
Ubiquitination in T-Cell Activation and Checkpoint Inhibition: New Avenues for Targeted Cancer Immunotherapy

Abstract
1. Introduction
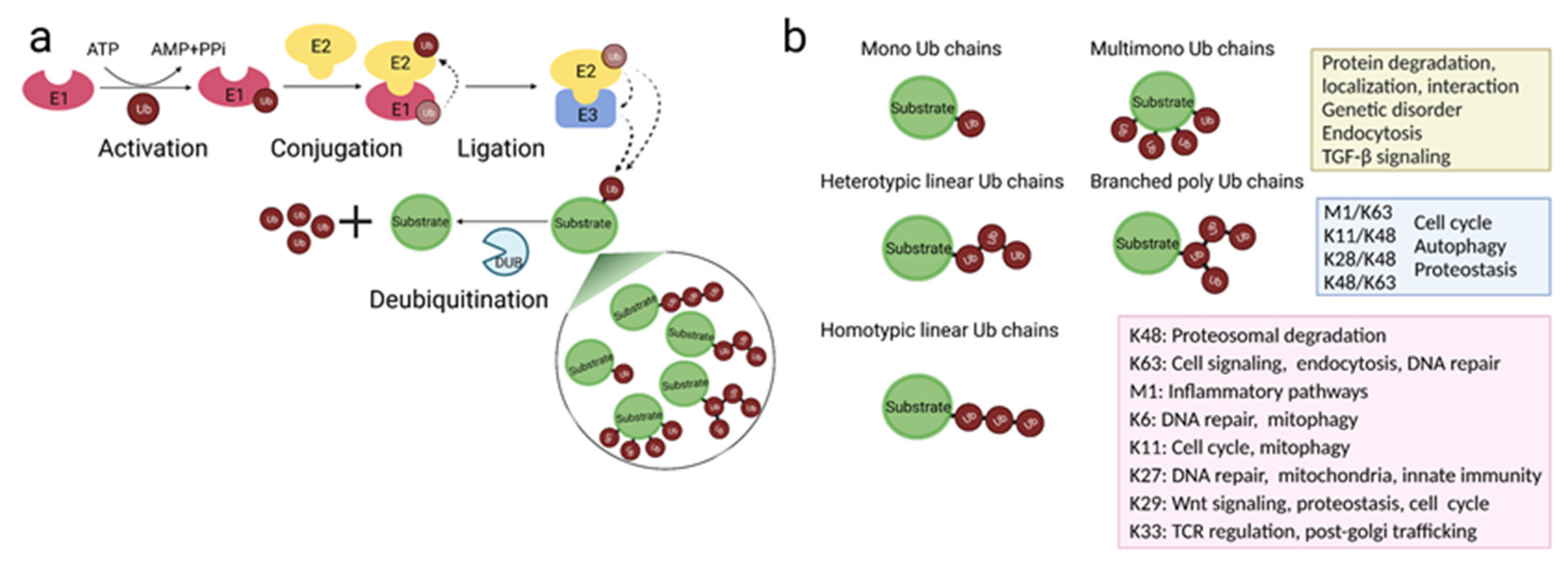
2. Protein Ubiquitination
3. Key Mechanism of T-Cell Activation and Inhibition and Its Use in Immunotherapy
3.1. T-Cell Activation
3.2. T-Cell Inhibition
3.3. T-Cell Targeted Cancer Immunotherapy
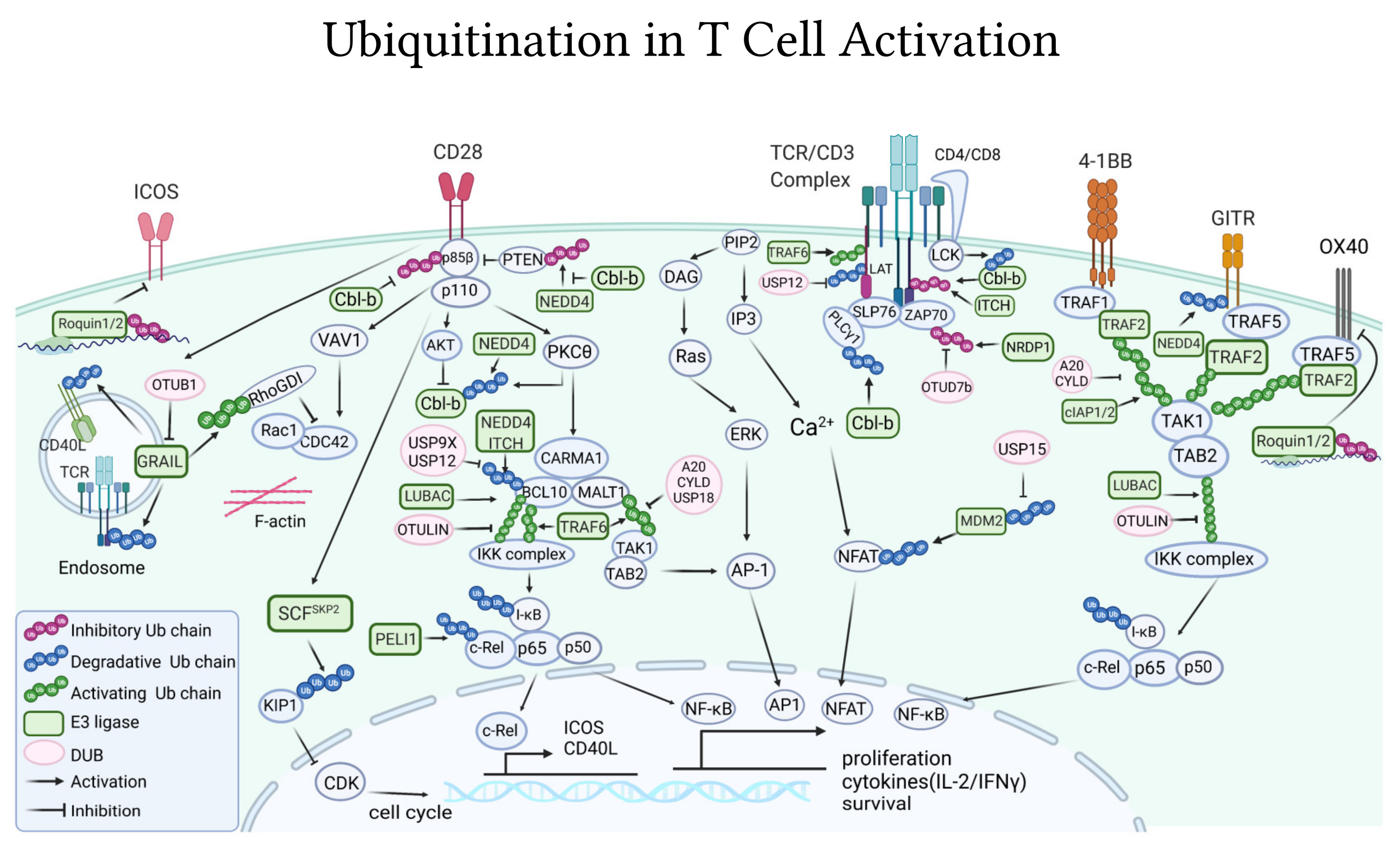
4. Ubiquitination Is Essential to Regulate T Cell Activating and Inhibitory Signaling
4.1. Ubiquitin-Pathways in TCR/CD3 Activation
4.1.1. E3 Ligases
4.1.2. DUBs
4.1.3. TCR Internalization
4.2. Ubiquitin-Pathways Downstream of Co-Stimulatory Receptors
4.2.1. CD28
4.2.2. Other Co-Stimulatory Receptors
4.3. Ubiquitin-Pathways Downstream CTLA-4 and PD-1/PD-L1 Co-Inhibitory Receptors
5. Exploiting Ubiquitin-Dependent Pathways for Cancer Treatment

{kind=link}
{kind=link}
{kind=link}
| Proteasome (Total = 39) | E3 Ligases (Total = 94) | DUBs (Total = 109) | |||
|---|---|---|---|---|---|
| 20S (total = 6) | RING-type (total = 70) | USPs (total = 77) | |||
| 20S | 6 (FDA = 3, C = 3) | MDM2 | 25 (C = 13, PC = 8) | USP7 | 30 (PC = 18) |
| 19S (total = 31) | SKP2 | 9 (C = 1, PC = 8) | unspecific-USP | 27 (C-1, PC = 15) | |
| USP14 | 14(PC = 4) | IAPs | 5 (C = 4, PC = 1) | USP1 | 8 (C = 1, PC = 6) |
| unspecific-USP14 | 10(C = 2, PC = 4) | Unspecific- RING | 5 (PC = 2) | USP2 | 3 (C = 1) |
