gazit, E.; Mezzenga, R. Self-assembling peptide and protein amyloids: From structure to tailored function in nanotechnology. Chem. Soc. Rev. 2017, 46, 4661–4708. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Yao, Y.; Wei, G. Expanding the structural diversity of peptide assemblies by coassembling dipeptides with diphenylalanine. Nanoscale 2020, 12, 3038–3049. [Google Scholar] [CrossRef] [PubMed]
- Moitra, P.; Kumar, K.; Kondaiah, P.; Bhattacharya, S. Efficacious anticancer drug delivery mediated by a pH-sensitive self-assembly of a conserved tripeptide derived from tyrosine kinase NGF receptor. Angew. Chem.-Int. Ed. 2014, 53, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.; Hakala, T.A.; Schnaider, L.; Bernardes, G.J.L.; Gazit, E.; Knowles, T.P.J. Biomimetic peptide self-assembly for functional materials. Nat. Rev. Chem. 2020, 1–20. [Google Scholar] [CrossRef]
- Sun, B.; Tao, K.; Jia, Y.; Yan, X.; Zou, Q.; Gazit, E.; Li, J. Photoactive properties of supramolecular assembled short peptides. Chem. Soc. Rev. 2019, 48, 4387–4400. [Google Scholar] [CrossRef]
- Frederix, P.W.J.M.; Patmanidis, I.; Marrink, S.J. Molecular simulations of self-assembling bio-inspired supramolecular systems and their connection to experiments. Chem. Soc. Rev. 2018, 47, 3470–3489. [Google Scholar] [CrossRef]
- Lampel, A.; Ulijn, R.V.; Tuttle, T. Guiding principles for peptide nanotechnology through directed discovery. Chem. Soc. Rev. 2018, 47, 3737–3758. [Google Scholar] [CrossRef]
- Wang, J.; Liu, K.; Xing, R.; Yan, X. Peptide self-assembly: Thermodynamics and kinetics. Chem. Soc. Rev. 2016, 45, 5589–5604. [Google Scholar] [CrossRef]
- Lakshmanan, A.; Hauser, C.A.E. Ultrasmall Peptides Self-Assemble into Diverse Nanostructures: Morphological Evaluation and Potential Implications. Int. J. Mol. Sci. 2011, 12, 5736–5746. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, A.; Cheong, D.W.; Accardo, A.; Di Fabrizio, E.; Riekel, C.; Hauser, C.A.E. Aliphatic peptides show similar self-assembly to amyloid core sequences, challenging the importance of aromatic interactions in amyloidosis. Proc. Natl. Acad. Sci. USA 2013, 110, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Yeh, M.Y.; Huang, C.T.; Lai, T.S.; Chen, F.Y.; Chu, N.T.; Tseng, D.T.H.; Hung, S.C.; Lin, H.C. Effect of Peptide Sequences on Supramolecular Interactions of Naphthaleneimide/Tripeptide Conjugates. Langmuir 2016, 32, 7630–7638. [Google Scholar] [CrossRef] [PubMed]
- Vargiu, A.V.; Iglesias, D.; Styan, K.E.; Waddington, L.J.; Easton, C.D.; Marchesan, S. Design of a hydrophobic tripeptide that self-assembles into amphiphilic superstructures forming a hydrogel biomaterial. Chem. Commun. 2016, 52, 5912–5915. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, J.K.; Nazareth, C.; Vandenberg, M.A.; Webber, M.J. Self-assembly of amphiphilic tripeptides with sequence-dependent nanostructure. Biomater. Sci. 2017, 5, 1526–1530. [Google Scholar] [CrossRef] [PubMed]
- Lampel, A.; McPhee, S.A.; Park, H.A.; Scott, G.G.; Humagain, S.; Hekstra, D.R.; Yoo, B.; Frederix, P.W.J.M.; Li, T.-D.; Abzalimov, R.R.; et al. Polymeric peptide pigments with sequence-encoded properties. Science 2017, 356, 1064–1068. [Google Scholar] [CrossRef]
