
ADP-Ribosylation as Post-Translational Modification of Proteins: Use of Inhibitors in Cancer Control

Abstract
1. Introduction
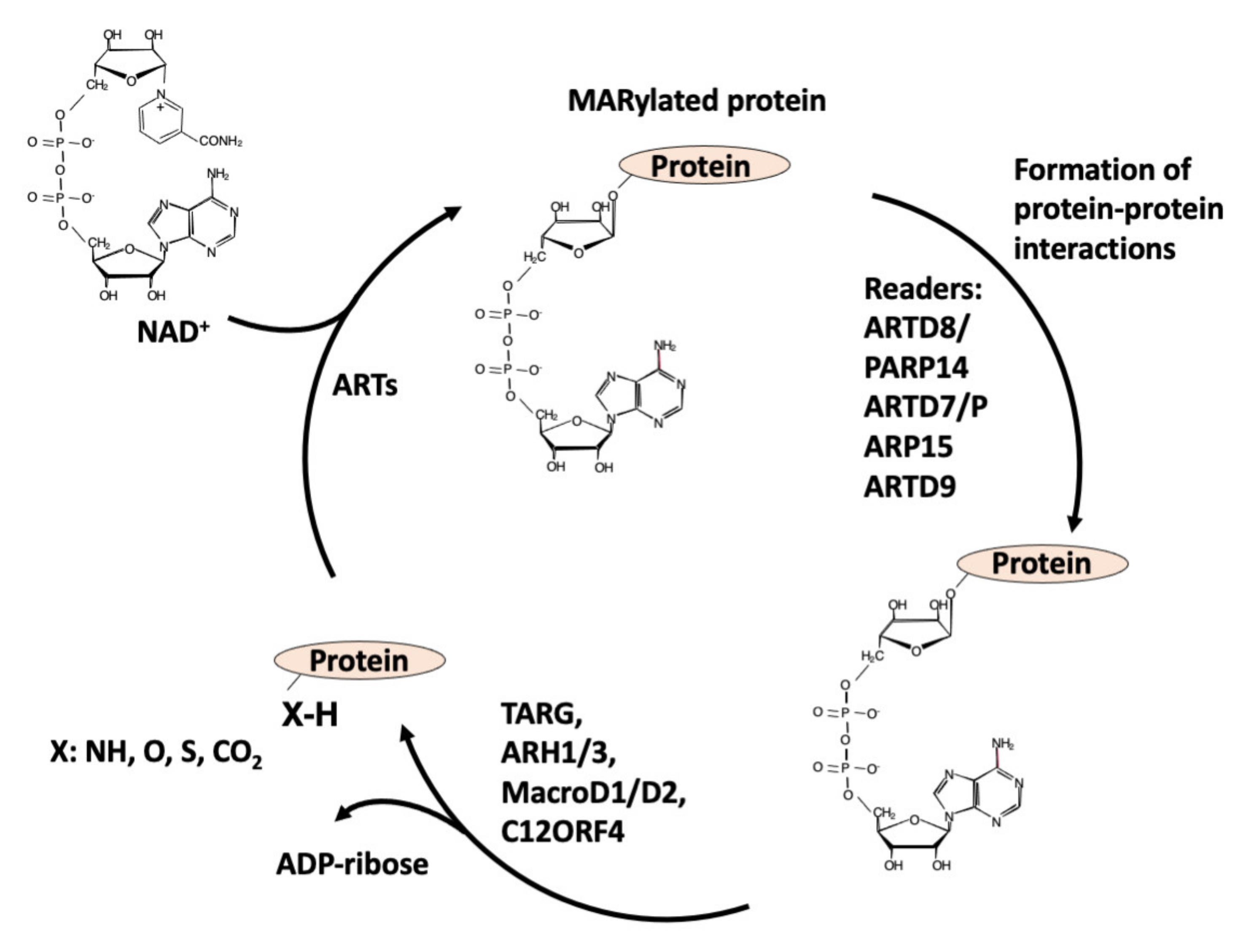
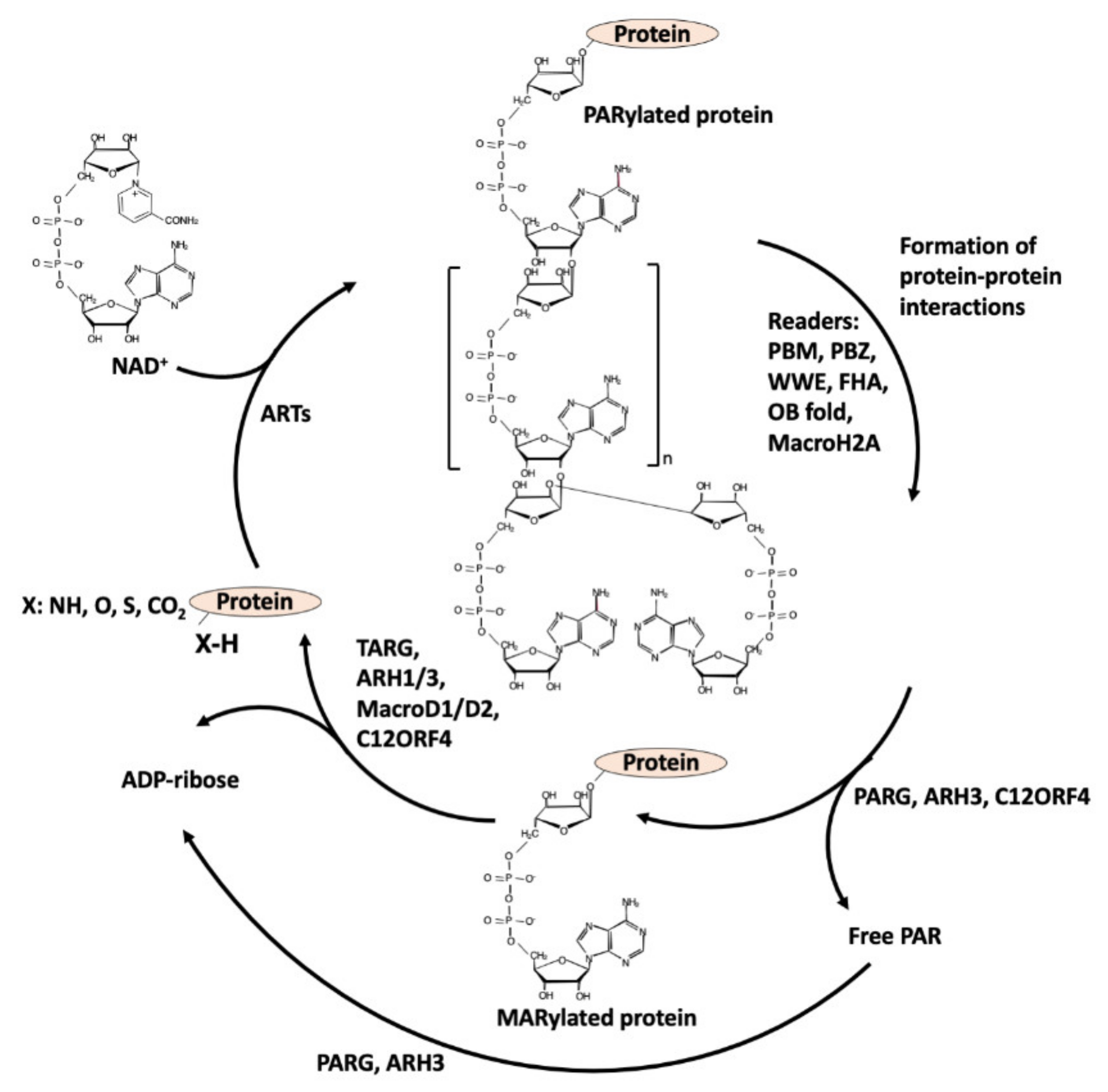
2. ADP-Ribosylation
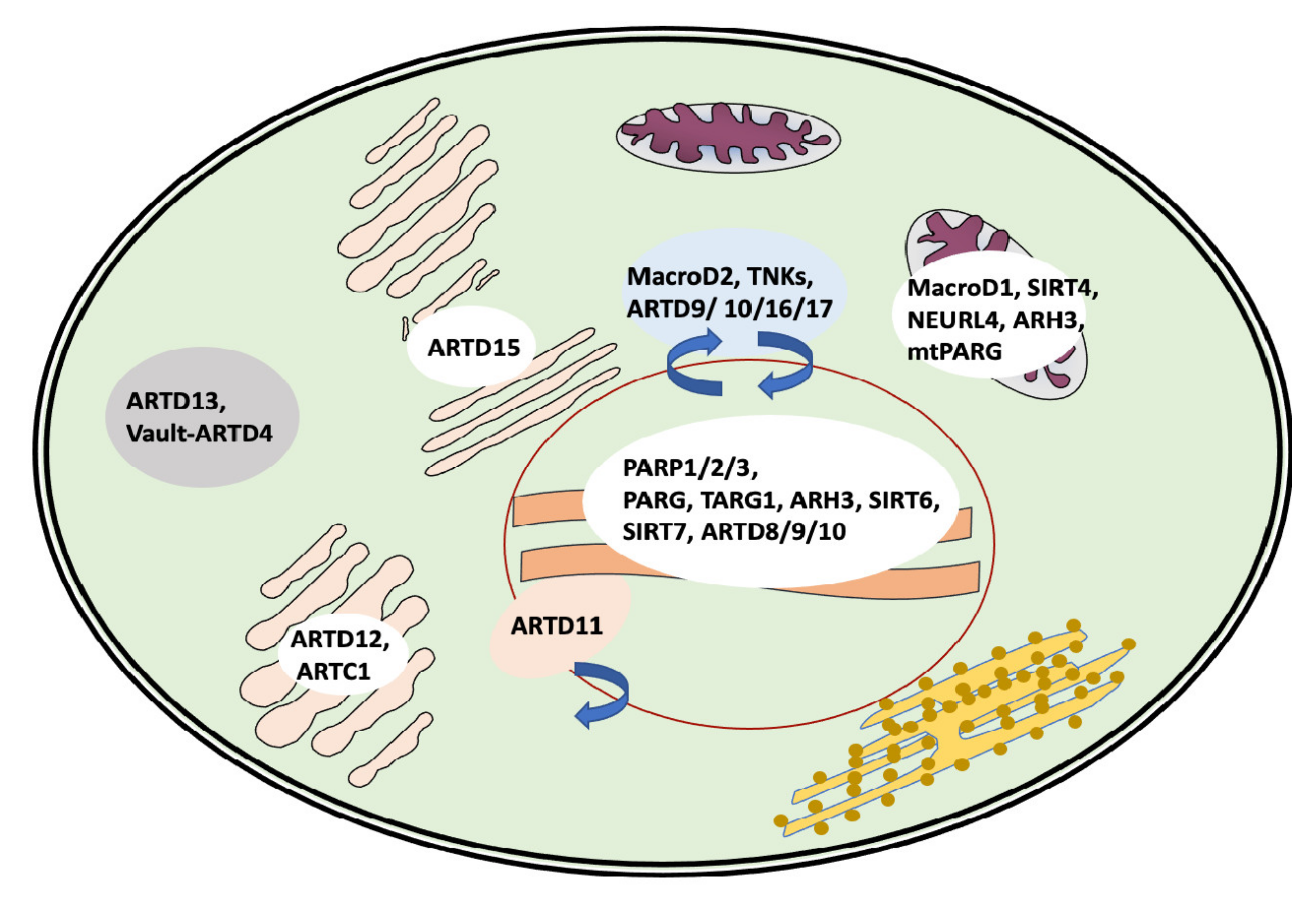
2.1. Enzymes Involved in ADP-Ribosylation of Proteins: The Writers
2.2. Amino Acids Modified by MAR/PAR
3. Readers
4. Erasers
5. Various Types of Post-Translational Modification of PARP1 and Competition on Amino Acid Substrates
6. Cancer Proliferation, Metastasis, Angiogenesis: The Role of ADP-Ribosylating Enzymes According to a Deregulated Expression or Use of ART Specific Inhibitors
6.1. PARP1/2/3 Inhibitors in Cancer Treatment
6.2. Tankyrases: Cell Functions and TNKS Inhibitors in Cancer Treatment
6.3. Inhibitors Specific for MARylating Enzymes with A Role in Cancer Development
6.3.1. Macro-PARPs: ARTD7, ARTD8, ARTD9
6.3.2. Other MARylating ART Enzymes Linked to Cancer and Potential Enzyme Inhibitors
6.4. Sirtuin Inhibitors Addressing the Roles of MARylating Sirtuins in Cancer
7. Discussion and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poltronieri, P.; Celetti, A.; Palazzo, L. Mono(ADP-ribosyl)ation Enzymes and NAD+ Metabolism: A Focus on Diseases and Therapeutic Perspectives. Cells 2021, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Poltronieri, P.; Miwa, M. Overview on ADP Ribosylation and PARP Superfamily of Proteins. Editorial. Curr. Protein Peptide Sci. 2016, 17, 630–632. [Google Scholar] [CrossRef]
- Perina, D.; Mikoč, A.; Ahel, J.; Ćetković, H.; Žaja, R.; Ahel, I. Distribution of protein poly(ADP-ribosyl)ation systems across all domains of life. DNA Repair 2014, 23, 4–16. [Google Scholar] [CrossRef]
- Citarelli, M.; Teotia, S.; Lamb, R.S. Evolutionary history of the poly(ADP-ribose) polymerase gene family in eukaryotes. BMC Evol. Biol. 2010, 10, 308. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Barbieri, J. Molecular mechanisms of the cytotoxicity of ADP-ribosylating toxins. Annu. Rev. MicroBiol. 2008, 62, 271–288. [Google Scholar] [CrossRef] [PubMed]
- Di Girolamo, M.; Fabrizio, G. The ADP-Ribosyl-Transferases Diphtheria Toxin-Like (ARTDs) Family: An Overview. Challenges 2018, 9, 24. [Google Scholar] [CrossRef]
- Kraus, W.L. PARPs and ADP-ribosylation: 50 years and counting. Mol. Cell 2015, 58, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Miwa, M.; Masutani, M. PolyADP-ribosylation and cancer. Cancer Sci. 2007, 98, 1528–1535. [Google Scholar] [CrossRef]
- Loseva, O.; Jemth, A.S.; Bryant, H.E.; Schüler, H.; Lehtiö, L.; Karlberg, T.; Helleday, T. PARP-3 is a mono-ADP-ribosylase that activates PARP-1 in the absence of DNA. J. Biol. Chem. 2010, 285, 8054–8060. [Google Scholar] [CrossRef]
- Van Beek, L.; McClay, É.; Patel, S.; Schimpl, M.; Spagnolo, L.; Maia de Oliveira, T. PARP Power: A Structural Perspective on PARP1, PARP2, and PARP3 in DNA Damage Repair and Nucleosome Remodelling. Int. J. Mol. Sci. 2021, 22, 5112. [Google Scholar] [CrossRef]
- Beck, C.; Robert, I.; Reina-San-Martin, B.; Schreiber, V.; Dantzer, F. Poly(ADP-ribose) polymerases in double-strand break repair: Focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 2014, 329, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, B.; Ahel, I.; Altmeyer, M.; Ashworth, A.; Bai, P.; Chang, P.; Cohen, M.; Corda, D.; Dantzer, F.; Daugherty, M.D.; et al. ADP-ribosyltransferases, an update on function and nomenclature. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Chatrin, C.; Gabrielsen, M.; Buetow, L.; Nakasone, M.A.; Ahmed, S.F.; Sumpton, D.; Sibbet, G.J.; Smith, B.O.; Huang, D.T. Structural insights into ADP-ribosylation of ubiquitin by Deltex family E3 ubiquitin ligases. Sci. Adv. 2020, 6, eabc0418. [Google Scholar] [CrossRef] [PubMed]
- Challa, S.; Stokes, M.S.; Kraus, W.L. MARTs and MARylation in the Cytosol: Biological Functions, Mechanisms of Action, and Therapeutic Potential. Cells 2021, 10, 313. [Google Scholar] [CrossRef] [PubMed]
- Di Girolamo, M.; Fabrizio, G. Overview of the mammalian ADP-ribosyl-transferases clostridia toxin-like (ARTCs) family. Biochem. Pharmacol. 2019, 167, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Poltronieri, P.; Mezzolla, V.; Farooqi, A.A.; Di Girolamo, M. NAD precursors, mitochondria targeting compounds and ADP-ribosylation inhibitors in treatment of inflammatory diseases and cancer. Curr. Med. Chem. 2021. [Google Scholar] [CrossRef]
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic β cells. Cell 2006, 126, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Bonfiglio, J.J.; Fontana, P.; Zhang, Q.; Colby, T.; Gibbs-Seymour, I.; Atanassov, I.; Bartlett, E.; Zaja, R.; Ahel, I.; Matic, I. Serine ADP-Ribosylation Depends on HPF1. Mol. Cell 2017, 65, 932–940.e6. [Google Scholar] [CrossRef]
- Suskiewicz, M.J.; Zobel, F.; Ogden, T.E.H.; Fontana, P.; Ariza, A.; Yang, J.-C.; Zhu, K.; Bracken, L.; Hawthorne, W.J.; Ahel, D.; et al. HPF1 completes the PARP active site for DNA damage-induced ADP-ribosylation. Nature 2020, 579, 598–602. [Google Scholar] [CrossRef]
- Larsen, S.C.; Hendriks, I.A.; Lodge, J.M.; Rykær, M.; Furtwängler, B.; Shishkova, E.; Westphall, M.S.; Coon, J.J.; Nielsen, M.L. Mapping Physiological ADP-Ribosylation Using Activated Ion Electron Transfer Dissociation. Cell Rep. 2020, 32, 108176. [Google Scholar] [CrossRef]
- Larsen, S.C.; Hendriks, I.A.; Lyon, D.; Jensen, L.J.; Nielsen, M.L. Systems-wide Analysis of Serine ADP-Ribosylation Reveals Widespread Occurrence and Site-Specific Overlap with Phosphorylation. Cell Rep. 2018, 24, 2493–2505.e4. [Google Scholar] [CrossRef]
- Bilan, V.; Leutert, M.; Nanni, P.; Panse, C.; Hottiger, M.O. Combining Higher-Energy Collision Dissociation and Electron-Transfer/Higher-Energy Collision Dissociation Fragmentation in a Product-Dependent Manner Confidently Assigns Proteomewide ADP-Ribose Acceptor Sites. Anal. Chem. 2017, 89, 1523–1530. [Google Scholar] [CrossRef]
- Crawford, K.; Bonfiglio, J.J.; Mikoč, A.; Matic, I.; Ahel, I. Specificity of reversible ADP-ribosylation and regulation of cellular processes. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 64–82. [Google Scholar] [CrossRef]
- Laing, S.; Unger, M.; Koch-Nolte, F.; Haag, F. ADP-ribosylation of arginine. Amino Acids 2011, 41, 257–269. [Google Scholar] [CrossRef]
- McDonald, L.J.; Moss, J. Enzymatic and nonenzymatic ADP-ribosylation of cysteine. Mol. Cell. Biochem. 1994, 138, 221–226. [Google Scholar] [CrossRef]
- Jacobson, E.L.; Cervantes-Laurean, D.; Jacobson, M.K. ADP-ribose in glycation and glycoxidation reactions. Adv. Exp. Med. Biol. 1997, 419, 371–379. [Google Scholar] [CrossRef]
- Lüscher, B.; Bütepage, M.; Eckei, L.; Krieg, S.; Verheugd, P.; Shilton, B.H. ADP-Ribosylation, a Multifaceted Posttranslational Modification Involved in the Control of Cell Physiology in Health and Disease. Chem. Rev. 2018, 118, 1092–1136. [Google Scholar] [CrossRef] [PubMed]
- DaRosa, P.A.; Wang, Z.; Jiang, X.; Pruneda, J.N.; Cong, F.; Klevit, R.E.; Xu, W. Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP-ribosyl)ation signal. Nature 2015, 517, 223–226. [Google Scholar] [CrossRef] [PubMed]
- DaRosa, P.A.; Klevit, R.E.; Xu, W. Structural basis for tankyrase-RNF146 interaction reveals noncanonical tankyrase-binding motifs. Protein Sci. 2018, 27, 1057–1067. [Google Scholar] [CrossRef]
- Chandrakumar, A.A.; Coyaud, É.; Marshall, C.B.; Ikura, M.; Raught, B.; Rottapel, R. Tankyrase regulates epithelial lumen formation via suppression of Rab11 GEFs. J. Cell Biol. 2021, 220, e202008037. [Google Scholar] [CrossRef] [PubMed]
