
Improving CAR T-Cell Persistence

 , ,
, ,
Abstract
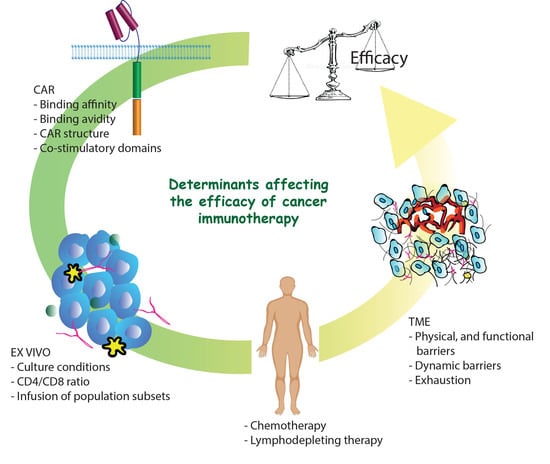
{kind=link}
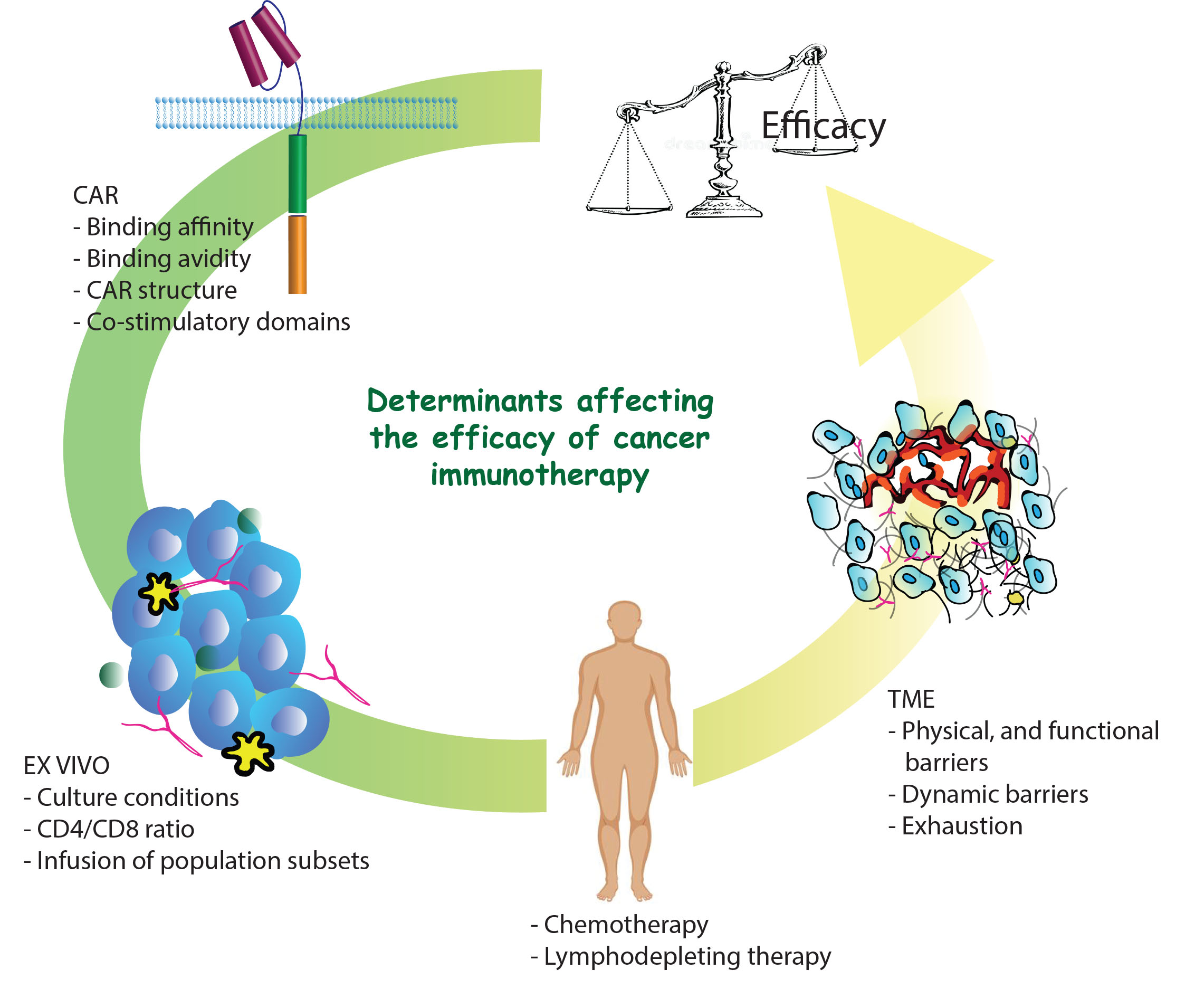
{kind=link}
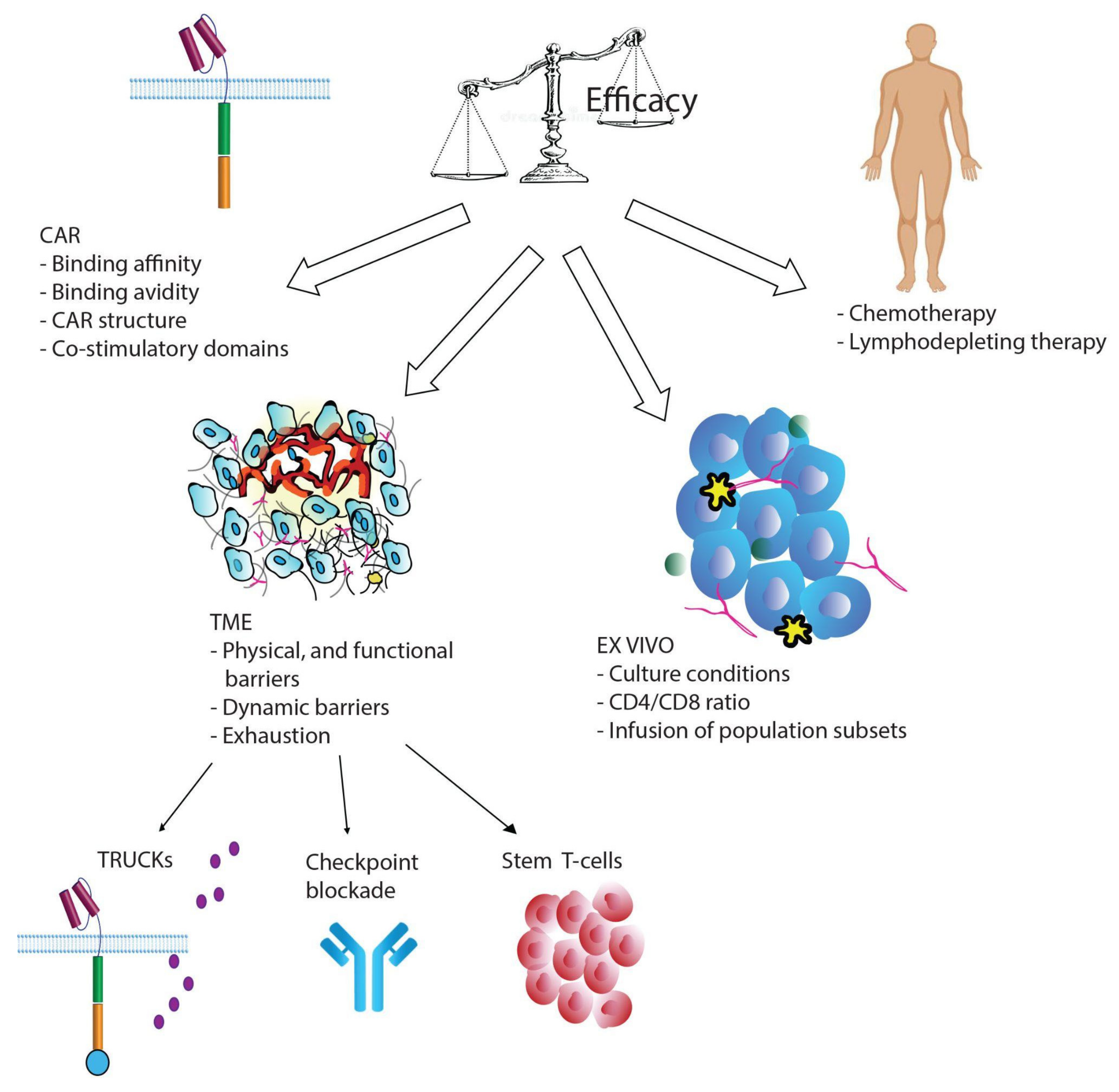
{kind=link}
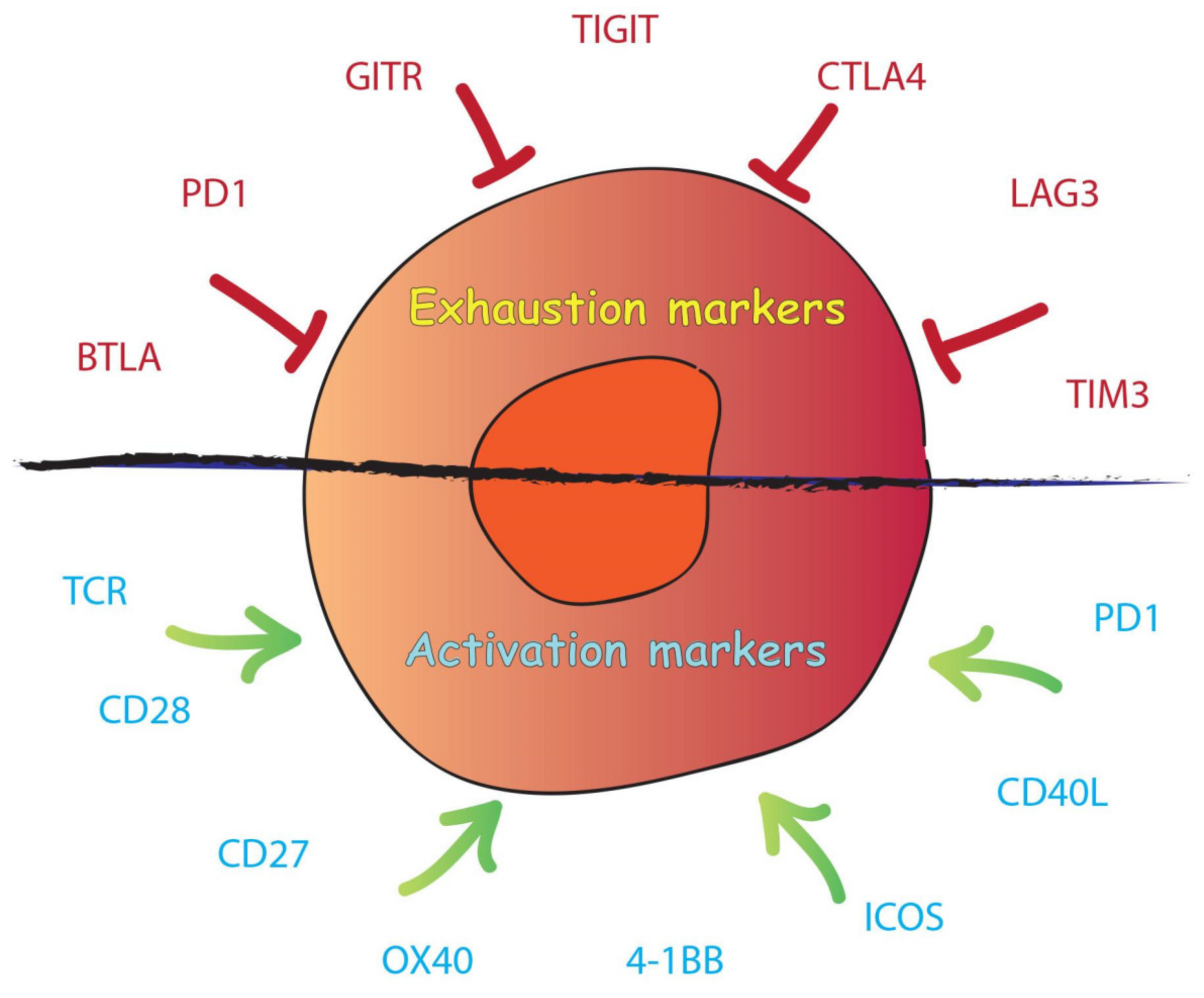
{kind=link}
1. Introduction
2. Factors Influencing T-Cell Persistence in Clinical Settings
2.1. CAR Construct
2.2. Ex Vivo Manipulation and Patient Lymphodepletion
2.3. T-Cell Exhaustion
3. Strategies to Improve Car T-Cell Persistence and Potency
3.1. Car-T-Cells Engineered to Express Cytokine and Their Receptors
3.2. Checkpoint Blockade Therapy
3.3. T-Cell Stemness
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| 4,6-Disubstituted pyrrolopyrimidine (TWS119) |
| activation-induced cell death (AICD) |
| activator protein 1 (AP1) |
| adoptive cell therapy (ACT) |
| basic leucine zipper transcription factor (BATF) |
| B-cell acute lymphoblastic leukemia (B-ALL) |
| B-cell non-Hodgkin lymphoma (B-NHL) |
| bendamustine (ben) |
| cancer-associated fibroblasts (CAFs) |
| central memory T-cells (TCMs) |
| chimeric antigen receptors (CARs) |
| chronic lymphocytic leukemia (CLL) |
| cyclophosphamide (cy) |
| cytotoxic T lymphocytes (CTLs) |
| diffuse Large B-Cell Lymphoma (DLBCL) |
| DNA methyltransferases (DNMT) |
| effector memory T (TEM) |
| fatty acid oxidation (FAO) |
| fludarabine (flu) |
| glycogen synthase kinase-3β (GSK-3β) |
| human-derived anti-CD19 CAR (HuCAR-19) |
| immunoreceptor tyrosine-based activation motifs (ITAMs) |
| mammalian target of rapamycin (mTOR) |
| mitogen-activated protein kinase (MAPK) |
| myeloid-derived suppressor cells (MDSCs) |
| natural killer (NK) |
| nuclear factor of activated t-cells (NFAT) |
| nuclear receptor subfamily 4 group A member (NR4A) |
| peripheral blood mononuclear cell (PBMC) |
| programmed cell death ligand 1 (PD-L1) |
| programmed cell death protein 1 (PD-1) |
| progression-free survival (PFS) |
| regulatory T-cells (Tregs) |
| severe cytokine release syndrome (CRS) |
| single chain variable fragment (scFv) |
| stem memory T cell (TSCM)T-cell receptor (TCR) |
| T-cells redirected for antigen-unrestricted cytokine-initiated killing (TRUCKs) |
| ten eleven translocation (TET2) |
| TNF receptor-associated factor 1 (TRAF1) |
| tumor microenvironment (TME) |
| tumor-associated macrophages (TAMs) |
| tumor-infiltrating lymphocytes (TILs) |
References
- Li, D.; Li, X.; Zhou, W.-L.; Huang, Y.; Liang, X.; Jiang, L.; Yang, X.; Sun, J.; Li, Z.; Han, W.-D.; et al. Genetically engineered T cells for cancer immunotherapy. Signal Transduct. Target. Ther. 2019, 4, 1–17. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. 1993, 90, 720–724. [Google Scholar] [CrossRef]
- Krause, A.; Guo, H.-F.; Latouche, J.-B.; Tan, C.; Cheung, N.-K.V.; Sadelain, M. Antigen-dependent CD28 Signaling Selectively Enhances Survival and Proliferation in Genetically Modified Activated Human Primary T Lymphocytes. J. Exp. Med. 1998, 188, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.-L.; Schmitt, A.; Neuber, B.; Hückelhoven-Krauss, A.; Kunz, A.; Wang, L.; Gern, U.; Michels, B.; Sellner, L.; Hofmann, S.; et al. Third-Generation CAR T Cells Targeting CD19 Are Associated with an Excellent Safety Profile and Might Improve Persistence of CAR T Cells in Treated Patients. Blood 2019, 134, 51. [Google Scholar] [CrossRef]
- Zhao, Z.; Condomines, M.; Van Der Stegen, S.J.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.-S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric Antigen Receptors Combining 4-1BB and CD28 Signaling Domains Augment PI3kinase/AKT/Bcl-XL Activation and CD8+ T Cell–mediated Tumor Eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Pule, M.; Straathof, K.C.; Dotti, G.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol. Ther. 2005, 12, 933–941. [Google Scholar] [CrossRef]
- Hombach, A.A.; Heiders, J.; Foppe, M.; Chmielewski, M.; Abken, H. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4+T cells. OncoImmunology 2012, 1, 458–466. [Google Scholar] [CrossRef]
- Mackay, M.; Afshinnekoo, E.; Rub, J.; Hassan, C.; Khunte, M.; Baskaran, N.; Owens, B.; Liu, L.; Roboz, G.J.; Guzman, M.L.; et al. The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat. Biotechnol. 2020, 38, 233–244. [Google Scholar] [CrossRef]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef] [PubMed]
- Halim, L.; Maher, J. CAR T-cell immunotherapy of B-cell malignancy: The story so far. Ther. Adv. Vaccines Immunother. 2020, 8, 2515135520927164. [Google Scholar] [CrossRef]
- Greenbaum, U.; Mahadeo, K.M.; Kebriaei, P.; Shpall, E.J.; Saini, N.Y. Chimeric Antigen Receptor T-Cells in B-Acute Lymphoblastic Leukemia: State of the Art and Future Directions. Front. Oncol. 2020, 10, 1594. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.C.; Yassine, F.; Sandoval-Sus, J.; A Kharfan-Dabaja, M. Anti-CD19 chimeric antigen receptor T-cell therapy in B-cell lymphomas: Current status and future directions. Int. J. Hematol. Oncol. 2021, 10, IJH33. [Google Scholar] [CrossRef] [PubMed]
- Al-Mansour, M.; Al-Foheidi, M.; Ibrahim, E. Efficacy and safety of second-generation CAR T-cell therapy in diffuse large B-cell lymphoma: A meta-analysis. Mol. Clin. Oncol. 2020, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Sun, Q.; Liang, X.; Chen, Z.; Zhang, X.; Zhou, X.; Li, M.; Tu, H.; Liu, Y.; Tu, S.; et al. Mechanisms of Relapse After CD19 CAR T-Cell Therapy for Acute Lymphoblastic Leukemia and Its Prevention and Treatment Strategies. Front. Immunol. 2019, 10, 2664. [Google Scholar] [CrossRef]
- Mancikova, V.; Smida, M. Current State of CAR T-Cell Therapy in Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2021, 22, 5536. [Google Scholar] [CrossRef]
- Chow, V.A.; Gopal, A.K.; Maloney, D.G.; Turtle, C.J.; Smith, S.D.; Ujjani, C.S.; Shadman, M.; Cassaday, R.D.; Till, B.G.; Tseng, Y.D.; et al. Outcomes of patients with large B-cell lymphomas and progressive disease following CD19-specific CAR T-cell therapy. Am. J. Hematol. 2019, 94, E209–E213. [Google Scholar] [CrossRef]
- Chow, V.A.; Gopal, A.K.; Gauthier, J.; Tseng, Y.D.; Turtle, C.J.; Maloney, D.G.; Shadman, M. Axicabtagene ciloleucel for relapsed or refractory lymphoma after prior treatment with a different CD19-directed CAR T-cell therapy. Blood Adv. 2020, 4, 4869–4872. [Google Scholar] [CrossRef]
- Hirayama, A.V.; Gauthier, J.; Hay, K.A.; Voutsinas, J.M.; Wu, Q.; Gooley, T.; Li, D.; Cherian, S.; Chen, X.; Pender, B.S.; et al. The response to lymphodepletion impacts PFS in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR T cells. Blood 2019, 133, 1876–1887. [Google Scholar] [CrossRef] [PubMed]
- Hucks, G.; Rheingold, S.R. The journey to CAR T cell therapy: The pediatric and young adult experience with relapsed or refractory B-ALL. Blood Cancer J. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Nie, Y.; Lu, W.; Chen, D.; Tu, H.; Guo, Z.; Zhou, X.; Li, M.; Tu, S.; Li, Y. Mechanisms underlying CD19-positive ALL relapse after anti-CD19 CAR T cell therapy and associated strategies. Biomark. Res. 2020, 8, 1–17. [Google Scholar] [CrossRef]
- Srivastava, S.; Riddell, S.R. Chimeric Antigen Receptor T Cell Therapy: Challenges to Bench-to-Bedside Efficacy. J. Immunol. 2018, 200, 459–468. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Ma, S.; Li, X.; Wang, X.; Cheng, L.; Li, Z.; Zhang, C.; Ye, Z.; Qian, Q. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int. J. Biol. Sci. 2019, 15, 2548–2560. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. eLife 2020, 9, 55185. [Google Scholar] [CrossRef] [PubMed]
- Pietrobon, V.; Marincola, F.M. Hypoxia and the phenomenon of immune exclusion. J. Transl. Med. 2021, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Xia, A.; Zhang, Y.; Xu, J.; Yin, T.; Lu, X.-J. T Cell Dysfunction in Cancer Immunity and Immunotherapy. Front. Immunol. 2019, 10, 1719. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Jiang, W.; He, Y.; He, W.; Wu, G.; Zhou, X.; Sheng, Q.; Zhong, W.; Lu, Y.; Ding, Y.; Lu, Q.; et al. Exhausted CD8+T Cells in the Tumor Immune Microenvironment: New Pathways to Therapy. Front. Immunol. 2021, 11, 622509. [Google Scholar] [CrossRef]