| unspecific- RPN11 | 4(PC = 3) | Cul4-DCAF15 | C = 4 | USP30 | 3 |
| RPN11 (PSMD14) | 3(PC = 1) | MDMX | 3 (PC = 2) | USP8 | 2 |
| ATPase (total = 2) | RNF4 | PC = 3 | USP9X | PC = 1 | |
| p97 | C = 2 | XIAP | C = 2 | USP19 | 1 |
| KEAP1 | C = 2 | USP20 | 1 | ||
| APC/C | PC = 2 | USP28 | 1 | ||
| β-TrCP1 | PC = 2 | UCHs (total = 18) | |||
| TRAF6 | PC = 2 | UCHL1 | 14 (PC = 4) | ||
| FBW7 | PC = 2 | UCHL3 | 3 (PC = 1) | ||
| Met30 | PC = 1 | unspecific-UCH | 1 | ||
| VHL | 3 | OTUs (total = 3) | |||
| HECT-type (total = 17) | TRABID | 1 | |||
| E6AP | PC = 8 | OTUB2 | 1 | ||
| HUWE1 | PC = 2 | Cezanne | 1 | ||
| SMURF1/2 | PC = 1 | JAMMs (total = 3) | |||
| unspecific-HECT | PC = 1 | CSN5 | 2 (PC = 1) | ||
| WWP2 | 5 | STAMBP(AMSH) | PC = 1 | ||
| RBR-type (total = 7) | Other DUBs (total = 8) | ||||
| HOIP | 7 (FDA = 1, PC = 3) | ADRM1(RPN13) | 3 (PC = 1) | ||
| SARS PLPro | 3 | ||||
| Ataxin | 1 | ||||
| Unspecific | 1 | ||||
6. Concluding Remarks and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T Cell Activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef] [PubMed]
- Pentcheva-Hoang, T.; Corse, E.; Allison, J.P. Negative Regulators of T-Cell Activation: Potential Targets for Therapeutic Intervention in Cancer, Autoimmune Disease, and Persistent Infections. Immunol. Rev. 2009, 229, 67–87. [Google Scholar] [CrossRef]