- Zamudio-Martinez, E.; Herrera-Campos, A.B.; Muñoz, A.; Rodríguez-Vargas, J.M.; Oliver, F.J. Tankyrases as modulators of pro-tumoral functions: Molecular insights and therapeutic opportunities. J. Exp. Clin. Cancer Res. 2021, 40, 144. [Google Scholar] [CrossRef]
- Verheugd, P.; Bütepage, M.; Eckei, L.; Lüscher, B. Players in ADP-ribosylation: Readers and Erasers. Curr. Protein Pept. Sci. 2016, 17, 654–667. [Google Scholar] [CrossRef]
- Zhang, F.; Shi, J.; Chen, S.H.; Bian, C.; Yu, X. The PIN domain of EXO1 recognizes poly(ADP-ribose) in DNA damage response. Nucleic Acids Res. 2015, 43, 10782–10794. [Google Scholar] [CrossRef]
- Poltronieri, P.; Čerekovic, N. Roles of Nicotinamide Adenine Dinucleotide (NAD+) in Biological Systems. Challenges 2018, 9, 3. [Google Scholar] [CrossRef]
- Dani, N.; Stilla, A.; Marchegiani, A.; Tamburro, A.; Till, S.; Ladurner, A.G.; Corda, D.; Di Girolamo, M. Combining affinity purification by ADP-ribose-binding macro domains with mass spectrometry to define the mammalian ADP-ribosyl proteome. Proc. Natl. Acad. Sci. USA 2009, 106, 4243–4248. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.; Rosenthal, F.; Karlberg, T.; Bütepage, M.; Thorsell, A.G.; Dreier, B.; Grossmann, J.; Sobek, J.; Imhof, R.; Lüscher, B.; et al. Engineering Af1521 improves ADP-ribose binding and identification of ADP-ribosylated proteins. Nat. Commun. 2020, 11, 5199. [Google Scholar] [CrossRef] [PubMed]
- García-Saura, A.G.; Herzog, L.K.; Dantuma, N.P.; Schüler, H. MacroGreen, a simple tool for detection of ADP-ribosylated proteins. Commun. Biol. 2021, 4, 919. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, P.D.; Hamilton, G.A.; Park, J.W.; Gamble, M.J. MacroH2A1 Regulation of Poly(ADP-Ribose) Synthesis and Stability Prevents Necrosis and Promotes DNA Repair. Mol. Cell. Biol. 2019, 40, e00230-19. [Google Scholar] [CrossRef]
- Simonet, N.G.; Thackray, J.K.; Vazquez, B.N.; Ianni, A.; Espinosa-Alcantud, M.; Morales-Sanfrutos, J.; Hurtado-Bagès, S.; Sabidó, E.; Buschbeck, M.; Tischfield, J.; et al. SirT7 auto-ADP-ribosylation regulates glucose starvation response through mH2A1. Sci. Adv. 2020, 6, eaaz2590. [Google Scholar] [CrossRef]
- Verma, P.; Zhou, Y.; Cao, Z.; Deraska, P.V.; Deb, M.; Arai, E.; Li, W.; Shao, Y.; Puentes, L.; Li, Y.; et al. ALC1 links chromatin accessibility to PARP inhibitor response in homologous recombination-deficient cells. Nat. Cell Biol. 2021, 23, 160–171. [Google Scholar] [CrossRef]
- Chen, S.H.; Yu, X. Targeting dePARylation selectively suppresses DNA repair-defective and PARP inhibitor-resistant malignancies. Sci. Adv. 2019, 5, eaav4340. [Google Scholar] [CrossRef]
- Wei, L.; Nakajima, S.; Hsieh, C.L.; Kanno, S.; Masutani, M.; Levine, A.S.; Yasui, A.; Lan, L. Damage response of XRCC1 at sites of DNA single strand breaks is regulated by phosphorylation and ubiquitylation after degradation of poly(ADP-ribose). J. Cell Sci. 2013, 126, 4414–4423. [Google Scholar] [CrossRef]
- Sasaki, Y.; Fujimori, H.; Hozumi, M.; Onodera, T.; Nozaki, T.; Murakami, Y.; Ashizawa, K.; Inoue, K.; Koizumi, F.; Masutani, M. Dysfunction of Poly (ADP-Ribose) Glycohydrolase Induces a Synthetic Lethal Effect in Dual Specificity Phosphatase 22-Deficient Lung Cancer Cells. Cancer Res. 2019, 79, 3851–3861. [Google Scholar] [CrossRef]
- Houl, J.H.; Ye, Z.; Brosey, C.A.; Balapiti-Modarage, L.P.F.; Namjoshi, S.; Bacolla, A.; Laverty, D.; Walker, B.L.; Pourfarjam, Y.; Warden, L.S.; et al. Selective small molecule PARG inhibitor causes replication fork stalling and cancer cell death. Nat. Commun. 2019, 10, 5654. [Google Scholar] [CrossRef] [PubMed]
- Pillay, N.; Tighe, A.; Nelson, L.; Littler, S.; Coulson-Gilmer, C.; Bah, N.; Golder, A.; Bakker, B.; Spierings, D.C.J.; James, D.I.; et al. DNA Replication Vulnerabilities Render Ovarian Cancer Cells Sensitive to Poly(ADP-Ribose) Glycohydrolase Inhibitors. Cancer Cell 2019, 35, 519–533.e8. [Google Scholar] [CrossRef]
- Sonoda, Y.; Sasaki, Y.; Gunji, A.; Shirai, H.; Araki, T.; Imamichi, S.; Onodera, T.; Rydén, A.M.; Watanabe, M.; Itami, J.; et al. Reduced Tumorigenicity of Mouse ES Cells and the Augmented Anti-Tumor Therapeutic Effects under Parg Deficiency. Cancers 2020, 12, 1056. [Google Scholar] [CrossRef]
- Janisiw, E.; Raices, M.; Balmir, F.; Paulin, L.F.; Baudrimont, A.; von Haeseler, A.; Yanowitz, J.L.; Jantsch, V.; Silva, N. Poly(ADP-ribose) glycohydrolase coordinates meiotic DNA double-strand break induction and repair independent of its catalytic activity. Nat. Commun. 2020, 11, 4869. [Google Scholar] [CrossRef]
- Fontana, P.; Bonfiglio, J.J.; Palazzo, L.; Bartlett, E.; Matic, I.; Ahel, I. Serine ADP-ribosylation reversal by the hydrolase ARH3. Elife 2017, 6, e28533. [Google Scholar] [CrossRef]
- Feijs, K.L.H.; Cooper, C.D.O.; Žaja, R. The Controversial Roles of ADP-Ribosyl Hydrolases MACROD1, MACROD2 and TARG1 in Carcinogenesis. Cancers 2020, 12, 604. [Google Scholar] [CrossRef] [PubMed]
- Rack, J.G.M.; Liu, Q.; Zorzini, V.; Voorneveld, J.; Ariza, A.; Honarmand Ebrahimi, K.; Reber, J.M.; Krassnig, S.C.; Ahel, D.; van der Marel, G.A.; et al. Mechanistic insights into the three steps of poly(ADP-ribosylation) reversal. Nat. Commun. 2021, 12, 4581. [Google Scholar] [CrossRef] [PubMed]
- Barkauskaite, E.; Jankevicius, G.; Ladurner, A.; Ahel, I.; Timinszky, G. The recognition and removal of cellular poly(ADP-ribose) signals. FEBS J. 2013, 280, 3491–3507. [Google Scholar] [CrossRef]
- Palazzo, L.; Daniels, C.M.; Nettleship, J.E.; Rahman, N.; McPherson, R.L.; Ong, S.E.; Kato, K.; Nureki, O.; Leung, A.K.; Ahel, I. ENPP1 processes protein ADP-ribosylation in vitro. FEBS J. 2016, 283, 3371–3388. [Google Scholar] [CrossRef]
- Wright, R.H.G.; Beato, M. Role of the NUDT Enzymes in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 2267. [Google Scholar] [CrossRef]
- Bu, X.; Kato, J.; Moss, J. Emerging roles of ADP-ribosyl-acceptor hydrolases (ARHs) in tumorigenesis and cell death pathways. Biochem. Pharmacol. 2019, 167, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Maruta, H.; Matsumura, N.; Tanuma, S. Role of (ADP-ribose)n catabolism in DNA repair. Biochem. Biophys. Res. Commun. 1997, 236, 265–269. [Google Scholar] [CrossRef]
- Mohseni, M.; Cidado, J.; Croessmann, S.; Cravero, K.; Cimino-Mathews, A.; Wong, H.Y.; Scharpf, R.; Zabransky, D.J.; Abukhdeir, A.M.; Garay, J.P.; et al. MACROD2 overexpression mediates estrogen independent growth and tamoxifen resistance in breast cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 17606–17611. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Fujioka, K.; Nakakido, M.; Hamamoto, R. Regulation of poly(ADP-Ribose) polymerase 1 functions by post-translational modifications. Front. Biosci. 2018, 23, 13–26. [Google Scholar] [CrossRef]
- Mao, Z.; Hine, C.; Tian, X.; Van Meter, M.; Au, M.; Vaidya, A.; Seluanov, A.; Gorbunova, V. SIRT6 promotes DNA repair under stress by activating PARP1. Science 2011, 332, 1443–1446. [Google Scholar] [CrossRef] [PubMed]
- Kukita, A.; Sone, K.; Oda, K.; Hamamoto, R.; Kaneko, S.; Komatsu, M.; Wada, M.; Honjoh, H.; Kawata, Y.; Kojima, M.; et al. Histone methyltransferase SMYD2 selective inhibitor LLY-507 in combination with poly ADP ribose polymerase inhibitor has therapeutic potential against high-grade serous ovarian carcinomas. Biochem. Biophys. Res. Commun. 2019, 513, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Kang, D.; Suzuki, T.; Masuda, A.; Dohmae, N.; Nakamura, Y.; Hamamoto, R. The histone methyltransferase SMYD2 methylates PARP1 and promotes poly(ADP-ribosyl)ation activity in cancer cells. Neoplasia 2014, 16, 257–264.e2. [Google Scholar] [CrossRef] [PubMed]