- Collier, J.L.; Weiss, S.A.; Pauken, K.E.; Sen, D.R.; Sharpe, A.H. Not-so-opposite ends of the spectrum: CD8+ T cell dysfunction across chronic infection, cancer and autoimmunity. Nat. Immunol. 2021, 22, 809–819. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef]
- Nüssing, S.; Trapani, J.A.; Parish, I.A. Revisiting T Cell Tolerance as a Checkpoint Target for Cancer Immunotherapy. Front. Immunol. 2020, 11, 589641. [Google Scholar] [CrossRef]
- Hansen, M.B. Interleukin-6 signaling requires only few IL-6 molecules: Relation to physiological concentrations of extracellular IL-6. Immunity, Inflamm. Dis. 2020, 8, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Rogovskii, V. Immune Tolerance as the Physiologic Counterpart of Chronic Inflammation. Front. Immunol. 2020, 11, 2061. [Google Scholar] [CrossRef]
- Wang, E.; Cesano, A.; Butterfield, L.H.; Marincola, F. Improving the therapeutic index in adoptive cell therapy: Key factors that impact efficacy. J. Immunother. Cancer 2020, 8, e001619. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Geyer, M.B.; Brentjens, R.J. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: Interpreting clinical outcomes to date. Blood 2016, 127, 3312–3320. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Das, J.K.; Xiong, X.; Chen, H.; Song, J. Development of CAR-T Cell Persistence in Adoptive Immunotherapy of Solid Tumors. Front. Oncol. 2021, 10, 574860. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, L.; Masoumi, E.; Mehrjardi, K.F.; Mirzaei, H.R.; Hadjati, J. Prolonged Persistence of Chimeric Antigen Receptor (CAR) T Cell in Adoptive Cancer Immunotherapy: Challenges and Ways Forward. Front. Immunol. 2020, 11, 702. [Google Scholar] [CrossRef]
- Guedan, S.; Calderon, H.; Posey, J.A.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther. - Methods Clin. Dev. 2019, 12, 145–156. [Google Scholar] [CrossRef]
- Fujiwara, K.; Tsunei, A.; Kusabuka, H.; Ogaki, E.; Tachibana, M.; Okada, N. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells 2020, 9, 1182. [Google Scholar] [CrossRef]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. EBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef] [PubMed]
- Lindner, S.E.; Johnson, S.M.; Brown, C.E.; Wang, L.D. Chimeric antigen receptor signaling: Functional consequences and design implications. Sci. Adv. 2020, 6, eaaz3223. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.V.; Haas, A.R.; Beatty, G.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T Cells Expressing Chimeric Antigen Receptors Can Cause Anaphylaxis in Humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.J.; Willemsen, R.; Van Elzakker, P.; Van Steenbergen-Langeveld, S.; Broertjes, M.; Oosterwijk-Wakka, J.; Oosterwijk, E.; Sleijfer, S.; Debets, R.; Gratama, J.W. Immune responses to transgene and retroviral vector in patients treated with ex vivo–engineered T cells. Blood 2011, 117, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.C.; Popplewell, L.; Cooper, L.J.; DiGiusto, D.; Kalos, M.; Ostberg, J.R.; Forman, S.J. Antitransgene Rejection Responses Contribute to Attenuated Persistence of Adoptively Transferred CD20/CD19-Specific Chimeric Antigen Receptor Redirected T Cells in Humans. Biol. Blood Marrow Transplant. 2010, 16, 1245–1256. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A Phase I Study on Adoptive Immunotherapy Using Gene-Modified T Cells for Ovarian Cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.L.; Fritsche, E.; Pulsipher, M.A.; Ahmed, N.; Hamieh, M.; Hegde, M.; Ruella, M.; Savoldo, B.; Shah, N.N.; Turtle, C.J.; et al. Immunogenicity of CAR T cells in cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 379–393. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef]
- Brudno, J.N.; Shi, V.; Stroncek, D.; Pittaluga, S.; Kanakry, J.A.; Curtis, L.M.; Gea-Banacloche, J.C.; Pavletic, S.; Bagheri, M.-H.; Rose, M.J.J.; et al. T Cells Expressing a Novel Fully-Human Anti-CD19 Chimeric Antigen Receptor Induce Remissions of Advanced Lymphoma in a First-in-Humans Clinical Trial. Blood 2016, 128, 999. [Google Scholar] [CrossRef]
- Mirzaei, H.R.; Jamali, A.; Jafarzadeh, L.; Masoumi, E.; Alishah, K.; Mehrjardi, K.F.; Emami, S.A.H.; Noorbakhsh, F.; Till, B.G.; Hadjati, J. Construction and functional characterization of a fully human anti-CD19 chimeric antigen receptor (huCAR)-expressing primary human T cells. J. Cell. Physiol. 2019, 234, 9207–9215. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, L.; Masoumi, E.; Alishah, K.; Mirzaei, H.R.; Jamali, A.; Mehrjardi, K.F.; Rostamian, H.; Khakpoor-Koosheh, M.; Meshkani, R.; Noorbakhsh, F.; et al. Construction and Functional Characterization of a Fully Human Anti-mesothelin Chimeric Antigen Receptor (CAR)-expressing T Cell. Iran. J. Allergy, Asthma Immunol. 2020, 19, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, N.R.; A Padlan, E.; De Pascalis, R.; Schuck, P.; Schlom, J.; Kashmiri, S.V.S. Minimizing immunogenicity of the SDR-grafted humanized antibody CC49 by genetic manipulation of the framework residues. Mol. Immunol. 2003, 40, 337–349. [Google Scholar] [CrossRef]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric Receptors Containing CD137 Signal Transduction Domains Mediate Enhanced Survival of T Cells and Increased Antileukemic Efficacy In Vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef]
- Dai, Q.; Han, P.; Qi, X.; Li, F.; Li, M.; Fan, L.; Zhang, H.; Zhang, X.; Yang, X. 4-1BB Signaling Boosts the Anti-Tumor Activity of CD28-Incorporated 2nd Generation Chimeric Antigen Receptor-Modified T Cells. Front. Immunol. 2020, 11, 539654. [Google Scholar] [CrossRef]
- Majzner, R.G.; Rietberg, S.P.; Sotillo, E.; Dong, R.; Vachharajani, V.T.; Labanieh, L.; Myklebust, J.H.; Kadapakkam, M.; Weber, E.W.; Tousley, A.M.; et al. Tuning the Antigen Density Requirement for CAR T-cell Activity. Cancer Discov. 2020, 10, 702–723. [Google Scholar] [CrossRef]
- Cheng, Z.; Wei, R.; Ma, Q.; Shi, L.; He, F.; Shi, Z.; Jin, T.; Xie, R.; Wei, B.; Chen, J.; et al. In Vivo Expansion and Antitumor Activity of Coinfused CD28- and 4-1BB-Engineered CAR-T Cells in Patients with B Cell Leukemia. Mol. Ther. 2018, 26, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Boucher, J.C.; Kotani, H.; Park, K.; Zhang, Y.; Shrestha, B.; Wang, X.; Guan, L.; Beatty, N.; Abate-Daga, D.; et al. 4-1BB enhancement of CAR T function requires NF-κB and TRAFs. JCI Insight 2018, 3, e121322. [Google Scholar] [CrossRef] [PubMed]
- Hamieh, M.; Dobrin, A.; Cabriolu, A.; Van Der Stegen, S.J.C.; Giavridis, T.; Mansilla-Soto, J.; Eyquem, J.; Zhao, Z.; Whitlock, B.M.; Miele, M.M.; et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019, 568, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Amatya, C.; Pegues, M.A.; Lam, N.; Vanasse, D.; Geldres, C.; Choi, S.; Hewitt, S.M.; Feldman, S.A.; Kochenderfer, J.N. Development of CAR T Cells Expressing a Suicide Gene Plus a Chimeric Antigen Receptor Targeting Signaling Lymphocytic-Activation Molecule F7. Mol. Ther. 2021, 29, 702–717. [Google Scholar] [CrossRef] [PubMed]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; Mcgettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, C.; McCaffrey, P.G.; Rao, A.; Sen, R. Physiologic activation of T cells via the T cell receptor induces NF-kappa B. J. Immunol. 1991, 147, 416–420. [Google Scholar]
- Kang, S.; Tran, A.; Grilli, M.; Lenardo, M. NF-kappa B subunit regulation in nontransformed CD4+ T lymphocytes. Science 1992, 256, 1452–1456. [Google Scholar] [CrossRef]
- Fraser, J.; Irving, B.; Crabtree, G.; Weiss, A. Regulation of interleukin-2 gene enhancer activity by the T cell accessory molecule CD28. Science 1991, 251, 313–316. [Google Scholar] [CrossRef]