- Vasudev, P.G.; Aravinda, S.; Shamala, N. Crystal structure of a tripeptide containing aminocyclododecane carboxylic acid: A supramolecular twisted parallel β-sheet in crystals. J. Pept. Sci. 2016, 22, 166–173. [Google Scholar] [CrossRef]
- Cringoli, M.C.; Bellotto, O.; De Zorzi, R.; Vargiu, A.V.; Marchesan, S. Self-Assembling l-d-l -Tripeptides Dance the Twist. Synlett 2020, 31, 434–438. [Google Scholar] [CrossRef]
- Hauser, C.A.E.; Deng, R.; Mishra, A.; Loo, Y.; Khoe, U.; Zhuang, F.; Cheong, D.W.; Accardo, A.; Sullivan, M.B.; Riekel, C.; et al. Natural tri- to hexapeptides self-assemble in water to amyloid beta-type fiber aggregates by unexpected alpha-helical intermediate structures. Proc. Natl. Acad. Sci. USA 2011, 108, 1361–1366. [Google Scholar] [CrossRef]
- Li, J.; Zhan, Z.; Du, X.; Wang, J.; Hong, B.; Xu, B. Selection of Secondary Structures of Heterotypic Supramolecular Peptide Assemblies by an Enzymatic Reaction. Angew. Chem. Int. Ed. 2018, 57, 11716–11721. [Google Scholar] [CrossRef]
- Bera, S.; Mondal, S.; Xue, B.; Shimon, L.J.W.; Cao, Y.; Gazit, E. Rigid helical-like assemblies from a self-aggregating tripeptide. Nat. Mater. 2019, 18, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, Y.; Sato, H.; Kayano, Y.; Yamaki, N.; Izato, Y.I.; Miyake, A.; Naito, A.; Kawamura, I. Self-assembly of tripeptides into γ-turn nanostructures. Phys. Chem. Chem. Phys. 2019, 21, 10879–10883. [Google Scholar] [CrossRef] [PubMed]
- Frederix, P.W.J.M.; Scott, G.G.; Abul-Haija, Y.M.; Kalafatovic, D.; Pappas, C.G.; Javid, N.; Hunt, N.T.; Ulijn, R.V.; Tuttle, T. Exploring the sequence space for (tri-)peptide self-assembly to design and discover new hydrogels. Nat. Chem. 2015, 7, 30–37. [Google Scholar] [CrossRef]
- ten Brummelhuis, N.; Wilke, P.; Börner, H.G. Identification of Functional Peptide Sequences to Lead the Design of Precision Polymers. Macromol. Rapid Commun. 2017, 38, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Varenik, M.; Bloch, D.N.; Atsmon-Raz, Y.; Jacoby, G.; Adler-Abramovich, L.; Shimon, L.J.W.; Beck, R.; Miller, Y.; Regev, O.; et al. A minimal length rigid helical peptide motif allows rational design of modular surfactants. Nat. Commun. 2017, 8, e14018. [Google Scholar] [CrossRef] [PubMed]
- Bera, S.; Mondal, S.; Tang, Y.; Jacoby, G.; Arad, E.; Guterman, T.; Jelinek, R.; Beck, R.; Wei, G.; Gazit, E. Deciphering the Rules for Amino Acid Co-Assembly Based on Interlayer Distances. ACS Nano 2019, 13, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Sahibbeyli, V.; Yildiz, D.B.; Papir, G.; Dede, Y.; Demirel, G. The Role of Molecular Structure of Phenylalanine Peptides on the Formation of Vertically Aligned Ordered Bionanostructures: Implications for Sensing Application. ACS Appl. Nano Mater. 2020, 3, 4305–4313. [Google Scholar] [CrossRef]
- Bera, S.; Mondal, S.; Rencus-Lazar, S.; Gazit, E. Organization of Amino Acids into Layered Supramolecular Secondary Structures. Acc. Chem. Res. 2018, 51, 2187–2197. [Google Scholar] [CrossRef]
- Sahoo, J.K.; Nazareth, C.; VandenBerg, M.A.; Webber, M.J. Aromatic identity, electronic substitution, and sequence in amphiphilic tripeptide self-assembly. Soft Matter 2018, 14, 9168–9174. [Google Scholar] [CrossRef]
- Brown, N.; Lei, J.; Zhan, C.; Shimon, L.J.W.; Adler-Abramovich, L.; Wei, G.; Gazit, E. Structural Polymorphism in a Self-Assembled Tri-Aromatic Peptide System. ACS Nano 2018, 12, 3253–3262. [Google Scholar] [CrossRef]