- Kassner, I.; Barandun, M.; Fey, M.; Rosenthal, F.; Hottiger, M.O. Crosstalk between SET7/9-dependent methylation and ARTD1-mediated ADP-ribosylation of histone H1.4. Epigenetics Chromatin 2013, 6, 1. [Google Scholar] [CrossRef]
- Kauppinen, T.M.; Chan, W.Y.; Suh, S.W.; Wiggins, A.K.; Huang, E.J.; Swanson, R.A. Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases 1/2. Proc. Natl. Acad. Sci. USA 2006, 103, 7136–7141. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Yamaguchi, H.; Wei, Y.; Hsu, J.L.; Wang, H.L.; Hsu, Y.H.; Lin, W.C.; Yu, W.H.; Leonard, P.G.; Lee, G.R., 4th; et al. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat. Med. 2016, 22, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Demény, M.A.; Virág, L. The PARP Enzyme Family and the Hallmarks of Cancer Part 1. Cell Intrinsic Hallmarks. Cancers 2021, 13, 2042. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Poltronieri, P.; Di Girolamo, M. ADP-ribosylation inhibitors in treatment of diseases. Biochem. Pharmacol. 2019, 167, 1–2. [Google Scholar] [CrossRef]
- Scarpa, E.S.; Fabrizio, G.; Di Girolamo, M. A role of intracellular mono-ADP-ribosylation in cancer biology. FEBS J. 2013, 280, 3551–3562. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Chang, P. New PARP targets for cancer therapy. Nat. Rev. Cancer 2014, 14, 502–509. [Google Scholar] [CrossRef]
- Curtin, N.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drug Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef]
- Wang, L.; Liang, C.; Li, F.; Guan, D.; Wu, X.; Fu, X.; Lu, A.; Zhang, G. PARP1 in Carcinomas and PARP1 Inhibitors as Antineoplastic Drugs. Int. J. Mol. Sci. 2017, 18, 2111. [Google Scholar] [CrossRef]
- Fu, L.; Wang, S.; Wang, X.; Wang, P.; Zheng, Y.; Yao, D.; Guo, M.; Zhang, L.; Ouyang, L. Crystal structure-based discovery of a novel synthesized PARP1 inhibitor (OL-1) with apoptosis-inducing mechanisms in triple-negative breast cancer. Sci. Rep. 2016, 6, 3. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Palazzo, L.; Ahel, I. PARPs in genome stability and signal transduction: Implications for cancer therapy. Biochem. Soc. Trans. 2018, 46, 1681–1695. [Google Scholar] [CrossRef]
- Cerrato, A.; Morra, F.; Celetti, A. Use of poly ADP-ribose polymerase [PARP] inhibitors in cancer cells bearing DDR defects: The rationale for their inclusion in the clinic. J. Exp. Clin. Cancer Res. 2016, 35, 179. [Google Scholar] [CrossRef] [PubMed]
- Janysek, D.C.; Kim, J.; Duijf, P.H.G.; Dray, E. Clinical use and mechanisms of resistance for PARP inhibitors in homologous recombination-deficient cancers. Transl. Oncol. 2021, 14, 101012. [Google Scholar] [CrossRef]
- Li, H.; Liu, Z.Y.; Wu, N.; Chen, Y.C.; Cheng, Q.; Wang, J. PARP inhibitor resistance: The underlying mechanisms and clinical implications. Mol. Cancer 2020, 19, 107. [Google Scholar] [CrossRef] [PubMed]
- Dias, M.P.; Moser, S.C.; Ganesan, S.; Jonkers, J. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Du, Y.; Nakai, K.; Ding, M.; Chang, S.S.; Hsu, J.L.; Yao, J.; Wei, Y.; Nie, L.; Jiao, S.; et al. EZH2 contributes to the response to PARP inhibitors through its PARP-mediated poly-ADP ribosylation in breast cancer. Oncogene 2018, 37, 208–217. [Google Scholar] [CrossRef]
- Tan, J.; Zheng, X.; Li, M.; Ye, F.; Song, C.; Xu, C.; Zhang, X.; Li, W.; Wang, Y.; Zeng, S.; et al. C/EBPβ promotes poly(ADP-ribose) polymerase inhibitor resistance by enhancing homologous recombination repair in high-grade serous ovarian cancer. Oncogene 2021, 40, 3845–3858. [Google Scholar] [CrossRef]
- Zimmermann, M.; Murina, O.; Reijns, M.A.M.; Agathanggelou, A.; Challis, R.; Tarnauskaitė, Ž.; Muir, M.; Fluteau, A.; Aregger, M.; McEwan, A.; et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 2018, 559, 285–289. [Google Scholar] [CrossRef]
- Hurley, R.M.; McGehee, C.D.; Nesic, K.; Correia, C.; Weiskittel, T.M.; Kelly, R.L.; Venkatachalam, A.; Hou, X.; Pathoulas, N.M.; Meng, X.W.; et al. Characterization of a RAD51C-silenced high-grade serous ovarian cancer model during development of PARP inhibitor resistance. NAR Cancer 2021, 3, zcab028. [Google Scholar] [CrossRef]
- Prokhorova, E.; Agnew, T.; Wondisford, A.R.; Tellier, M.; Kaminski, N.; Beijer, D.; Holder, J.; Groslambert, J.; Suskiewicz, M.J.; Zhu, K.; et al. Unrestrained poly-ADP-ribosylation provides insights into chromatin regulation and human disease. Mol. Cell 2021, 81, 2640–2655.e8. [Google Scholar] [CrossRef]
- Pulliam, N.; Fang, F.; Ozes, A.R.; Tang, J.; Adewuyi, A.; Keer, H.; Lyons, J.; Baylin, S.B.; Matei, D.; Nakshatri, H.; et al. An Effective Epigenetic-PARP Inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA Mutations. Clin. Cancer Res. 2018, 24, 3163–3175. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Yuan, B.; Chen, H.D.; Xu, L.; Tian, Y.N.; Zhang, A.; He, J.X.; Miao, Z.H. Acquired resistance of phosphatase and tensin homolog-deficient cells to poly(ADP-ribose) polymerase inhibitor and Ara-C mediated by 53BP1 loss and SAMHD1 overexpression. Cancer Sci. 2018, 109, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Clements, K.E.; Thakar, T.; Nicolae, C.M.; Liang, X.; Wang, H.G.; Moldovan, G.L. Loss of E2F7 confers resistance to poly-ADP-ribose polymerase (PARP) inhibitors in BRCA2-deficient cells. Nucleic Acids Res. 2018, 46, 8898–8907. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.; Rodriguez-Vargas, J.M.; Boehler, C.; Robert, I.; Heyer, V.; Hanini, N.; Gauthier, L.R.; Tissier, A.; Schreiber, V.; Elofsson, M.; et al. PARP3, a new therapeutic target to alter Rictor/mTORC2 signaling and tumor progression in BRCA1-associated cancers. Cell Death Differ. 2019, 26, 1615–1630. [Google Scholar] [CrossRef]
- Lindgren, A.E.; Karlberg, T.; Thorsell, A.G.; Hesse, M.; Spjut, S.; Ekblad, T.; Andersson, C.D.; Pinto, A.F.; Weigelt, J.; Hottiger, M.O.; et al. PARP inhibitor with selectivity toward ADP-ribosyltransferase ARTD3/PARP3. ACS Chem. Biol. 2013, 8, 1698–1703. [Google Scholar] [CrossRef]
- Karpova, Y.; Wu, C.; Divan, A.; McDonnell, M.E.; Hewlett, E.; Makhov, P.; Gordon, J.; Ye, M.; Reitz, A.B.; Childers, W.E.; et al. Non-NAD-like PARP-1 inhibitors in prostate cancer treatment. Biochem. Pharmacol. 2019, 167, 149–162. [Google Scholar] [CrossRef]
- Sherstyuk, Y.V.; Ivanisenko, N.V.; Zakharenko, A.L.; Sukhanova, M.V.; Peshkov, R.Y.; Eltsov, I.V.; Kutuzov, M.M.; Kurgina, T.A.; Belousova, E.A.; Ivanisenko, V.A.; et al. Design, Synthesis and Molecular Modeling Study of Conjugates of ADP and Morpholino Nucleosides as A Novel Class of Inhibitors of PARP-1, PARP-2 and PARP-3. Int. J. Mol. Sci. 2019, 21, 214. [Google Scholar] [CrossRef]
- Karlberg, T.; Hammarström, M.; Schütz, P.; Svensson, L.; Schüler, H. Crystal structure of the catalytic domain of human PARP2 in complex with PARP inhibitor ABT-888. Biochemistry 2010, 49, 1056–1058. [Google Scholar] [CrossRef]
- Pai Bellare, G.; Saha, B.; Patro, B.S. Targeting autophagy reverses de novo resistance in homologous recombination repair proficient breast cancers to PARP inhibition. Br. J. Cancer 2021, 124, 1260–1274. [Google Scholar] [CrossRef]