- Verweij, C.; Geerts, M.; Aarden, L. Activation of interleukin-2 gene transcription via the T-cell surface molecule CD28 is mediated through an NF-kB-like response element. J. Biol. Chem. 1991, 266, 14179–14182. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Dholaria, B.R.; Bachmeier, C.A.; Locke, F. Mechanisms and Management of Chimeric Antigen Receptor T-Cell Therapy-Related Toxicities. BioDrugs 2018, 33, 45–60. [Google Scholar] [CrossRef]
- Kroon, H.M.; Li, Q.; Teitz-Tennenbaum, S.; Whitfield, J.R.; Noone, A.-M.; Chang, A.E. 4-1BB Costimulation of Effector T Cells for Adoptive Immunotherapy of Cancer: Involvement of Bcl Gene Family Members. J. Immunother. 2007, 30, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Stärck, L.; Scholz, C.; Dörken, B.; Daniel, P.T. Costimulation by CD137/4-1BB inhibits T cell apoptosis and induces Bcl-xLand c-FLIPshortvia phosphatidylinositol 3-kinase and AKT/protein kinase B. Eur. J. Immunol. 2005, 35, 1257–1266. [Google Scholar] [CrossRef]
- Khoshnan, A.; Tindell, C.; Laux, I.; Bae, D.; Bennett, B.L.; Nel, A.E. The NF-κB Cascade Is Important in Bcl-xLExpression and for the Anti-Apoptotic Effects of the CD28 Receptor in Primary Human CD4+Lymphocytes. J. Immunol. 2000, 165, 1743–1754. [Google Scholar] [CrossRef]
- Saoulli, K.; Lee, S.Y.; Cannons, J.L.; Yeh, W.C.; Santana, A.; Goldstein, M.D.; Bangia, N.; Debenedette, M.A.; Mak, T.W.; Choi, Y.; et al. CD28-independent, TRAF2-dependent Costimulation of Resting T Cells by 4-1BB Ligand. J. Exp. Med. 1998, 187, 1849–1862. [Google Scholar] [CrossRef]
- Jang, I.K.; Lee, Z.H.; Kim, Y.J.; Kim, S.H.; Kwon, B.S. Human 4-1BB (CD137) Signals Are Mediated by TRAF2 and Activate Nuclear Factor-κB. Biochem. Biophys. Res. Commun. 1998, 242, 613–620. [Google Scholar] [CrossRef]
- Arch, R.H.; Thompson, C.B. 4-1BB and Ox40 Are Members of a Tumor Necrosis Factor (TNF)-Nerve Growth Factor Receptor Subfamily That Bind TNF Receptor-Associated Factors and Activate Nuclear Factor κB. Mol. Cell. Biol. 1998, 18, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Shou, P.; Du, H.; Hirabayashi, K.; Chen, Y.; Herring, L.E.; Ahn, S.; Xu, Y.; Suzuki, K.; Li, G.; et al. THEMIS-SHP1 Recruitment by 4-1BB Tunes LCK-Mediated Priming of Chimeric Antigen Receptor-Redirected T Cells. Cancer Cell 2020, 37, 216–225.e6. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Madar, A.; Casado-Medrano, V.; Shaw, C.E.; Wing, A.; Liu, F.; Young, R.M.; June, C.H.; Posey, A.D., Jr. Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J. Clin. Investig. 2020, 130, 3087–3097. [Google Scholar] [CrossRef]
- Shen, C.; Zhang, Z.; Zhang, Y. Chimeric Antigen Receptor T Cell Exhaustion during Treatment for Hematological Malignancies. BioMed Res. Int. 2020, 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. New Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, L. Rewiring Mitochondrial Metabolism for CD8+ T Cell Memory Formation and Effective Cancer Immunotherapy. Front. Immunol. 2020, 11, 1834. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.K.; Lee, D.Y.; Lee, D.G.; Kim, Y.H.; Kim, S.-H.; Oh, H.S.; Han, C.; Kwon, B.S. 4-1BB signaling activates glucose and fatty acid metabolism to enhance CD8+ T cell proliferation. Cell. Mol. Immunol. 2017, 14, 748–757. [Google Scholar] [CrossRef] [PubMed]
- A Frauwirth, K.; Riley, J.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.; Thompson, C.B. The CD28 Signaling Pathway Regulates Glucose Metabolism. Immunology 2002, 16, 769–777. [Google Scholar] [CrossRef]
- Beckermann, K.E.; Hongo, R.; Ye, X.; Young, K.; Carbonell, K.; Healey, D.C.C.; Siska, P.J.; Barone, S.; Roe, C.E.; Smith, C.C.; et al. CD28 costimulation drives tumor-infiltrating T cell glycolysis to promote inflammation. JCI Insight 2020, 5, e138729. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, J.A.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; Van Der Stegen, S.J.C.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.-J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef]
- Drent, E.; Poels, R.; Ruiter, R.; Van De Donk, N.W.C.J.; Zweegman, S.; Yuan, H.; de Bruijn, J.; Sadelain, M.; Lokhorst, H.M.; Groen, R.W.; et al. Combined CD28 and 4-1BB Costimulation Potentiates Affinity-tuned Chimeric Antigen Receptor–engineered T Cells. Clin. Cancer Res. 2019, 25, 4014–4025. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef]
- Das, R.K.; Storm, J.; Barrett, D.M. Abstract 1631: T cell dysfunction in pediatric cancer patients at diagnosis and after chemotherapy can limit chimeric antigen receptor potential. Clin. Res. 2018, 78, 1631. [Google Scholar] [CrossRef]
- Singh, N.; Perazzelli, J.; Grupp, S.A.; Barrett, D.M. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci. Transl. Med. 2016, 8, 320ra3. [Google Scholar] [CrossRef] [PubMed]
- Ghassemi, S.; Bedoya, F.; Nunez-Cruz, S.; June, C.; Melenhorst, J.; Milone, M. 203. Shortened T Cell Culture with IL-7 and IL-15 Provides the Most Potent Chimeric Antigen Receptor (CAR)-Modified T Cells for Adoptive Immunotherapy. Mol. Ther. 2016, 24, S79. [Google Scholar] [CrossRef]
- Wallace, D.L.; Bérard, M.; Soares, M.V.D.; Oldham, J.; Cook, J.E.; Akbar, A.N.; Tough, D.F.; Beverley, P.C.L. Prolonged exposure of naïve CD8+T cells to interleukin-7 or interleukin-15 stimulates proliferation without differentiation or loss of telomere length. Immunology 2006, 119, 243–253. [Google Scholar] [CrossRef]
- Cieri, N.; Camisa, B.; Cocchiarella, F.; Forcato, M.; Oliveira, G.; Provasi, E.; Bondanza, A.; Bordignon, C.; Peccatori, I.; Ciceri, F.; et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 2013, 121, 573–584. [Google Scholar] [CrossRef]
- Oh, S.; Perera, L.P.; Terabe, M.; Ni, L.; Waldmann, T.A.; Berzofsky, J.A. IL-15 as a mediator of CD4+help for CD8+T cell longevity and avoidance of TRAIL-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 5201–5206. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, M.; Ramos, C.A.; Durett, A.; Liu, E.; Dakhova, O.; Liu, H.; Creighton, C.J.; Gee, A.P.; Heslop, H.E.; et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 2014, 123, 3750–3759. [Google Scholar] [CrossRef]
- Coppola, C.; Hopkins, B.; Huhn, S.; Du, Z.; Huang, Z.; Kelly, W.J. Investigation of the Impact from IL-2, IL-7, and IL-15 on the Growth and Signaling of Activated CD4+ T Cells. Int. J. Mol. Sci. 2020, 21, 7814. [Google Scholar] [CrossRef]
- Pilipow, K.; Scamardella, E.; Puccio, S.; Gautam, S.; De Paoli, F.; Mazza, E.M.C.; De Simone, G.; Polletti, S.; Buccilli, M.; Zanon, V.; et al. Antioxidant metabolism regulates CD8+ T memory stem cell formation and antitumor immunity. JCI Insight 2018, 3, e122299. [Google Scholar] [CrossRef]
- Kesarwani, P.; Al-Khami, A.A.; Scurti, G.; Thyagarajan, K.; Kaur, N.; Husain, S.; Fang, Q.; Naga, O.S.; Simms, P.; Beeson, G.; et al. Promoting Thiol Expression Increases the Durability of Antitumor T-cell Functions. Cancer Res. 2014, 74, 6036–6047. [Google Scholar] [CrossRef] [PubMed]
- Kesarwani, P.; Thyagarajan, K.; Chatterjee, S.; Palanisamy, V.; Mehrotra, S. Anti-oxidant capacity and anti-tumor T cell function: A direct correlation. OncoImmunology 2015, 4, e985942. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gattinoni, L.; Finkelstein, S.E.; Klebanoff, C.; Antony, P.A.; Palmer, D.; Spiess, P.J.; Hwang, L.N.; Yu, Z.; Wrzesinski, C.; Heimann, D.M.; et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J. Exp. Med. 2005, 202, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.; Khong, H.T.; Antony, P.A.; Palmer, D.C.; Restifo, N.P. Sinks, suppressors and antigen presenters: How lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005, 26, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Turtle, M.C.J.; Berger, C.; Sommermeyer, D.; Hanafi, L.-A.; Pender, M.B.; Robinson, B.E.M.; Melville, K.; Budiarto, B.T.M.; Steevens, N.N.; Chaney, R.C.; et al. Anti-CD19 Chimeric Antigen Receptor-Modified T Cell Therapy for B Cell Non-Hodgkin Lymphoma and Chronic Lymphocytic Leukemia: Fludarabine and Cyclophosphamide Lymphodepletion Improves In Vivo Expansion and Persistence of CAR-T Cells and Clinical Outcomes. Blood 2015, 126, 184. [Google Scholar] [CrossRef]