- Abul-Haija, Y.M.; Scott, G.G.; Sahoo, J.K.; Tuttle, T.; Ulijn, R.V. Cooperative, ion-sensitive co-assembly of tripeptide hydrogels. Chem. Commun. 2017, 53, 9562–9565. [Google Scholar] [CrossRef] [PubMed]
- Frederix, P.W.J.M.; Ulijn, R.V.; Hunt, N.T.; Tuttle, T. Virtual screening for dipeptide aggregation: Toward predictive tools for peptide self-Assembly. J. Phys. Chem. Lett. 2011, 2, 2380–2384. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Schulten, K. Further optimization of a hybrid united-atom and coarse-grained force field for folding simulations: Improved backbone hydration and interactions between charged side chains. J. Chem. Theory Comput. 2012, 8, 4413–4424. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xiong, Q.; Jiang, Y.; Cai, X.; Yang, F.; Li, Z.; Han, W. Conformation Dependence of Diphenylalanine Self-Assembly Structures and Dynamics: Insights from Hybrid-Resolution Simulations. ACS Nano 2019, 13, 4455–4468. [Google Scholar] [CrossRef] [PubMed]
- Smadbeck, J.; Chan, K.H.; Khoury, G.A.; Xue, B.; Robinson, R.C.; Hauser, C.A.E.; Floudas, C.A. De Novo Design and Experimental Characterization of Ultrashort Self-Associating Peptides. PLoS Comput. Biol. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.H.; Xue, B.; Robinson, R.C.; Hauser, C.A.E. Systematic Moiety Variations of Ultrashort Peptides Produce Profound Effects on Self-Assembly, Nanostructure Formation, Hydrogelation, and Phase Transition. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Auer, S.; Dobson, C.M.; Vendruscolo, M. Characterization of the nucleation barriers for protein aggregation and amyloid formation. HFSP J. 2007, 1, 137–146. [Google Scholar] [CrossRef]
- Nicholas, S.; Rizzoli, C. Crystal structure of the tripeptide N-(benzyloxycarbonyl) glycylglycyl-L-norvaline. Acta Crystallogr. Sect. E Struct. Rep. Online 2015, 71, o216–o217. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| all-anti | ||||
| Sequence [ref ID] | DYF [16] | Ac-YLD [35] | Boc-Leu-Val-Ac12c-OMe [17] | |
| Conformation states a | βββ | βββ | ββαR | |
| RMSD (Å) | 0.291 | 0.822 | 0.744 | |
| all-syn | ||||
| Sequence [ref ID] | Ac-LLE [36] | YFD [16] | hyp-FF [21] | pro-FF [21] |
| Conformation states | αRαRβ | αLαLPPII | βαRβ | βαRβ |
| RMSD (Å) | 0.497 | 1.623 | 1.063 | 1.021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, Q.; Liu, Z.; Han, W. Sequence-Dependent Nanofiber Structures of Phenylalanine and Isoleucine Tripeptides. Int. J. Mol. Sci. 2020, 21, 8431. https://doi.org/10.3390/ijms21228431
Xiong Q, Liu Z, Han W. Sequence-Dependent Nanofiber Structures of Phenylalanine and Isoleucine Tripeptides. International Journal of Molecular Sciences. 2020; 21(22):8431. https://doi.org/10.3390/ijms21228431
Chicago/Turabian StyleXiong, Qinsi, Ziye Liu, and Wei Han. 2020. "Sequence-Dependent Nanofiber Structures of Phenylalanine and Isoleucine Tripeptides" International Journal of Molecular Sciences 21, no. 22: 8431. https://doi.org/10.3390/ijms21228431
APA StyleXiong, Q., Liu, Z., & Han, W. (2020). Sequence-Dependent Nanofiber Structures of Phenylalanine and Isoleucine Tripeptides. International Journal of Molecular Sciences, 21(22), 8431. https://doi.org/10.3390/ijms21228431