- Marzi, L.; Szabova, L.; Gordon, M.; Weaver Ohler, Z.; Sharan, S.K.; Beshiri, M.L.; Etemadi, M.; Murai, J.; Kelly, K.; Pommier, Y. The Indenoisoquinoline TOP1 Inhibitors Selectively Target Homologous Recombination-Deficient and Schlafen 11-Positive Cancer Cells and Synergize with Olaparib. Clin. Cancer Res. 2019, 25, 6206–6216. [Google Scholar] [CrossRef]
- Eldhose, E.; Lakshmanan, K.; Krishnamurthy, P.T.; Rajagopal, K.; Mohammed, M.; Prudviraj, P.; Byran, G. 1,3,4-Thiadiazolo (3,2-A) Pyrimidine-6-Carbonitrile Scaffold as PARP1 Inhibitors. Anticancer Agents Med. Chem. 2020, 21, 2050–2065. [Google Scholar] [CrossRef]
- Yin, L.; Liu, Y.; Peng, Y.; Peng, Y.; Yu, X.; Gao, Y.; Yuan, B.; Zhu, Q.; Cao, T.; He, L.; et al. PARP inhibitor veliparib and HDAC inhibitor SAHA synergistically co-target the UHRF1/BRCA1 DNA damage repair complex in prostate cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 153. [Google Scholar] [CrossRef] [PubMed]
- Kirby, I.T.; Cohen, M.S. Small-Molecule Inhibitors of PARPs: From Tools for Investigating ADP-Ribosylation to Therapeutics. Curr. Top. Microbiol. Immunol. 2019, 420, 211–231. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, B.N.; Thackray, J.K.; Simonet, N.G.; Kane-Goldsmith, N.; Martinez-Redondo, P.; Nguyen, T.; Bunting, S.; Vaquero, A.A.; Tischfield, J.; Serrano, L. SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. EMBO J. 2016, 35, 1488–1503. [Google Scholar] [CrossRef] [PubMed]
- Palve, V.; Knezevic, C.E.; Bejan, D.S.; Luo, Y.; Li, X.; Novakova, S.; Welsh, E.A.; Fang, B.; Kinose, F.; Haura, E.B.; et al. The non-canonical target PARP16 contributes to polypharmacology of the PARP inhibitor talazoparib and its synergy with WEE1 inhibitors. Cell Chem. Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, R.W. Novel Azaquinolones as PARP1 Inhibitors for Treating Cancer. ACS Med. Chem. Lett. 2021, 12, 524–525. [Google Scholar] [CrossRef]
- Patel, P.R.; Senyuk, V.; Rodriguez, N.S.; Oh, A.L.; Bonetti, E.; Mahmud, D.; Barosi, G.; Mahmud, N.; Rondelli, D. Synergistic Cytotoxic Effect of Busulfan and the PARP Inhibitor Veliparib in Myeloproliferative Neoplasms. Biol. Blood Marrow Transplant. 2019, 25, 855–860. [Google Scholar] [CrossRef]
- Boussios, S.; Karihtala, P.; Moschetta, M.; Abson, C.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Ryan, J.E.; Sheriff, M.; Rassy, E.; Pavlidis, N. Veliparib in ovarian cancer: A new synthetically lethal therapeutic approach. Investig. New Drugs 2020, 38, 181–193. [Google Scholar] [CrossRef]
- Lau, T.; Chan, E.; Callow, M.; Waaler, J.; Boggs, J.; Blake, R.A.; Magnuson, S.; Sambrone, A.; Schutten, M.; Firestein, R.; et al. A novel tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res. 2013, 73, 3132–3144. [Google Scholar] [CrossRef]
- Ma, L.; Wang, X.; Jia, T.; Wei, W.; Chua, M.S.; So, S. Tankyrase inhibitors attenuate WNT/ß-catenin signaling and inhibit growth of hepatocellular carcinoma cells. Oncotarget 2015, 6, 25390–25401. [Google Scholar] [CrossRef]
- Huang, S.-M.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [Google Scholar] [CrossRef]
- Callow, M.G.; Tran, H.; Phu, L.; Lau, T.; Lee, J.; Sandoval, W.N.; Liu, P.S.; Bheddah, S.; Tao, J.; Lill, J.R.; et al. Ubiquitin ligase RNF146 regulates tankyrase and Axin to promote Wnt signaling. PLoS ONE 2011, 6, e22595. [Google Scholar] [CrossRef]
- Mygland, L.; Brinch, S.A.; Strand, M.F.; Olsen, P.A.; Aizenshtadt, A.; Lund, K.; Solberg, N.T.; Lycke, M.; Thorvaldsen, T.E.; Espada, S.; et al. Identification of response signatures for tankyrase inhibitor treatment in tumor cell lines. iScience 2021, 24, 102807. [Google Scholar] [CrossRef] [PubMed]
- Ferri, M.; Liscio, P.; Carotti, A.; Asciutti, S.; Sardella, R.; Macchiarulo, A.; Camaioni, E. Targeting Wnt-driven cancers: Discovery of novel tankyrase inhibitors. Eur. J. Med. Chem. 2017, 142, 506–522. [Google Scholar] [CrossRef] [PubMed]
- Martins-Neves, S.R.; Paiva-Oliveira, D.I.; Fontes-Ribeiro, C.; Bovée, J.V.M.G.; Cleton-Jansen, A.M.; Gomes, C.M.F. IWR-1, a tankyrase inhibitor, attenuates Wnt/β-catenin signaling in cancer stem-like cells and inhibits in vivo the growth of a subcutaneous human osteosarcoma xenograft. Cancer Lett. 2018, 414, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Nam, K.Y.; Kim, H.J.; Song, J.Y.; Hwang, S.G.; Kim, J.S.; Kim, J.; Ahn, J. Discovery of a Novel Triazolopyridine Derivative as a Tankyrase Inhibitor. Int. J. Mol. Sci. 2021, 22, 7330. [Google Scholar] [CrossRef]
- Buchstaller, H.P.; Anlauf, U.; Dorsch, D.; Kögler, S.; Kuhn, D.; Lehmann, M.; Leuthner, B.; Lodholz, S.; Musil, D.; Radtki, D.; et al. Optimization of a Screening Hit toward M2912, an Oral Tankyrase Inhibitor with Antitumor Activity in Colorectal Cancer Models. J. Med. Chem. 2021, 64, 10371–10392. [Google Scholar] [CrossRef]
- Shirai, F.; Tsumura, T.; Yashiroda, Y.; Yuki, H.; Niwa, H.; Sato, S.; Chikada, T.; Koda, Y.; Washizuka, K.; Yoshimoto, N.; et al. Discovery of Novel Spiroindoline Derivatives as Selective Tankyrase Inhibitors. J. Med. Chem. 2019, 62, 3407–3427. [Google Scholar] [CrossRef]
- Menon, M.; Elliott, R.; Bowers, L.; Balan, N.; Rafiq, R.; Costa-Cabral, S.; Munkonge, F.; Trinidade, I.; Porter, R.; Campbell, A.D.; et al. A novel tankyrase inhibitor, MSC2504877, enhances the effects of clinical CDK4/6 inhibitors. Sci. Rep. 2019, 9, 201. [Google Scholar] [CrossRef]
- Li, B.; Liang, J.; Lu, F.; Zeng, G.; Zhang, J.; Ma, Y.; Liu, P.; Wang, Q.; Zhou, Q.; Chen, L. Discovery of Novel Inhibitor for WNT/β-Catenin Pathway by Tankyrase 1/2 Structure-Based Virtual Screening. Molecules 2020, 25, 1680. [Google Scholar] [CrossRef]
- Solberg, N.T.; Waaler, J.; Lund, K.; Mygland, L.; Olsen, P.A.; Krauss, S. TANKYRASE Inhibition Enhances the Antiproliferative Effect of PI3K and EGFR Inhibition, Mutually Affecting β-CATENIN and AKT Signaling in Colorectal Cancer. Mol. Cancer Res. 2018, 16, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Haikarainen, T.; Narwal, M.; Joensuu, P.; Lehtiö, L. Evaluation and Structural Basis for the Inhibition of Tankyrases by PARP Inhibitors. ACS Med. Chem. Lett. 2013, 5, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Bregman, H.; Chakka, N.; Guzman-Perez, A.; Gunaydin, H.; Gu, Y.; Huang, X.; Berry, V.; Liu, J.; Teffera, Y.; Huang, L.; et al. Discovery of novel, induced-pocket binding oxazolidinones as potent, selective, and orally bioavailable tankyrase inhibitors. J. Med. Chem. 2013, 56, 4320–4342. [Google Scholar] [CrossRef] [PubMed]
- Shultz, M.D.; Cheung, A.K.; Kirby, C.A.; Firestone, B.; Fan, J.; Chen, C.H.T.; Chen, Z.; Chin, D.N.; Dipietro, L.; Fazal, A.; et al. Identification of NVP-TNKS656: The use of structure-efficiency relationships to generate a highly potent, selective, and orally active tankyrase inhibitor. J. Med. Chem. 2013, 56, 6495–6511. [Google Scholar] [CrossRef]
- Vilchez Larrea, S.; Valsecchi, W.M.; Fernández Villamil, S.H.; Lafon Hughes, L.I. First body of evidence suggesting a role of a tankyrase-binding motif (TBM) of vinculin (VCL) in epithelial cells. PeerJ 2021, 9, e11442. [Google Scholar] [CrossRef]
- Guettler, S.; LaRose, J.; Petsalaki, E.; Gish, G.; Scotter, A.; Pawson, T.; Rottapel, R.; Sicheri, F. Structural basis and sequence rules for substrate recognition by Tankyrase explain the basis for cherubism disease. Cell 2011, 147, 1340–1354. [Google Scholar] [CrossRef]
- Pollock, K.; Liu, M.; Zaleska, M.; Meniconi, M.; Pfuhl, M.; Collins, I.; Guettler, S. Fragment-based screening identifies molecules targeting the substrate-binding ankyrin repeat domains of tankyrase. Sci. Rep. 2019, 9, 19130. [Google Scholar] [CrossRef]