- Turtle, M.C.J.; Hanafi, L.-A.; Berger, C.; Sommermeyer, D.; Pender, M.B.; Robinson, B.E.M.; Melville, K.; Budiarto, B.T.M.; Steevens, N.N.; Chaney, R.C.; et al. Addition of Fludarabine to Cyclophosphamide Lymphodepletion Improves In Vivo Expansion of CD19 Chimeric Antigen Receptor-Modified T Cells and Clinical Outcome in Adults with B Cell Acute Lymphoblastic Leukemia. Blood 2015, 126, 3773. [Google Scholar] [CrossRef]
- Ramos, C.A.; Grover, N.S.; Beaven, A.W.; Lulla, P.D.; Wu, M.-F.; Ivanova, A.; Wang, T.; Shea, T.C.; Rooney, C.M.; Dittus, C.; et al. Anti-CD30 CAR-T Cell Therapy in Relapsed and Refractory Hodgkin Lymphoma. J. Clin. Oncol. 2020, 38, 3794–3804. [Google Scholar] [CrossRef]
- Pai, S.I.; Cesano, A.; Marincola, F.M. The Paradox of Cancer Immune Exclusion: Immune Oncology Next Frontier. Precis. Med. Cancer Ther. 2020, 180, 173–195. [Google Scholar] [CrossRef]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.-F.; Han, W.; Wang, H. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 2020, 5, e133977. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, Y.; Hu, Y.; Mei, H. T Cell Exhaustion and CAR-T Immunotherapy in Hematological Malignancies. BioMed Res. Int. 2021, 2021, 1–8. [Google Scholar] [CrossRef]
- Chapman, N.M.; Chi, H. mTOR Links Environmental Signals to T Cell Fate Decisions. Front. Immunol. 2015, 5, 686. [Google Scholar] [CrossRef]
- Zheng, W.; O’Hear, C.E.; Alli, R.; Basham, J.H.; Abdelsamed, H.A.; Palmer, L.E.; Jones, L.L.; Youngblood, B.; Geiger, T.L. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia 2018, 32, 1157–1167. [Google Scholar] [CrossRef]
- Sengal, A.; Velazquez, J.; Hahne, M.V.; Burke, T.M.; Abhyankar, H.; Reyes, R.W.I.; Olea, W.; Scull, B.P.; Eckstein, O.S.; Bigenwald, C.; et al. Overcoming T-cell exhaustion in LCH: PD-1 blockade and targeted MAPK inhibition are synergistic in a mouse model of LCH. Blood 2021, 137, 1777–1791. [Google Scholar] [CrossRef]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.-H.; Jones, R.G. Fueling Immunity: Insights into Metabolism and Lymphocyte Function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef]
- Gennert, D.G.; Lynn, R.C.; Granja, J.M.; Weber, E.W.; Mumbach, M.R.; Zhao, Y.; Duren, Z.; Sotillo, E.; Greenleaf, W.J.; Wong, W.H.; et al. Dynamic chromatin regulatory landscape of human CAR T cell exhaustion. Proc. Natl. Acad. Sci. USA 2021, 118, e2104758118. [Google Scholar] [CrossRef] [PubMed]
- LaFleur, M.W.; Nguyen, T.H.; Coxe, M.A.; Miller, B.; Yates, K.B.; Gillis, J.E.; Sen, D.R.; Gaudiano, E.F.; Al Abosy, R.; Freeman, G.J.; et al. PTPN2 regulates the generation of exhausted CD8+ T cell subpopulations and restrains tumor immunity. Nat. Immunol. 2019, 20, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- A Teft, W.; A Chau, T.; Madrenas, J. Structure-Function analysis of the CTLA-4 interaction with PP2A. BMC Immunol. 2009, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Scharl, M.; Hruz, P.; McCole, D.F. Protein Tyrosine Phosphatase non-Receptor Type 2 regulates IFN-γ-induced cytokine signaling in THP-1 monocytes. Inflamm. Bowel Dis. 2010, 16, 2055–2064. [Google Scholar] [CrossRef]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral Persistence Alters CD8 T-Cell Immunodominance and Tissue Distribution and Results in Distinct Stages of Functional Impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef]
- Fuller, M.J.; Zajac, A.J. Ablation of CD8 and CD4 T Cell Responses by High Viral Loads. J. Immunol. 2003, 170, 477–486. [Google Scholar] [CrossRef]
- Wherry, E.J.; Ha, S.-J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef]
- Hwang, J.-R.; Byeon, Y.; Kim, D.; Park, S.-G. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp. Mol. Med. 2020, 52, 750–761. [Google Scholar] [CrossRef]
- Poorebrahim, M.; Melief, J.; de Coaña, Y.P.; Wickström, S.L.; Cid-Arregui, A.; Kiessling, R. Counteracting CAR T cell dysfunction. Oncogene 2021, 40, 421–435. [Google Scholar] [CrossRef]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.-S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef]
- Sears, J.D.; Waldron, K.J.; Wei, J.; Chang, C. Targeting metabolism to reverse T-cell exhaustion in chronic viral infections. Immunology 2021, 162, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Pearce, E.L. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat. Immunol. 2016, 17, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Shyer, J.A.; Flavell, R.A.; Bailis, W. Metabolic signaling in T cells. Cell Res. 2020, 30, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Gerriets, V.A.; Kishton, R.J.; Nichols, A.G.; Macintyre, A.; Inoue, M.; Ilkayeva, O.; Winter, P.S.; Liu, X.; Priyadharshini, B.; Slawinska, M.E.; et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Investig. 2015, 125, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Ramstead, A.G.; Robison, A.; Blackwell, A.; Jerome, M.; Freedman, B.; Lubick, K.J.; Hedges, J.F.; Jutila, M.A. Roles of Toll-Like Receptor 2 (TLR2), TLR4, and MyD88 during Pulmonary Coxiella burnetii Infection. Infect. Immun. 2016, 84, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Angela, M.; Endo, Y.; Asou, H.K.; Yamamoto, T.; Tumes, D.J.; Tokuyama, H.; Yokote, K.; Nakayama, T. Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARγ directs early activation of T cells. Nat. Commun. 2016, 7, 13683. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-L.; Ou, D.-L.; Bai, L.-Y.; Chen, C.-W.; Lin, L.; Huang, S.-F.; Cheng, A.-L.; Jeng, Y.-M.; Hsu, C. Exploring Markers of Exhausted CD8 T Cells to Predict Response to Immune Checkpoint Inhibitor Therapy for Hepatocellular Carcinoma. Liver Cancer 2021, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Baitsch, L.; Baumgaertner, P.; Devêvre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Pagliano, O.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Olive, D.; Kuchroo, V.K.; Zarour, H.M. CD8+ T Cells Specific for Tumor Antigens Can Be Rendered Dysfunctional by the Tumor Microenvironment through Upregulation of the Inhibitory Receptors BTLA and PD-1. Cancer Res. 2012, 72, 887–896. [Google Scholar] [CrossRef]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-cell Function to Promote Tumoral Immune Escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1–specific CD8+T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. 2010, 107, 7875–7880. [Google Scholar] [CrossRef]
- Cai, J.; Wang, D.; Zhang, G.; Guo, X. The Role Of PD-1/PD-L1 Axis In Treg Development And Function: Implications For Cancer Immunotherapy. OncoTargets Ther. 2019, 12, 8437–8445. [Google Scholar] [CrossRef]
- Dong, Y.; Sun, Q.; Zhang, X. PD-1 and its ligands are important immune checkpoints in cancer. Oncotarget 2017, 8, 2171–2186. [Google Scholar] [CrossRef]
- Diskin, B.; Adam, S.; Cassini, M.F.; Sanchez, G.; Liria, M.; Aykut, B.; Buttar, C.; Li, E.; Sundberg, B.; Salas, R.D.; et al. PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nat. Immunol. 2020, 21, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Gianchecchi, E.; Fierabracci, A. Inhibitory Receptors and Pathways of Lymphocytes: The Role of PD-1 in Treg Development and Their Involvement in Autoimmunity Onset and Cancer Progression. Front. Immunol. 2018, 9, 2374. [Google Scholar] [CrossRef] [PubMed]
- Terawaki, S.; Chikuma, S.; Shibayama, S.; Hayashi, T.; Yoshida, T.; Okazaki, T.; Honjo, T. IFN-α Directly Promotes Programmed Cell Death-1 Transcription and Limits the Duration of T Cell-Mediated Immunity. J. Immunol. 2011, 186, 2772–2779. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhang, Y.; Zheng, L.; Zheng, C.; Song, J.; Zhang, Q.; Kang, B.; Liu, Z.; Jin, L.; Xing, R.; et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018, 24, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Kallies, A.; Zehn, D.; Utzschneider, D. Precursor exhausted T cells: Key to successful immunotherapy? Nat. Rev. Immunol. 2019, 20, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Im, S.J.; Konieczny, B.T.; Hudson, W.H.; Masopust, D.; Ahmed, R. PD-1+ stemlike CD8 T cells are resident in lymphoid tissues during persistent LCMV infection. Proc. Natl. Acad. Sci. 2020, 117, 4292–4299. [Google Scholar] [CrossRef]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nat. Cell Biol. 2016, 537, 417–421. [Google Scholar] [CrossRef]