- Cheng, H.; Li, X.; Wang, C.; Chen, Y.; Li, S.; Tan, J.; Tan, B.; He, Y. Inhibition of tankyrase by a novel small molecule significantly attenuates prostate cancer cell proliferation. Cancer Lett. 2019, 443, 80–90. [Google Scholar] [CrossRef]
- Barbarulo, A.; Iansante, V.; Chaidos, A.; Naresh, K.; Rahemtulla, A.; Franzoso, G.; Karadimitris, A.; Haskard, D.O.; Papa, S.; Bubici, C. Poly(ADP-ribose) polymerase family member 14 (PARP14) is a novel effector of the JNK2-dependent pro-survival signal in multiple myeloma. Oncogene 2013, 32, 4231–4242. [Google Scholar] [CrossRef]
- Dhoonmoon, A.; Schleicher, E.M.; Clements, K.E.; Nicolae, C.M.; Moldovan, G.L. Genome-wide CRISPR synthetic lethality screen identifies a role for the ADP-ribosyltransferase PARP14 in DNA replication dynamics controlled by ATR. Nucleic Acids Res. 2020, 48, 7252–7264. [Google Scholar] [CrossRef]
- Peng, B.; Thorsell, A.-G.; Karlberg, T.; Schüler, H.; Yao, S.Q. Small Molecule Microarray Based Discovery of PARP14 Inhibitors. Angew Chem. Int. Ed. Engl. 2017, 56, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Upton, K.; Meyers, M.; Thorsell, A.-G.; Karlberg, T.; Holechek, J.; Lease, R.; Schey, G.; Wolf, E.; Lucente, A.; Schüler, H.; et al. Design and synthesis of potent inhibitors of the mono(ADP-ribosyl)transferase, PARP14. Bioorg. Med. Chem. Lett. 2017, 27, 2907–2911. [Google Scholar] [CrossRef]
- Qin, W.; Wu, H.-J.; Cao, L.-Q.; Li, H.-J.; He, C.-X.; Zhao, D.; Xing, L.; Li, P.-Q.; Jin, X.; Cao, H.-L. Research Progress on PARP14 as a Drug Target. Front. Pharmacol. 2019, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Holechek, J.; Lease, R.; Thorsell, A.G.; Karlberg, T.; McCadden, C.; Grant, R.; Keen, A.; Callahan, E.; Schüler, H.; Ferraris, D. Design, synthesis and evaluation of potent and selective inhibitors of mono-(ADP-ribosyl)transferases PARP10 and PARP14. Bioorg. Med. Chem. Lett. 2018, 28, 2050–2054. [Google Scholar] [CrossRef] [PubMed]
- Moustakim, M.; Riedel, K.; Schuller, M.; Gehring, A.P.; Monteiro, O.P.; Martin, S.P.; Fedorov, O.; Heer, J.; Dixon, D.J.; Elkins, J.M.; et al. Discovery of a novel allosteric inhibitor scaffold for polyadenosine-diphosphate-ribose polymerase 14 (PARP14) macrodomain 2. Bioorg. Med. Chem. 2018, 26, 2965–2972. [Google Scholar] [CrossRef]
- Wigle, T.J.; Ren, Y.; Molina, J.R.; Blackwell, D.J.; Schenkel, L.B.; Swinger, K.K.; Kuplast-Barr, K.; Majer, C.R.; Church, W.D.; Lu, A.Z.; et al. Targeted Degradation of PARP14 Using a Heterobifunctional Small Molecule. ChemBioChem 2021, 22, 2107–2110. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, L.B.; Molina, J.R.; Swinger, K.K.; Abo, R.; Blackwell, D.J.; Lu, A.Z.; Cheung, A.E.; Church, W.D.; Kunii, K.; Kuplast-Barr, K.G.; et al. A potent and selective PARP14 inhibitor decreases protumor macrophage gene expression and elicits inflammatory responses in tumor explants. Cell. Chem. Biol. 2021, 28, 1158–1168.e13. [Google Scholar] [CrossRef] [PubMed]
- Murthy, S.; Desantis, J.; Verheugd, P.; Maksimainen, M.M.; Venkannagari, H.; Massari, S.; Ashok, Y.; Obaji, E.; Nkizinkinko, Y.; Lüscher, B.; et al. 4-(Phenoxy) and 4-(benzyloxy)benzamides as potent and selective inhibitors of mono-ADP-ribosyltransferase PARP10/ARTD10. Eur. J. Med. Chem. 2018, 156, 93–102. [Google Scholar] [CrossRef]
- Morgan, R.K.; Kirby, I.T.; Schmaedick, A.V.; Rodriguez, K.; Cohen, M.S. Rational Design of Cell-Active Inhibitors of PARP10. ACS Med. Chem. Lett. 2018, 10, 74–79. [Google Scholar] [CrossRef]
- Venkannagari, H.; Verheugd, P.; Koivunen, J.; Haikarainen, T.; Obaji, E.; Ashok, Y.; Narwal, M.; Pihlajaniemi, T.; Lüscher, B.; Lehtiö, L. Small-Molecule Chemical Probe Rescues Cells from Mono-ADP-Ribosyltransferase ARTD10/PARP10-Induced Apoptosis and Sensitizes Cancer Cells to DNA Damage. Cell Chem. Biol. 2016, 23, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liang, X.; Wei, L.; Liu, Y.; Liu, J.; Feng, H.; Zheng, F.; Wang, Y.; Ma, H.; Wu, J. RNF114 suppresses metastasis through regulation of PARP10 in cervical cancer cells. Cancer Commun. 2021, 41, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Kirby, I.T.; Kojic, A.; Arnold, M.R.; Thorsell, A.G.; Karlberg, T.; Vermehren-Schmaedick, A.; Sreenivasan, R.; Schultz, C.; Schüler, H.; Cohen, M.S. A Potent and Selective PARP11 Inhibitor Suggests Coupling between Cellular Localization and Catalytic Activity. Cell Chem. Biol. 2018, 25, 1547–1553.e12. [Google Scholar] [CrossRef]
- Guo, T.; Zuo, Y.; Qian, L.; Liu, J.; Yuan, Y.; Xu, K.; Miao, Y.; Feng, Q.; Chen, X.; Jin, L.; et al. ADP-ribosyltransferase PARP11 modulates the interferon antiviral response by mono-ADP-ribosylating the ubiquitin E3 ligase β-TrCP. Nat. Microbiol. 2019, 4, 1872–1884. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Ficca, M.L.; Ihara, M.; Bader, J.J.; Leu, N.A.; Beneke, S.; Meyer, R.G. Spermatid head elongation with normal nuclear shaping requires ADP-ribosyltransferase PARP11 (ARTD11) in mice. Biol. Reprod. 2015, 92, 80. [Google Scholar] [CrossRef][Green Version]
- Li, L.; Shi, Y.; Li, S.; Liu, J.; Zu, S.; Xu, X.; Gao, M.; Sun, N.; Pan, C.; Peng, L.; et al. ADP-ribosyltransferase PARP11 suppresses Zika virus in synergy with PARP12. Cell Biosci. 2021, 11, 116. [Google Scholar] [CrossRef]
- Lu, A.Z.; Abo, R.; Ren, Y.; Gui, B.; Mo, J.R.; Blackwell, D.; Wigle, T.; Keilhack, H.; Niepel, M. Enabling drug discovery for the PARP protein family through the detection of mono-ADP-ribosylation. Biochem. Pharmacol. 2019, 167, 97–106. [Google Scholar] [CrossRef]
- Wigle, T.J.; Church, W.D.; Majer, C.R.; Swinger, K.K.; Aybar, D.; Schenkel, L.B.; Vasbinder, M.M.; Brendes, A.; Beck, C.; Prahm, M.; et al. Forced Self-Modification Assays as a Strategy to Screen MonoPARP Enzymes. SLAS Discov. 2020, 25, 241–252. [Google Scholar] [CrossRef]
- Wigle, T.J.; Blackwell, D.J.; Schenkel, L.B.; Ren, Y.; Church, W.D.; Desai, H.J.; Swinger, K.K.; Santospago, A.G.; Majer, C.R.; Lu, A.Z.; et al. In Vitro and Cellular Probes to Study PARP Enzyme Target Engagement. Cell. Chem. Biol. 2020, 27, 877–887.e14. [Google Scholar] [CrossRef]
- Palavalli Parsons, L.H.; Challa, S.; Gibson, B.A.; Nandu, T.; Stokes, M.S.; Huang, D.; Lea, J.S.; Kraus, W.L. Identification of PARP-7 substrates reveals a role for MARylation in microtubule control in ovarian cancer cells. Elife 2021, 10, e60481. [Google Scholar] [CrossRef]
- Roper, S.J.; Chrysanthou, S.; Senner, C.E.; Sienerth, A.; Gnan, S.; Murray, A.; Masutani, M.; Latos, P.; Hemberger, M. ADP-ribosyltransferases Parp1 and Parp7 safeguard pluripotency of ES cells. Nucleic Acids Res. 2014, 42, 8914–8927. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, K.M.; Buch-Larsen, S.C.; Kirby, I.T.; Siordia, I.R.; Hutin, D.; Rasmussen, M.; Grant, D.M.; David, L.L.; Matthews, J.; Nielsen, M.L.; et al. Chemical genetics and proteome-wide site mapping reveal cysteine MARylation by PARP-7 on immune-relevant protein targets. Elife 2021, 10, e60480. [Google Scholar] [CrossRef] [PubMed]
- Hopp, A.K.; Hottiger, M.O. Uncovering the Invisible: Mono-ADP-ribosylation Moved into the Spotlight. Cells 2021, 10, 680. [Google Scholar] [CrossRef] [PubMed]
- Vasbinder, M.M.; Schenkel, L.B.; Swinger, K.K.; Kuntz, K.W. Pyridazinones as PARP7 Inhibitors. International Patent WO/2019/212937, 11 July 2019. [Google Scholar]
- Vasbinder, M.M.; Gozgit, J.M.; Abo, R.P.; Kunii, K.; Kuplast-Barr, K.G.; Gui, B.; Lu, A.Z.; Swinger, K.K.; Wigle, T.J.; Blackwell, D.J.; et al. Abstract DDT02-01: RBN-2397: A first-in-class PARP7 inhibitor targeting a newly discovered cancer vulnerability in stress-signaling pathways. Tumor Biol. 2020, 80, DDT02-01. [Google Scholar]
- Gozgit, J.M.; Vasbinder, M.M.; Abo, R.P.; Kunii, K.; Kuplast-Barr, K.G.; Gui, B.; Lu, A.Z.; Molina, J.R.; Minissale, E.; Swinger, K.K.; et al. PARP7 negatively regulates the type I interferon response in cancer cells and its inhibition triggers antitumor immunity. Cancer Cell 2021, 39, 1214–1226.e10. [Google Scholar] [CrossRef]
- Schleicher, E.M.; Galvan, A.M.; Imamura-Kawasawa, Y.; Moldovan, G.L.; Nicolae, C.M. PARP10 promotes cellular proliferation and tumorigenesis by alleviating replication stress. Nucleic Acids Res. 2018, 46, 8908–8916. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, C.; Song, D.; Xia, R.; Yu, W.; Dang, Y.; Fei, Y.; Yu, L.; Wu, J. Epigallocatechin-3-gallate enhances ER stress-induced cancer cell apoptosis by directly targeting PARP16 activity. Cell Death Discov. 2017, 3, 17034. [Google Scholar] [CrossRef]
- Di Paola, S.; Micaroni, M.; Di Tullio, G.; Buccione, R.; Di Girolamo, M. PARP16/ARTD15 is a novel endoplasmic-reticulum-associated mono-ADP-ribosyltransferase that interacts with, and modifies karyopherin-ß1. PLoS ONE 2012, 7, e37352. [Google Scholar] [CrossRef]
- Jwa, M.; Chang, P. PARP16 is a tail-anchored endoplasmic reticulum protein required for the PERK- and IRE1α-mediated unfolded protein response. Nat. Cell Biol. 2012, 14, 1223–1230. [Google Scholar] [CrossRef]
- Di Girolamo, M.; Fabrizio, G.; Scarpa, E.S.; Di Paola, S. NAD⁺-dependent enzymes at the endoplasmic reticulum. Curr. Top. Med. Chem. 2013, 13, 3001–3010. [Google Scholar] [CrossRef]
- Carlile, G.W.; Robert, R.; Matthes, E.; Yang, Q.; Solari, R.; Hatley, R.; Edge, C.M.; Hanrahan, J.W.; Andersen, R.; Thomas, D.Y.; et al. Latonduine Analogs Restore F508del-Cystic Fibrosis Transmembrane Conductance Regulator Trafficking through the Modulation of Poly-ADP Ribose Polymerase 3 and Poly-ADP Ribose Polymerase 16 Activity. Mol. Pharmacol. 2016, 90, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Centko, R.M.; Carlile, G.W.; Barne, I.; Patrick, B.O.; Blagojevic, P.; Thomas, D.Y.; Andersen, R.J. Combination of Selective PARP3 and PARP16 Inhibitory Analogues of Latonduine A Corrects F508del-CFTR Trafficking. ACS Omega 2020, 5, 25593–25604. [Google Scholar] [CrossRef]
- Tuncel, H.; Tanaka, S.; Oka, S.; Nakai, S.; Fukutomi, R.; Okamoto, M.; Ota, T.; Kaneko, H.; Tatsuka, M.; Shimamoto, F. PARP6, a mono(ADP-ribosyl) transferase and a negative regulator of cell proliferation, is involved in colorectal cancer development. Int. J. Oncol. 2012, 41, 2079–2086. [Google Scholar] [CrossRef] [PubMed]
- Vermehren-Schmaedick, A.; Huang, J.Y.; Levinson, M.; Pomaville, M.B.; Reed, S.; Bellus, G.A.; Gilbert, F.; Keren, B.; Heron, D.; Haye, D.; et al. Characterization of PARP6 Function in Knockout Mice and Patients with Developmental Delay. Cells 2021, 10, 1289. [Google Scholar] [CrossRef]
- Wang, Z.; Grosskurth, S.E.; Cheung, T.; Petteruti, P.; Zhang, J.; Wang, X.; Wang, W.; Gharahdaghi, F.; Wu, J.; Su, N.; et al. Pharmacological Inhibition of PARP6 Triggers Multipolar Spindle Formation and Elicits Therapeutic Effects in Breast Cancer. Cancer Res. 2018, 78, 6691–6702. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Zhang, Y.; Wang, W.; Qi, G.; Shimamoto, F. PARP6 suppresses the proliferation and metastasis of hepatocellular carcinoma by degrading XRCC6 to regulate the Wnt/β-catenin pathway. Am. J. Cancer Res. 2020, 10, 2100–2113. [Google Scholar] [PubMed]
- Fabrizio, G.; Di Paola, S.; Stilla, A.; Giannotta, M.; Ruggiero, C.; Menzel, S.; Koch-Nolte, F.; Sallese, M.; Di Girolamo, M. ARTC1-mediated ADP-ribosylation of GRP78/BiP: A new player in endoplasmic-reticulum stress responses. Cell. Mol. Life Sci. 2015, 72, 1209–1225. [Google Scholar] [CrossRef]
- Yang, L.; Xiao, M.; Li, X.; Tang, Y.; Wang, Y.L. Arginine ADP-ribosyltransferase 1 promotes angiogenesis in colorectal cancer via the PI3K/Akt pathway. Int. J. Mol. Med. 2016, 37, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wang, Y.L.; Yang, L.; Xu, J.X.; Xiong, W.; Xiao, M.; Li, M. Inhibition of arginine ADP-ribosyltransferase 1 reduces the expression of poly(ADP-ribose) polymerase-1 in colon carcinoma. Int. J. Mol. Med. 2013, 32, 130–136. [Google Scholar] [CrossRef]
- Wei, X.; Tang, Y.; Wang, Y.; Xu, J.-X. Effects of ART1 gene silencing on the ability of CT26 cellular matrix adhesion and migration. Fudan Univ. J. Med. Sci. 2013, 40, 328–334. [Google Scholar]
- Song, G.L.; Jin, C.C.; Zhao, W.; Tang, Y.; Wang, Y.L.; Li, M.; Xiao, M.; Li, X.; Li, Q.S.; Lin, X.; et al. Regulation of the RhoA/ROCK/AKT/β-catenin pathway by arginine-specific ADP-ribosytransferases 1 promotes migration and epithelial-mesenchymal transition in colon carcinoma. Int. J. Oncol. 2016, 49, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Bütepage, M.; Eckei, L.; Verheugd, P.; Lüscher, B. Intracellular Mono-ADP-Ribosylation in Signaling and Disease. Cells 2015, 4, 569–595. [Google Scholar] [CrossRef]
- Hoch, N.C.; Polo, L.M. ADP-ribosylation: From molecular mechanisms to human disease. Genet. Mol. Biol. 2019, 43, e20190075. [Google Scholar] [CrossRef]
- Ekblad, T.; Schüler, H. Sirtuins are Unaffected by PARP Inhibitors Containing Planar Nicotinamide Bioisosteres. Chem. Biol. Drug Des. 2016, 87, 478–482. [Google Scholar] [CrossRef]
- Hawse, W.F.; Wolberger, C. Structure-based mechanism of ADP-ribosylation by sirtuins. J. Biol. Chem. 2009, 284, 33654–33661. [Google Scholar] [CrossRef]
- Fiorentino, F.; Carafa, V.; Favale, G.; Altucci, L.; Mai, A.; Rotili, D. The Two-Faced Role of SIRT6 in Cancer. Cancers 2021, 13, 1156. [Google Scholar] [CrossRef]
- Bai, L.; Lin, G.; Sun, L.; Liu, Y.; Huang, X.; Cao, C.; Guo, Y.; Xie, C. Upregulation of SIRT6 predicts poor prognosis and promotes metastasis of non-small cell lung cancer via the ERK1/2/MMP9 pathway. Oncotarget 2016, 7, 40377–40386. [Google Scholar] [CrossRef]
- Lin, H.; Hao, Y.; Zhao, Z.; Tong, Y. Sirtuin 6 contributes to migration and invasion of osteosarcoma cells via the ERK1/2/MMP9 pathway. FEBS Open Bio 2017, 7, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.H.; Zhang, C.L.; Zhang, J.Y.; Gao, P.; He, M.; Li, Y.L. Overexpression of Sirt6 is a novel biomarker of malignant human colon carcinoma. J. Cell Biochem. 2018, 119, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, J.; Huang, R.; Fang, L.; Yu, S. SIRT4 and SIRT6 Serve as Novel Prognostic Biomarkers With Competitive Functions in Serous Ovarian Cancer. Front. Genet. 2021, 12, 666630. [Google Scholar] [CrossRef]
- Tan, Y.; Li, B.; Peng, F.; Gong, G.; Li, N. Integrative Analysis of Sirtuins and Their Prognostic Significance in Clear Cell Renal Cell Carcinoma. Front. Oncol. 2020, 10, 218. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.A.; Denu, J.M. Biological and catalytic functions of sirtuin 6 as targets for small-molecule modulators. J. Biol. Chem. 2020, 295, 11021–11041. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Peterson, L.M.; Ndiaye, M.A.; Chhabra, G.; Singh, C.K.; Guzmán-Pérez, G.; Iczkowski, K.A.; Ahmad, N. CRISPR/Cas9-mediated Knockout of SIRT6 Imparts Remarkable Antiproliferative Response in Human Melanoma Cells in vitro and in vivo. Photochem. Photobiol. 2020, 96, 1314–1320. [Google Scholar] [CrossRef]
- Beauharnois, J.M.; Bolívar, B.E.; Welch, J.T. Sirtuin 6: A review of biological effects and potential therapeutic properties. Mol. Biosyst. 2013, 9, 1789–1806. [Google Scholar] [CrossRef]