- Utzschneider, D.; Charmoy, M.; Chennupati, V.; Pousse, L.; Ferreira, D.P.; Calderon-Copete, S.; Danilo, M.; Alfei, F.; Hofmann, M.; Wieland, D.; et al. T Cell Factor 1-Expressing Memory-like CD8+ T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 2016, 45, 415–427. [Google Scholar] [CrossRef]
- Jadhav, R.R.; Im, S.J.; Hu, B.; Hashimoto, M.; Li, P.; Lin, J.-X.; Leonard, W.J.; Greenleaf, W.J.; Ahmed, R.; Goronzy, J.J. Epigenetic signature of PD-1+ TCF1+ CD8 T cells that act as resource cells during chronic viral infection and respond to PD-1 blockade. Proc. Natl. Acad. Sci. 2019, 116, 14113–14118. [Google Scholar] [CrossRef]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Marraco, S.A.F.; Calderon-Copete, S.; Ferreira, D.P.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e10. [Google Scholar] [CrossRef]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef]
- Kartikasari, A.; Prakash, M.D.; Cox, M.; Wilson, K.; Boer, J.; Cauchi, J.A.; Plebanski, M. Therapeutic Cancer Vaccines—T Cell Responses and Epigenetic Modulation. Front. Immunol. 2019, 9, 3109. [Google Scholar] [CrossRef]
- Schietinger, A.; Philip, M.; Krisnawan, V.; Chiu, E.Y.; Delrow, J.J.; Basom, R.S.; Lauer, P.; Brockstedt, D.G.; Knoblaugh, S.E.; Hämmerling, G.J.; et al. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity 2016, 45, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157.e19. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.M.; Gamper, C.J.; Ladle, B.H.; Powell, J.D.; Wells, A.D. De Novo DNA Methylation Is Required to Restrict T Helper Lineage Plasticity. J. Biol. Chem. 2012, 287, 22900–22909. [Google Scholar] [CrossRef] [PubMed]
- Henning, A.; Roychoudhuri, R.; Restifo, N.P. Epigenetic control of CD8+ T cell differentiation. Nat. Rev. Immunol. 2018, 18, 340–356. [Google Scholar] [CrossRef]
- Lee, M.; Li, J.; Li, J.; Fang, S.; Zhang, J.; Vo, A.T.T.; Han, W.; Zeng, H.; Isgandarova, S.; Martinez-Moczygemba, M.; et al. Tet2 Inactivation Enhances the Antitumor Activity of Tumor-Infiltrating Lymphocytes. Cancer Res. 2021, 81, 1965–1976. [Google Scholar] [CrossRef]
- Xu, Y.-P.; Lv, L.; Liu, Y.; Smith, M.D.; Li, W.-C.; Tan, X.-M.; Cheng, M.; Li, Z.; Bovino, M.; Aubé, J.; et al. Tumor suppressor TET2 promotes cancer immunity and immunotherapy efficacy. J. Clin. Investig. 2019, 129, 4316–4331. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef]
- Macian, F.; Garcia-Rodriguez, C.; Rao, A. Gene expression elicited by NFAT in the presence or absence of cooperative recruitment of Fos and Jun. EMBO J. 2000, 19, 4783–4795. [Google Scholar] [CrossRef] [PubMed]
- Macian, F.; Garcia-Cozar, F.; Im, S.-H.; Horton, H.F.; Byrne, M.C.; Rao, A. Transcriptional Mechanisms Underlying Lymphocyte Tolerance. Cell 2002, 109, 719–731. [Google Scholar] [CrossRef]
- Martinez, G.J.; Pereira, R.M.; Äijö, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The Transcription Factor NFAT Promotes Exhaustion of Activated CD8 + T Cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Bengsch, B.; Wherry, E.J. The Importance of Cooperation: Partnerless NFAT Induces T Cell Exhaustion. Immunity 2015, 42, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Scott-Browne, J.P.; López-Moyado, I.; Trifari, S.; Wong, V.; Chavez, L.; Rao, A.; Pereira, R. Dynamic Changes in Chromatin Accessibility Occur in CD8 + T Cells Responding to Viral Infection. Immunity 2016, 45, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Mognol, G.P.; Carneiro, F.; Robbs, B.; Faget, D.; Viola, J.P.B. Cell cycle and apoptosis regulation by NFAT transcription factors: New roles for an old player. Cell Death Dis. 2016, 7, e2199. [Google Scholar] [CrossRef]
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef]
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef]
- Chen, J.; López-Moyado, I.; Seo, H.; Lio, J.; Hempleman, L.J.; Sekiya, T.; Yoshimura, A.; Scott-Browne, J.P.; Rao, A. NR4A transcription factors limit CAR T cell function in solid tumours. Nat. Cell Biol. 2019, 567, 530–534. [Google Scholar] [CrossRef]
- Flemming, A. What’s driving T cell dysfunction? Nat. Rev. Cancer 2019, 19, 251. [Google Scholar] [CrossRef]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nat. Cell Biol. 2019, 576, 293–300. [Google Scholar] [CrossRef]
- Li, J.; He, Y.; Hao, J.; Ni, L.; Dong, C. High Levels of Eomes Promote Exhaustion of Anti-tumor CD8+ T Cells. Front. Immunol. 2018, 9, 2981. [Google Scholar] [CrossRef]
- Intlekofer, A.; Takemoto, N.; Wherry, E.J.; A Longworth, S.; Northrup, J.T.; Palanivel, V.R.; Mullen, A.C.; Gasink, C.R.; Kaech, S.M.; Miller, J.D.; et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat. Immunol. 2005, 6, 1236–1244. [Google Scholar] [CrossRef] [PubMed]
- Paley, M.; Gordon, S.; Bikoff, E.K.; Robertson, E.J.; Wherry, E.J.; Reiner, S.L. Technical Advance: Fluorescent reporter reveals insights into eomesodermin biology in cytotoxic lymphocytes. J. Leukoc. Biol. 2013, 93, 307–315. [Google Scholar] [CrossRef] [PubMed]
- McLane, L.M.; Ngiow, S.F.; Chen, Z.; Attanasio, J.; Manne, S.; Ruthel, G.; Wu, J.E.; Staupe, R.P.; Xu, W.; Amaravadi, R.K.; et al. Role of nuclear localization in the regulation and function of T-bet and Eomes in exhausted CD8 T cells. Cell Rep. 2021, 35, 109120. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Zhao, M.; Song, Z.; Wang, Z. T-bet interferes with PD-1/PD-L1-mediated suppression of CD4 + T cell inflammation and survival in Crohn’s disease. Clin. Exp. Pharmacol. Physiol. 2019, 46, 798–805. [Google Scholar] [CrossRef]
- Seo, W.; Jerin, C.; Nishikawa, H. Transcriptional regulatory network for the establishment of CD8+ T cell exhaustion. Exp. Mol. Med. 2021, 53, 202–209. [Google Scholar] [CrossRef]
- Collins, M.H.; Henderson, A.J. Transcriptional regulation and T cell exhaustion. Curr. Opin. HIV AIDS 2014, 9, 459–463. [Google Scholar] [CrossRef]
- Agnellini, P.; Wolint, P.; Rehr, M.; Cahenzli, J.; Karrer, U.; Oxenius, A. Impaired NFAT nuclear translocation results in split exhaustion of virus-specific CD8+ T cell functions during chronic viral infection. Proc. Natl. Acad. Sci. 2007, 104, 4565–4570. [Google Scholar] [CrossRef]
- Quigley, M.; Pereyra, F.; Nilsson, B.; Porichis, F.; Fonseca, C.; Eichbaum, Q.; Julg, B.; Jesneck, J.L.; Brosnahan, K.; Imam, S.; et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat. Med. 2010, 16, 1147–1151. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.; Oestreich, K.J.; Paley, M.; Crawford, A.; Angelosanto, J.M.; A Ali, M.-A.; Intlekofer, A.; Boss, J.M.; Reiner, S.L.; Weinmann, A.S.; et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat. Immunol. 2011, 12, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Paley, M.; Kroy, D.C.; Odorizzi, P.M.; Johnnidis, J.B.; Dolfi, D.V.; Barnett, B.E.; Bikoff, E.K.; Robertson, E.J.; Lauer, G.; Reiner, S.L.; et al. Progenitor and Terminal Subsets of CD8+ T Cells Cooperate to Contain Chronic Viral Infection. Science 2012, 338, 1220–1225. [Google Scholar] [CrossRef] [PubMed]
- Doedens, A.L.; Phan, A.T.; Stradner, M.H.; Fujimoto, J.K.; Nguyen, J.V.; Yang, E.; Johnson, R.S.; Goldrath, A.W. Hypoxia-inducible factors enhance the effector responses of CD8+ T cells to persistent antigen. Nat. Immunol. 2013, 14, 1173–1182. [Google Scholar] [CrossRef]
- Staron, M.M.; Gray, S.M.; Marshall, H.D.; Parish, I.A.; Chen, J.H.; Perry, C.J.; Cui, G.; Li, M.O.; Kaech, S.M. The Transcription Factor FoxO1 Sustains Expression of the Inhibitory Receptor PD-1 and Survival of Antiviral CD8+ T Cells during Chronic Infection. Immunity 2014, 41, 802–814. [Google Scholar] [CrossRef]
- Stephen, T.L.; Rutkowski, M.R.; Allegrezza, M.J.; Perales-Puchalt, A.; Tesone, A.J.; Svoronos, N.; Nguyen, J.M.; Sarmin, F.; Borowsky, M.E.; Tchou, J.; et al. Transforming Growth Factor β-Mediated Suppression of Antitumor T Cells Requires FoxP1 Transcription Factor Expression. Immunity 2014, 41, 427–439. [Google Scholar] [CrossRef]
- Wang, C.; Singer, M.; Anderson, A.C. Molecular Dissection of CD8 + T-Cell Dysfunction. Trends Immunol. 2017, 38, 567–576. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Y.; Lu, H.; Li, J.; Yan, X.; Xiao, M.; Hao, J.; Alekseev, A.; Khong, H.; Chen, T.; et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 2019, 567, 525–529. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKS, the fourth-generation CAR T cells: Current developments and clinical translation. Adv. CELL GENE Ther. 2020, 3. [Google Scholar] [CrossRef]
- Santomasso, B.; Bachier, C.; Westin, J.; Rezvani, K.; Shpall, E.J. The Other Side of CAR T-Cell Therapy: Cytokine Release Syndrome, Neurologic Toxicity, and Financial Burden. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 433–444. [Google Scholar] [CrossRef]
- Tokarew, N.; Ogonek, J.; Endres, S.; Von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: Next-generation CAR T cells. Br. J. Cancer 2019, 120, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, E.R.; D’Souza, R.R.; Klampatsa, A. Armored CAR T-Cells: The Next Chapter in T-Cell Cancer Immunotherapy. Biol. Targets Ther. 2021, 15, 95–105. [Google Scholar] [CrossRef]
- Trinchieri, G. Interleukin-12: A Proinflammatory Cytokine with Immunoregulatory Functions that Bridge Innate Resistance and Antigen-Specific Adaptive Immunity. Annu. Rev. Immunol. 1995, 13, 251–276. [Google Scholar] [CrossRef] [PubMed]
- Wojno, E.T.; Hunter, C.A.; Stumhofer, J.S. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 2019, 50, 851–870. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef]
- Kueberuwa, G.; Kalaitsidou, M.; Cheadle, E.; Hawkins, R.E.; Gilham, D.E. CD19 CAR T Cells Expressing IL-12 Eradicate Lymphoma in Fully Lymphoreplete Mice through Induction of Host Immunity. Mol. Ther. - Oncolytics 2018, 8, 41–51. [Google Scholar] [CrossRef]
- Nguyen, K.G.; Vrabel, M.R.; Mantooth, S.M.; Hopkins, J.J.; Wagner, E.S.; Gabaldon, T.A.; Zaharoff, D.A. Localized Interleukin-12 for Cancer Immunotherapy. Front. Immunol. 2020, 11, 575597. [Google Scholar] [CrossRef]
- Zhang, M.; Wen, B.; Anton, O.; Yao, Z.; Dubois, S.; Ju, W.; Sato, N.; DiLillo, D.J.; Bamford, R.N.; Ravetch, J.V.; et al. IL-15 enhanced antibody-dependent cellular cytotoxicity mediated by NK cells and macrophages. Proc. Natl. Acad. Sci. 2018, 115, E10915–E10924. [Google Scholar] [CrossRef]
- Hata, H.; Yoshimoto, T.; Hayashi, N.; Hada, T.; Nakanishi, K. IL-18 together with anti-CD3 antibody induces human Th1 cells to produce Th1- and Th2-cytokines and IL-8. Int. Immunol. 2004, 16, 1733–1739. [Google Scholar] [CrossRef][Green Version]
- Yasuda, K.; Nakanishi, K.; Tsutsui, H. Interleukin-18 in Health and Disease. Int. J. Mol. Sci. 2019, 20, 649. [Google Scholar] [CrossRef]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bethigh FoxO1low Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Hurton, L.V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.-A.; et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. 2016, 113, E7788–E7797. [Google Scholar] [CrossRef]
- Rathmell, J.C.; Farkash, E.A.; Gao, W.; Thompson, C.B. IL-7 Enhances the Survival and Maintains the Size of Naive T Cells. J. Immunol. 2001, 167, 6869–6876. [Google Scholar] [CrossRef]
- Lin, J.; Zhu, Z.; Xiao, H.; Wakefield, M.R.; Ding, V.A.; Bai, Q.; Fang, Y. The role of IL-7 in Immunity and Cancer. Anticancer. Res. 2017, 37, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Sportès, C.; Hakim, F.T.; Memon, S.; Zhang, H.; Chua, K.S.; Brown, M.R.; Fleisher, T.A.; Krumlauf, M.; Babb, R.R.; Chow, C.K.; et al. Administration of rhIL-7 in humans increases in vivo TCR repertoire diversity by preferential expansion of naive T cell subsets. J. Exp. Med. 2008, 205, 1701–1714. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Su, J.; Sun, R.; Sun, Y.; Wang, Y.; Dong, Y.; Shi, B.; Jiang, H.; Li, Z. Coexpression of IL7 and CCL21 Increases Efficacy of CAR-T Cells in Solid Tumors without Requiring Preconditioned Lymphodepletion. Clin. Cancer Res. 2020, 26, 5494–5505. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Sukumaran, S.; Bajgain, P.; Watanabe, N.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Fisher, W.E.; Leen, A.M.; Vera, J.F. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol. Ther. 2017, 25, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, H.; Luo, H.; Sun, Y.; Shi, B.; Sun, R.; Li, Z. An IL-4/21 Inverted Cytokine Receptor Improving CAR-T Cell Potency in Immunosuppressive Solid-Tumor Microenvironment. Front. Immunol. 2019, 10, 1691. [Google Scholar] [CrossRef] [PubMed]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zila, N.; Hoeller, C.; Paulitschke, V. Novel immune checkpoints beyond PD-1 in advanced melanoma. memo - Mag. Eur. Med Oncol. 2021, 14, 135–142. [Google Scholar] [CrossRef]
- Wei, F.; Zhong, S.; Ma, Z.; Kong, H.; Medvec, A.; Ahmed, R.; Freeman, G.J.; Krogsgaard, M.; Riley, J.L. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc. Natl. Acad. Sci. USA 2013, 110, E2480–E2489. [Google Scholar] [CrossRef]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubat, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef]
- Kinter, A.L.; Godbout, E.J.; McNally, J.P.; Sereti, I.; Roby, G.A.; O’Shea, M.A.; Fauci, A.S. The Common γ-Chain Cytokines IL-2, IL-7, IL-15, and IL-21 Induce the Expression of Programmed Death-1 and Its Ligands. J. Immunol. 2008, 181, 6738–6746. [Google Scholar] [CrossRef]
- Schneider, H.; Valk, E.; Leung, R.; Rudd, C.E. CTLA-4 Activation of Phosphatidylinositol 3-Kinase (PI 3-K) and Protein Kinase B (PKB/AKT) Sustains T-Cell Anergy without Cell Death. PLoS ONE 2008, 3, e3842. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed]
- Ikemizu, S.; Gilbert, R.; Fennelly, J.A.; Collins, A.V.; Harlos, K.; Jones, Y.; Stuart, D.; Davis, S.J. Structure and Dimerization of a Soluble Form of B7-1. Immunity 2000, 12, 51–60. [Google Scholar] [CrossRef]
- Bhatia, S.; Sun, K.; Almo, S.C.; Nathenson, S.G.; Hodes, R.J. Dynamic Equilibrium of B7-1 Dimers and Monomers Differentially Affects Immunological Synapse Formation and T Cell Activation in Response to TCR/CD28 Stimulation. J. Immunol. 2010, 184, 1821–1828. [Google Scholar] [CrossRef]
- Ha, D.; Tanaka, A.; Kibayashi, T.; Tanemura, A.; Sugiyama, D.; Wing, J.B.; Lim, E.L.; Teng, K.W.W.; Adeegbe, D.; Newell, E.; et al. Differential control of human Treg and effector T cells in tumor immunity by Fc-engineered anti–CTLA-4 antibody. Proc. Natl. Acad. Sci. 2019, 116, 609–618. [Google Scholar] [CrossRef]
- Zhu, Y.; Paniccia, A.; Schulick, A.C.; Chen, W.; Koenig, M.R.; Byers, J.T.; Yao, S.; Bevers, S.; Edil, B.H. Identification of CD112R as a novel checkpoint for human T cells. J. Exp. Med. 2016, 213, 167–176. [Google Scholar] [CrossRef]
- Bottino, C.; Castriconi, R.; Pende, D.; Rivera, P.; Nanni, M.; Carnemolla, B.; Cantoni, C.; Grassi, J.; Marcenaro, S.; Reymond, N.; et al. Identification of PVR (CD155) and Nectin-2 (CD112) as Cell Surface Ligands for the Human DNAM-1 (CD226) Activating Molecule. J. Exp. Med. 2003, 198, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; A Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2008, 10, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Seth, S.; Maier, M.; Qiu, Q.; Ravens, I.; Kremmer, E.; Forster, R.; Bernhardt, G. The murine pan T cell marker CD96 is an adhesion receptor for CD155 and nectin-1. Biochem. Biophys. Res. Commun. 2007, 364, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef]