- Miteva, Y.V.; Cristea, I.M. A proteomic perspective of Sirtuin 6 (SIRT6) phosphorylation and interactions and their dependence on its catalytic activity. Mol. Cell. Proteomics 2014, 13, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Van Meter, M.; Mao, Z.; Gorbunova, V.; Seluanov, A. SIRT6 overexpression induces massive apoptosis in cancer cells but not in normal cells. Cell Cycle 2011, 10, 3153–3158. [Google Scholar] [CrossRef] [PubMed]
- Van Meter, M.; Kashyap, M.; Rezazadeh, S.; Geneva, A.J.; Morello, T.D.; Seluanov, A.; Gorbunova, V. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat. Commun. 2014, 5, 5011. [Google Scholar] [CrossRef] [PubMed]
- Mellini, P.; Itoh, Y.; Elboray, E.E.; Tsumoto, H.; Li, Y.; Suzuki, M.; Takahashi, Y.; Tojo, T.; Kurohara, T.; Miyake, Y.; et al. Identification of Diketopiperazine-Containing 2-Anilinobenzamides as Potent Sirtuin 2 (SIRT2)-Selective Inhibitors Targeting the “Selectivity Pocket”, Substrate-Binding Site, and NAD+-Binding Site. J. Med. Chem. 2019, 62, 5844–5862. [Google Scholar] [CrossRef]
- Rezazadeh, S.; Yang, D.; Biashad, S.A.; Firsanov, D.; Takasugi, M.; Gilbert, M.; Tombline, G.; Bhanu, N.V.; Garcia, B.A.; Seluanov, A.; et al. SIRT6 mono-ADP ribosylates KDM2A to locally increase H3K36me2 at DNA damage sites to inhibit transcription and promote repair. Aging 2020, 12, 11165–11184. [Google Scholar] [CrossRef] [PubMed]
- Rezazadeh, S.; Yang, D.; Tombline, G.; Simon, M.; Regan, S.P.; Seluanov, A.; Gorbunova, V. SIRT6 promotes transcription of a subset of NRF2 targets by mono-ADP-ribosylating BAF170. Nucleic Acids Res. 2019, 47, 7914–7928. [Google Scholar] [CrossRef] [PubMed]
- Asaba, T.; Suzuki, T.; Ueda, R.; Tsumoto, H.; Nakagawa, H.; Miyata, N. Inhibition of human sirtuins by in situ generation of an acetylated lysine-ADP-ribose conjugate. J. Am. Chem. Soc. 2009, 131, 6989–6996. [Google Scholar] [CrossRef]
- Hu, J.; Jing, H.; Lin, H. Sirtuin inhibitors as anticancer agents. Future Med. Chem. 2014, 6, 945–966. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, L.; Zhang, W.; Meng, D.; Zhang, H.; Jiang, Y.; Xu, X.; Van Meter, M.; Seluanov, A.; Gorbunova, V.; et al. SIRT6 rescues the age related decline in base excision repair in a PARP1-dependent manner. Cell Cycle 2015, 14, 269–276. [Google Scholar] [CrossRef]
- Khan, R.I.; Nirzhor, S.S.R.; Akter, R. A Review of the Recent Advances Made with SIRT6 and its Implications on Aging Related Processes, Major Human Diseases, and Possible Therapeutic Targets. Biomolecules 2018, 8, 44. [Google Scholar] [CrossRef]
- McCord, R.A.; Michishita, E.; Hong, T.; Berber, E.; Boxer, L.D.; Kusumoto, R.; Guan, S.; Shi, X.; Gozani, O.; Burlingame, A.L.; et al. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging 2009, 1, 109–121. [Google Scholar] [CrossRef]
- Chen, W.; Liu, N.; Zhang, H.; Zhang, H.; Qiao, J.; Jia, W.; Zhu, S.; Mao, Z.; Kang, J. Sirt6 Promotes DNA End Joining in iPSCs Derived from Old Mice. Cell Rep. 2017, 18, 2880–2892. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, J.; Sun, X.; Yu, J.; Qian, Z.; Wu, L.; Xu, X.; Wan, X.; Jiang, Y.; Zhang, J.; et al. The SIRT6 activator MDL-800 improves genomic stability and pluripotency of old murine-derived iPS cells. Aging Cell 2020, 19, e13185. [Google Scholar] [CrossRef]
- Toiber, D.; Erdel, F.; Bouazoune, K.; Silberman, D.M.; Zhong, L.; Mulligan, P.; Sebastian, C.; Cosentino, C.; Martinez-Pastor, B.; Giacosa, S.; et al. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol. Cell 2013, 51, 454–468. [Google Scholar] [CrossRef] [PubMed]
- Damonte, P.; Sociali, G.; Parenti, M.D.; Soncini, D.; Bauer, I.; Boero, S.; Grozio, A.; Holtey, M.V.; Piacente, F.; Becherini, P.; et al. SIRT6 inhibitors with salicylate-like structure show immunosuppressive and chemosensitizing effects. Bioorg. Med. Chem. 2017, 25, 5849–5858. [Google Scholar] [CrossRef] [PubMed]
- Sociali, G.; Magnone, M.; Ravera, S.; Damonte, P.; Vigliarolo, T.; Von Holtey, M.; Vellone, V.G.; Millo, E.; Caffa, I.; Cea, M.; et al. Pharmacological Sirt6 inhibition improves glucose tolerance in a type 2 diabetes mouse model. FASEB J. 2017, 31, 3138–3149. [Google Scholar] [CrossRef]
- Cui, Y.; Yang, J.; Bai, Y.; Zhang, Y.; Yao, Y.; Zheng, T.; Liu, C.; Wu, F. miR-424-5p regulates cell proliferation and migration of esophageal squamous cell carcinoma by targeting SIRT4. J. Cancer 2020, 11, 6337–6347. [Google Scholar] [CrossRef]
- Brown, D.G.; Shorter, J.; Wobst, H.J. Emerging small-molecule therapeutic approaches for amyotrophic lateral sclerosis and frontotemporal dementia. Bioorg. Med. Chem. Lett. 2020, 30, 126942. [Google Scholar] [CrossRef]
- McGurk, L.; Mojsilovic-Petrovic, J.; Van Deerlin, V.M.; Shorter, J.; Kalb, R.G.; Lee, V.M.; Trojanowski, J.Q.; Lee, E.B.; Bonini, N.M. Nuclear poly(ADP-ribose) activity is a therapeutic target in amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2018, 6, 84. [Google Scholar] [CrossRef]
- Berger, N.A.; Besson, V.C.; Boulares, A.H.; Bürkle, A.; Chiarugi, A.; Clark, R.S.; Curtin, N.J.; Cuzzocrea, S.; Dawson, T.M.; Dawson, V.L.; et al. Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. Br. J. Pharmacol. 2018, 175, 192–222. [Google Scholar] [CrossRef]
- Jayabalan, A.K.; Adivarahan, S.; Koppula, A.; Abraham, R.; Batish, M.; Zenklusen, D.; Griffin, D.E.; Leung, A.K.L. Stress granule formation, disassembly, and composition are regulated by alphavirus ADP-ribosylhydrolase activity. Proc. Natl. Acad. Sci. USA 2021, 118, e2021719118. [Google Scholar] [CrossRef]
- Žaja, R.; Aydin, G.; Lippok, B.E.; Feederle, R.; Lüscher, B.; Feijs, K.L.H. Comparative analysis of MACROD1, MACROD2 and TARG1 expression, localisation and interactome. Sci. Rep. 2020, 10, 8286. [Google Scholar] [CrossRef]
- Rack, J.G.M.; Ariza, A.; Drown, B.S.; Henfrey, C.; Bartlett, E.; Shirai, T.; Hergenrother, P.J.; Ahel, I. (ADP-ribosyl)hydrolases: Structural Basis for Differential Substrate Recognition and Inhibition. Cell Chem. Biol. 2018, 25, 1533–1546.e12. [Google Scholar] [CrossRef] [PubMed]
- Drown, B.S.; Shirai, T.; Rack, J.G.M.; Ahel, I.; Hergenrother, P.J. Monitoring Poly (ADP-ribosyl) glycohydrolase Activity with a Continuous Fluorescent Substrate. Cell Chem. Biol. 2018, 25, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Wyżewski, Z.; Gradowski, M.; Krysińska, M.; Dudkiewicz, M.; Pawłowski, K. A novel predicted ADP-ribosyltransferase-like family conserved in eukaryotic evolution. PeerJ 2021, 9, e11051. [Google Scholar] [CrossRef] [PubMed]
- Cardamone, M.D.; Gao, Y.; Kwan, J.; Hayashi, V.; Sheeran, M.; Xu, J.; English, J.; Orofino, J.; Emili, A.; Perissi, V. ADP-ribosylation of mitochondrial proteins is mediated by Neuralized-like protein 4 (NEURL4). bioRxiv 2021. [Google Scholar] [CrossRef]
- Dudkiewicz, M.; Pawłowski, K. A novel conserved family of Macro-like domains—Putative new players in ADP-ribosylation signaling. PeerJ 2019, 7, e6863. [Google Scholar] [CrossRef] [PubMed]
- Ayyappan, V.; Wat, R.; Barber, C.; Vivelo, C.A.; Gauch, K.; Visanpattanasin, P.; Cook, G.; Sazeides, C.; Leung, A.K.L. ADPriboDB 2.0: An updated database of ADP-ribosylated proteins. Nucleic Acids Res. 2021, 49, D261–D265. [Google Scholar] [CrossRef]
- Mitra, N.; Dei, S. Understanding the mechanism of monoADP ribosylation in OsSRT1 and its linkage to DNA repair system under stress conditions. BioRxiv 2021. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drug | Target Molecules | Therapeutic Applications | References |