- John, L.B.; Devaud, C.P.; Duong, C.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 Antibody Therapy Potently Enhances the Eradication of Established Tumors By Gene-Modified T Cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; Van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Chun, J.Y.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Gestermann, N.; Saugy, D.; Martignier, C.; Tillé, L.; Marraco, S.A.F.; Zettl, M.; Tirapu, I.; Speiser, D.E.; Verdeil, G. LAG-3 and PD-1+LAG-3 inhibition promote anti-tumor immune responses in human autologous melanoma/T cell co-cultures. OncoImmunology 2020, 9, 1736792. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef]
- Zelba, H.; Bedke, J.; Hennenlotter, J.; Mostböck, S.; Zettl, M.; Zichner, T.; Chandran, P.A.; Stenzl, A.; Rammensee, H.-G.; Gouttefangeas, C. PD-1 and LAG-3 Dominate Checkpoint Receptor–Mediated T-cell Inhibition in Renal Cell Carcinoma. Cancer Immunol. Res. 2019, 7, 1891–1899. [Google Scholar] [CrossRef]
- Seidel, J.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Shen, L.; Li, H.; Bin, S.; Li, P.; Chen, J.; Gu, H.; Yuan, W. The efficacy of third generation anti-HER2 chimeric antigen receptor T cells in combination with PD1 blockade against malignant glioblastoma cells. Oncol. Rep. 2019, 42, 1549–1557. [Google Scholar] [CrossRef]
- Li, H.; Yuan, W.; Bin, S.; Wu, G.; Li, P.; Liu, M.; Yang, J.; Li, X.; Yang, K.; Gu, H. Overcome trastuzumab resistance of breast cancer using anti-HER2 chimeric antigen receptor T cells and PD1 blockade. Am. J. Cancer Res. 2020, 10, 688–703. [Google Scholar] [PubMed]
- Richards, R.M.; Sotillo, E.; Majzner, R.G. CAR T Cell Therapy for Neuroblastoma. Front. Immunol. 2018, 9, 2380. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-T.; Liu, J.-H.; Song, T.-T.; Ma, B.; Amidula, N.; Bai, C. EGLIF-CAR-T Cells Secreting PD-1 Blocking Antibodies Significantly Mediate the Elimination of Gastric Cancer. Cancer Manag. Res. 2020, 12, 8893–8902. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-E.; Yu, J.; Wang, Y.; Wang, H.; Wang, J.; Wang, Y.; Yu, L.; Yan, Z. ShRNA-mediated silencing of PD-1 augments the efficacy of chimeric antigen receptor T cells on subcutaneous prostate and leukemia xenograft. Biomed. Pharmacother. 2021, 137, 111339. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhang, Q.; Li, D.; Zhang, L.; Gu, Z.; Liu, J.; Liu, G.; Yang, M.; Gu, J.; Cui, X.; et al. PD-1 silencing improves anti-tumor activities of human mesothelin-targeted CAR T cells. Hum. Immunol. 2021, 82, 130–138. [Google Scholar] [CrossRef]
- Torikai, H.; Reik, A.; Liu, P.-Q.; Zhou, Y.; Zhang, L.; Maiti, S.; Huls, H.; Miller, J.C.; Kebriaei, P.; Rabinovitch, B.; et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 2012, 119, 5697–5705. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Meng, T.; Zhao, Z.; Han, J.; Zhang, W.; Gao, F.; Cai, J. CRISPR knock out CTLA-4 enhances the anti-tumor activity of cytotoxic T lymphocytes. Gene 2017, 636, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Gu, Y.-M.; Zhang, F.; Zhang, Z.-C.; Zhang, Y.-T.; He, Y.-D.; Wang, L.; Zhou, N.; Tang, F.-T.; Liu, H.-J.; et al. Construction of PD1/CD28 chimeric-switch receptor enhances anti-tumor ability of c-Met CAR-T in gastric cancer. OncoImmunology 2021, 10, 1901434. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lei, W.; Zhang, C.; Yang, C.; Wei, J.; Guo, Q.; Guo, X.; Chen, Z.; Lu, Y.; Young, K.H.; et al. CD19-specific CAR T Cells that Express a PD-1/CD28 Chimeric Switch-Receptor are Effective in Patients with PD-L1–positive B-Cell Lymphoma. Clin. Cancer Res. 2021, 27, 473–484. [Google Scholar] [CrossRef]
- Odorizzi, P.M.; Pauken, K.E.; Paley, M.A.; Sharpe, A.H.; Wherry, E.J. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med. 2015, 212, 1125–1137. [Google Scholar] [CrossRef]
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.E.; et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Investig. 2014, 124, 2246–2259. [Google Scholar] [CrossRef]
- Mognol, G.P.; Spreafico, R.; Wong, V.; Scott-Browne, J.P.; Togher, S.; Hoffmann, A.; Hogan, P.G.; Rao, A.; Trifari, S. Exhaustion-associated regulatory regions in CD8+ tumor-infiltrating T cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2776–E2785. [Google Scholar] [CrossRef]
- Verma, V.; Shrimali, R.K.; Ahmad, S.; Dai, W.; Wang, H.; Lu, S.; Nandre, R.; Gaur, P.; Lopez, J.; Sade-Feldman, M.; et al. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1+CD38hi cells and anti-PD-1 resistance. Nat. Immunol. 2019, 20, 1231–1243. [Google Scholar] [CrossRef]
- Gattinoni, L.; Speiser, D.E.; Lichterfeld, M.; Bonini, C. T memory stem cells in health and disease. Nat. Med. 2017, 23, 18–27. [Google Scholar] [CrossRef]
- Kueberuwa, G.; Gornall, H.; Alcantar-Orozco, E.M.; Bouvier, D.; Kapacee, Z.A.; Hawkins, R.E.; Gilham, D.E. CCR7+ selected gene-modified T cells maintain a central memory phenotype and display enhanced persistence in peripheral blood in vivo. J. Immunother. Cancer 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Biasco, L.; Scala, S.; Ricci, L.B.; Dionisio, F.; Baricordi, C.; Calabria, A.; Giannelli, S.; Cieri, N.; Barzaghi, F.; Pajno, R.; et al. In vivo tracking of T cells in humans unveils decade-long survival and activity of genetically modified T memory stem cells. Sci. Transl. Med. 2015, 7, 273ra13. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer Regression and Autoimmunity in Patients After Clonal Repopulation with Antitumor Lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated With Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Gattinoni, L.; Torabi-Parizi, P.; Kerstann, K.; Cardones, A.R.; Finkelstein, S.E.; Palmer, D.C.; Antony, P.A.; Hwang, S.T.; Rosenberg, S.A.; et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9571–9576. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Somerville, R.P.; Shi, V.; Rose, J.J.; Halverson, D.C.; Fowler, D.H.; Gea-Banacloche, J.C.; Pavletic, S.Z.; Hickstein, D.D.; Lu, T.L.; et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J. Clin. Oncol. 2016, 34, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Hudecek, M.; Pender, B.; Robinson, E.; Hawkins, R.; Chaney, C.; Cherian, S.; Chen, X.; et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+and CD4+CD19-specific chimeric antigen receptor–modified T cells. Sci. Transl. Med. 2016, 8, 355ra116. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Scott, C.D.; Leonardi, A.J.; Yamamoto, T.N.; Cruz, A.C.; Ouyang, C.; Ramaswamy, M.; Roychoudhuri, R.; Ji, Y.; Eil, R.L.; et al. Memory T cell–driven differentiation of naive cells impairs adoptive immunotherapy. J. Clin. Investig. 2015, 126, 318–334. [Google Scholar] [CrossRef]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor–positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Hinrichs, C.S.; Borman, Z.A.; Cassard, L.; Gattinoni, L.; Spolski, R.; Yu, Z.; Sanchez-Perez, L.; Muranski, P.; Kern, S.J.; Logun, C.; et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc. Natl. Acad. Sci. USA 2009, 106, 17469–17474. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen–specific CAR T cells are clinically active in multiple myeloma. J. Clin. Investig. 2019, 129, 2210–2221. [Google Scholar] [CrossRef]
- Gattinoni, L.; Zhong, X.-S.; Palmer, D.; Ji, Y.; Hinrichs, C.S.; Yu, Z.; Wrzesinski, C.; Boni, A.; Cassard, L.; Garvin, L.M.; et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med. 2009, 15, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Klebanoff, C.A.; Palmer, D.C.; Wrzesinski, C.; Kerstann, K.W.; Yu, Z.; Finkelstein, S.E.; Theoret, M.R.; Rosenberg, S.A.; Restifo, N.P. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J. Clin. Investig. 2005, 115, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wong, C.W.; Urak, R.; Taus, E.; Aguilar, B.; Chang, W.-C.; Mardiros, A.; Budde, L.E.; Brown, C.E.; Berger, C.; et al. Comparison of naïve and central memory derived CD8+effector cell engraftment fitness and function following adoptive transfer. OncoImmunology 2015, 5, e1072671. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Rossi, J.M.; Neelapu, S.S.; Jacobson, C.A.; Miklos, D.B.; Ghobadi, A.; Oluwole, O.O.; Reagan, P.M.; Lekakis, L.J.; Lin, Y.; et al. Tumor burden, inflammation, and product attributes determine outcomes of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. 2020, 4, 4898–4911. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pós, Z.; Paulos, C.M.; Quigley, M.F.; de Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell–like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, D.; Wong, R.A.; Yang, X.; Wang, D.; Pecoraro, J.R.; Kuo, C.-F.; Aguilar, B.; Qi, Y.; Ann, D.K.; Starr, R.; et al. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing mTORC1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol. Res. 2019, 7, 759–772. [Google Scholar] [CrossRef]
- Alvarez-Fernández, C.; Escribà-Garcia, L.; Vidal, S.; Sierra, J.; Briones, J. A short CD3/CD28 costimulation combined with IL-21 enhance the generation of human memory stem T cells for adoptive immunotherapy. J. Transl. Med. 2016, 14, 1–10. [Google Scholar] [CrossRef]
- Hinrichs, C.S.; Spolski, R.; Paulos, C.M.; Gattinoni, L.; Kerstann, K.W.; Palmer, D.C.; Klebanoff, C.A.; Rosenberg, S.A.; Leonard, W.J.; Restifo, N.P. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood 2008, 111, 5326–5333. [Google Scholar] [CrossRef]
- Pilipow, K.; Roberto, A.; Roederer, M.; Waldmann, T.A.; Mavilio, D.; Lugli, E. IL15 and T-cell Stemness in T-cell–Based Cancer Immunotherapy. Cancer Res. 2015, 75, 5187–5193. [Google Scholar] [CrossRef]
- Ding, Z.-C.; Liu, C.; Cao, Y.; Habtetsion, T.; Kuczma, M.; Pi, W.; Kong, H.; Cacan, E.; Greer, S.F.; Cui, Y.; et al. IL-7 signaling imparts polyfunctionality and stemness potential to CD4+ T cells. OncoImmunology 2016, 5, e1171445. [Google Scholar] [CrossRef] [PubMed]
- Konforte, D.; Paige, C.J. Identification of Cellular Intermediates and Molecular Pathways Induced by IL-21 in Human B Cells. J. Immunol. 2006, 177, 8381–8392. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Spolski, R.; Casas, E.; Zhu, W.; Levy, D.E.; Leonard, W.J. The molecular basis of IL-21–mediated proliferation. Blood 2007, 109, 4135–4142. [Google Scholar] [CrossRef]
- Kondo, T.; Imura, Y.; Chikuma, S.; Hibino, S.; Omata-Mise, S.; Ando, M.; Akanuma, T.; Iizuka, M.; Sakai, R.; Morita, R.; et al. Generation and application of human induced-stem cell memory T cells for adoptive immunotherapy. Cancer Sci. 2018, 109, 2130–2140. [Google Scholar] [CrossRef]
- Vodnala, S.K.; Eil, R.; Kishton, R.J.; Sukumar, M.; Yamamoto, T.N.; Ha, N.-H.; Lee, P.-H.; Shin, M.; Patel, S.J.; Yu, Z.; et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 2019, 363, eaau0135. [Google Scholar] [CrossRef] [PubMed]
- Scholz, G.; Jandus, C.; Zhang, L.; Grandclément, C.; Lopez-Mejia, I.C.; Soneson, C.; Delorenzi, M.; Fajas, L.; Held, W.; Dormond, O.; et al. Modulation of mTOR Signalling Triggers the Formation of Stem Cell-like Memory T Cells. EBioMedicine 2016, 4, 50–61. [Google Scholar] [CrossRef]
- Mousset, C.M.; Hobo, W.; Ji, Y.; Fredrix, H.; De Giorgi, V.; Allison, R.D.; Kester, M.G.D.; Falkenburg, J.H.F.; Schaap, N.P.M.; Jansen, J.H.; et al. Ex vivo AKT-inhibition facilitates generation of polyfunctional stem cell memory-like CD8+ T cells for adoptive immunotherapy. OncoImmunology 2018, 7, e1488565. [Google Scholar] [CrossRef]
- Lanzavecchia, A.; Sallusto, F. Progressive differentiation and selection of the fittest in the immune response. Nat. Rev. Immunol. 2002, 2, 982–987. [Google Scholar] [CrossRef]
- Schepers, K.; Swart, E.; Van Heijst, J.W.; Gerlach, C.; Castrucci, M.; Sie, D.; Heimerikx, M.; Velds, A.; Kerkhoven, R.M.; Arens, R.; et al. Dissecting T cell lineage relationships by cellular barcoding. J. Exp. Med. 2008, 205, 2309–2318. [Google Scholar] [CrossRef]
- Gerlach, C.; Rohr, J.C.; Perié, L.; van Rooij, N.; van Heijst, J.W.J.; Velds, A.; Urbanus, J.; Naik, S.H.; Jacobs, H.; Beltman, J.B.; et al. Heterogeneous Differentiation Patterns of Individual CD8+ T Cells. Science 2013, 340, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Jensen, M.C.; Lansdorp, P.M.; Gough, M.; Elliott, C.; Riddell, S.R. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Investig. 2008, 118, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Cieri, N.; Oliveira, G.; Greco, R.; Forcato, M.; Taccioli, C.; Cianciotti, B.; Valtolina, V.; Noviello, M.; Vago, L.; Bondanza, A.; et al. Generation of human memory stem T cells after haploidentical T-replete hematopoietic stem cell transplantation. Blood 2015, 125, 2865–2874. [Google Scholar] [CrossRef]
- Roberto, A.; Castagna, L.; Zanon, V.; Bramanti, S.; Crocchiolo, R.; McLaren, J.E.; Gandolfi, S.; Tentorio, P.; Sarina, B.; Timofeeva, I.; et al. Role of naive-derived T memory stem cells in T-cell reconstitution following allogeneic transplantation. Blood 2015, 125, 2855–2864. [Google Scholar] [CrossRef]
- Crompton, J.G.; Narayanan, M.; Cuddapah, S.; Roychoudhuri, R.; Ji, Y.; Yang, W.; Patel, S.J.; Sukumar, M.; Palmer, D.; Peng, W.; et al. Lineage relationship of CD8+ T cell subsets is revealed by progressive changes in the epigenetic landscape. Cell. Mol. Immunol. 2016, 13, 502–513. [Google Scholar] [CrossRef]
- Takeshita, M.; Suzuki, K.; Kassai, Y.; Takiguchi, M.; Nakayama, Y.; Otomo, Y.; Morita, R.; Miyazaki, T.; Yoshimura, A.; Takeuchi, T. Polarization diversity of human CD4+ stem cell memory T cells. Clin. Immunol. 2015, 159, 107–117. [Google Scholar] [CrossRef]
- Ando, M.; Ito, M.; Srirat, T.; Kondo, T.; Yoshimura, A. Memory T cell, exhaustion, and tumor immunity. Immunol. Med. 2019, 43, 1–9. [Google Scholar] [CrossRef]
- Tejera, M.M.; Kim, E.H.; Sullivan, J.A.; Plisch, E.H.; Suresh, M. FoxO1 Controls Effector-to-Memory Transition and Maintenance of Functional CD8 T Cell Memory. J. Immunol. 2013, 191, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.V.; Ouyang, W.; Liao, W.; Zhang, M.Q.; Li, M.O. The Transcription Factor Foxo1 Controls Central-Memory CD8+ T Cell Responses to Infection. Immunity 2013, 39, 286–297. [Google Scholar] [CrossRef]
- Kim, C.; Jin, J.; Weyand, C.M.; Goronzy, J.J. The Transcription Factor TCF1 in T Cell Differentiation and Aging. Int. J. Mol. Sci. 2020, 21, 6497. [Google Scholar] [CrossRef]
- Yu, Q.; Sharma, A.; Sen, J.M. TCF1 and β-catenin regulate T cell development and function. Immunol. Res. 2010, 47, 45–55. [Google Scholar] [CrossRef]
- Patrick, M.S.; Cheng, N.-L.; Kim, J.; An, J.; Dong, F.; Yang, Q.; Zou, I.; Weng, N.-P. Human T Cell Differentiation Negatively Regulates Telomerase Expression Resulting in Reduced Activation-Induced Proliferation and Survival. Front. Immunol. 2019, 10, 1993. [Google Scholar] [CrossRef] [PubMed]
- McLellan, A.D.; Rad, S.M.A.H. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol. Cell Biol. 2019, 97, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Fowell, D.J.; Kim, M. The spatio-temporal control of effector T cell migration. Nat. Rev. Immunol. 2021, 21, 582–596. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietrobon, V.; Todd, L.A.; Goswami, A.; Stefanson, O.; Yang, Z.; Marincola, F. Improving CAR T-Cell Persistence. Int. J. Mol. Sci. 2021, 22, 10828. https://doi.org/10.3390/ijms221910828
Pietrobon V, Todd LA, Goswami A, Stefanson O, Yang Z, Marincola F. Improving CAR T-Cell Persistence. International Journal of Molecular Sciences. 2021; 22(19):10828. https://doi.org/10.3390/ijms221910828
Chicago/Turabian StylePietrobon, Violena, Lauren Anne Todd, Anghsumala Goswami, Ofir Stefanson, Zhifen Yang, and Francesco Marincola. 2021. "Improving CAR T-Cell Persistence" International Journal of Molecular Sciences 22, no. 19: 10828. https://doi.org/10.3390/ijms221910828
APA StylePietrobon, V., Todd, L. A., Goswami, A., Stefanson, O., Yang, Z., & Marincola, F. (2021). Improving CAR T-Cell Persistence. International Journal of Molecular Sciences, 22(19), 10828. https://doi.org/10.3390/ijms221910828