|---|---|---|---|
| Olaparib, rucaparib, niraparib, veliparib, talazoparib, often associated with alkylating agents or platinum | PARP1/PARP2 inhibition and trapping on DNA | BRCA1, BRCA2, ATM, ATR, FANC, PALB2 mutated, CCDC6 inactivated, HR defective cancers; RNASEH2B deleted cancers; HER negative cancers, prostate cancer, 53BP1 deficient cancers | [14,67] |
| Non-NAD+-like PARP-1 inhibitor, 5F02 | PARP1 | Prostate cancers (hormone dependent and independent); PARG mutated cancers | [88] |
| Veliparib + SAHA or belinostat | PARP1 and HDAC | Prostate cancer | [94] |
| Guadecitabine + talazoparib | PARP1 and DNMT1 | Breast and ovarian cancers | [83] |
| SMYD2 inhibitor LLY-507 or BCI-121 + olaparib | PARP1 and SMYD2 | Serous ovarian cancer, high grade (HGSOC) | [57,58] |
| PARPi + EZH2 inhibitors | BRCA1-mutant cell sensitization to PARPi | Inhibition of PARP by PARPi attenuates alkylating DNA damage-induced EZH2 downregulation, promoting EZH2-mediated gene silencing and cancer stem cell control | [78] |
| PARPi as chemosensitizer in combination with temozolomide, DNA crosslinkers (cisplatin), or bleomycin | PARP1/2 | BRCAness in various cancers | [71,73] |
| PJ34 ((N-(6-oxo-5, 6-dihydrophenanthridin-2-yl)-N, N-dimethylacetamide), veliparib | Broad inhibitors | Protection against oxidative stress; suppression of inflammatory responses, ALS, TDP-43 degenerative processes | [95,96,97] |
| ME0328 | ARTD3/PARP3 | Exploits vulnerabilities in DNA repair in cancers | [87] |
| 5-Amino-7-(aldehyde)-2[(naphthalene-2-yloxy)methyl]-[1,3,4]thiadiazolo-[3,2-α]-pyrimidine-6-carbonitrile scaffolds | PARP1 | [11,93] | |
| Conjugates of ADP and morpholino nucleosides | PARP1/2/3 | Cancers | [89] |
| ABT-888 | PARP2 | Epithelial ovarian cancer | [90] |
| Azaquinolones | PARP1 | Cancers | [98] |
| Veliparib + busulfan | PARP1 | Myeloproliferative neoplasms | [99] |
| Olaparib + radiotherapy | PARP1 | Medulloblastoma | [100] |
| Drug | Target | Therapeutic Applications | References |
|---|---|---|---|
| IWR-1, AZ-6102 | ARTD5, ARTD6 (TNKS) | Wnt-driven cancers Colon, lung, brain, breast, ovarian, liver cancers. | [106,107] |
| N-([1,2,4]triazolo[4 ,3-a]pyridin-3-yl)-1-(2-cyanophenyl)piperidine-4-carboxamide (TI-12403) | TNKS | Combined 5-FU/TI-12403 treatment synergistically block proliferation of colorectal cancer (CRC) | [108] |
| M2912 | TNKS | CRC | [109] |
| Spiroindoline derivative 40c RK-287107) | TNKS | CRC | [110] |
| MSC2504877 + CDC4/6 inhibitors | TNKS | APC mutant CRC | [111] |
| NAM binders | |||
| XAV939, LZZ-02, AZ1366, RK-582, RK-287107 | TNKS | CRC with APC mutations, Lung cancers | [112] |
| In association with PI3K (BKM120) and epidermal growth factor receptor (EGFR) (erlotinib) inhibitors | CRC with APC mutations | [113] | |
| Adenosine binders | |||
| IWR-1, G007-LK, OD366, OM-1700, K-756 | TNKS: restoration of β-catenin degradation | CRC | [103] |
| Dual binders: binding to adenosine and nicotinamide subsites | |||
| CMP4, WIKI4, IWR-1, IWR-2, CMP4b puinazolinone, CMP24, [1,2,4]triazol-3-ylsulfanylmethyl)-3-phenyl-[1,2,4]oxadiazoles, NVP-TNKS656 | TNKS | CRC, mammary adenocarcinoma | [106,114] |
| EB-47 | TNKS, PARP1 | [114] | |
| Quinoxaline-based set of fragments Ankyrin repeat cluster (ARC)-TNKS binding modules (TBM) | TNKS | [118,119] | |
| C44 disrupts the protein–protein interaction (PPI) interface of TNKS and USP25 | TNKS | Prostate cancer, increase of Axin-1 levels | [120] |
| Drug | Target | Therapeutic Applications | References |
|---|---|---|---|
| H10 | ARTD8/PARP14 | In vitro | [123] |
| 4t and 8b, in a group of -4-(3-carbamoylphenylamino) 4 oxobut-2-enyl amides | ARTD8/PARP14 | Multiple myeloma, HCC, prostate cancer | [123,124,125] |
| N-(2(-9H-carbazol-1-yl)phenyl)acetamide/sulfonamide, GeA-69 | ARTD8/PARP14 macrodomain | Lymphoma (DLBCL), HCC | [125,127] |
| 8k and 8m diaryl ethers | ARTD8/PARP14 | In vitro | [123,124,125] |
| RBN012759, RBN011980, RBN012759, RBN012811, RBN012042, RBN010860, AZ12629495, RBN0120420; RBN013527 similar to RBN012811, but does not cause the degradation of PARP14 | ARTD8/PARP14 | In vitro | [128,129] |
| Drug | Target | Therapeutic Applications | References |
|---|---|---|---|
| 4-Benzyloxybenzimide derivatives, 4-4-cyanophenoxybenzamide, 3-4-carbamoylphenoxybenzamide, OUl35 | ARTD10 | Neurodegenerative disorder’ gene amplification in breast and ovarian cancer | [14,95,126,127,128,129,130,131,132,164,165] |
| ITK7 | ARTD11 | Causes PARP11 to dissociate from the nuclear envelope | [128,134,143] |
| RBN-2397 RBN011147 (FRET), RBN11198 (BRET), RBN010860 | ARTD14/ PARP7 | Metastatic solid tumors, lung cancer, anti-proliferative, macrophage polarization, immune signaling | [133,134,135,136,137,138] |
| EGCG RBN010860, RBN012148, talazoparib | ARTD15/ PARP16 | ER stress, intracellular trafficking, SCLC | [6,66,97,130,139,140] |
| RBN010860, AZ0108 | ARTD17/ PARP6 | CRC, various cancers | [138,139,140,141,142,143,144] |
| Cholera-like ARTs | |||
| Aspecific MARylation inhibitors FRET (RBN011147) and NanoBRET (RBN011198) probes | ARTC1 | Various cancers (CRC, glioma), angiogenesis, block of epithelial-to-mesenchymal transition (EMT) | [15,66,118,144,159,160,161,162,163] |
| Drug | Target | Therapeutic Applications | References |
|---|---|---|---|
| Acetylated lysine-ADP-ribose conjugates | SIRT7 | NSCLC, control of ERK1 activity | [183] |
| EX527 (selisistat), OSS_128167 | SIRT6 (and other SIRTs) target the NAD+ binding pocket | Chemosensitizing effects | [166,174] |
| Quinazolinedione derivatives 1-phenylpiperazine derivative | SIRT6, target the NAD+ binding pocket | Increase in DNA damage/cell death induced by olaparib in Capan-1 cells, silencing blocks proliferation and induces apoptosis in melanoma | [168,175] |
| Quinazolinediones, salicylate-like structure OSS_128167 | SIRT6 inhibition | Sensitivity of cancer to gemcitabine, pancreatic ductal adenocarcinoma | [191,192] |
| MDL-800, CL5D, MDL-801 | SIRT6 activators | Several cancer cells, antiproliferative | [172,189] |
| FK866 (NAMPT inhibitor) NAD salvage pathway block | SIRT4 inhibition by NAM | Improving aging and neurological diseases, decreasing oxidative stress | [1,27] |
| Nicotinamide riboside, miR-424-5p antagomiRs | SIRT4 activation/upregulation | Esophageal squamous cell carcinoma (ESCC) proliferation inhibition | [1,193] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poltronieri, P.; Miwa, M.; Masutani, M. ADP-Ribosylation as Post-Translational Modification of Proteins: Use of Inhibitors in Cancer Control. Int. J. Mol. Sci. 2021, 22, 10829. https://doi.org/10.3390/ijms221910829
Poltronieri P, Miwa M, Masutani M. ADP-Ribosylation as Post-Translational Modification of Proteins: Use of Inhibitors in Cancer Control. International Journal of Molecular Sciences. 2021; 22(19):10829. https://doi.org/10.3390/ijms221910829
Chicago/Turabian StylePoltronieri, Palmiro, Masanao Miwa, and Mitsuko Masutani. 2021. "ADP-Ribosylation as Post-Translational Modification of Proteins: Use of Inhibitors in Cancer Control" International Journal of Molecular Sciences 22, no. 19: 10829. https://doi.org/10.3390/ijms221910829
APA StylePoltronieri, P., Miwa, M., & Masutani, M. (2021). ADP-Ribosylation as Post-Translational Modification of Proteins: Use of Inhibitors in Cancer Control. International Journal of Molecular Sciences, 22(19), 10829. https://doi.org/10.3390/ijms221910829