- Lennerz, V.; Fatho, M.; Gentilini, C.; Frye, R.A.; Lifke, A.; Ferel, D.; Wolfel, C.; Huber, C.; Wolfel, T. The Response of Autologous T Cells to a Human Melanoma Is Dominated by Mutated Neoantigens. Proc. Natl. Acad. Sci. USA 2005, 102, 16013–16018. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, I.; Sasieni, P.; Bodmer, W. How Many Mutations in a Cancer? Am. J. Pathol. 2002, 160, 755–758. [Google Scholar] [CrossRef]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive Strategies That Are Mediated by Tumor Cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef]
- Garner, H.; de Visser, K.E. Immune Crosstalk in Cancer Progression and Metastatic Spread: A Complex Conversation. Nat. Rev. Immunol. 2020, 20, 483–497. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, Y.; Zhang, X.; Fu, J.; Xing, X.; Wang, C.; Gao, L.; Liu, Y.; Shi, L. Potential Applications of Nanoparticles for Tumor Microenvironment Remodeling to Ameliorate Cancer Immunotherapy. Int. J. Pharm. 2019, 570, 118636. [Google Scholar] [CrossRef]
- Johnson, D.B.; Jakubovic, B.D.; Sibaud, V.; Sise, M.E. Balancing Cancer Immunotherapy Efficacy and Toxicity. J. Allergy Clin. Immunol. Pract. 2020, 8, 2898–2906. [Google Scholar] [CrossRef]
- Grigor, E.J.M.; Fergusson, D.; Kekre, N.; Montroy, J.; Atkins, H.; Seftel, M.D.; Daugaard, M.; Presseau, J.; Thavorn, K.; Hutton, B.; et al. Risks and Benefits of Chimeric Antigen Receptor T-Cell (CAR-T) Therapy in Cancer: A Systematic Review and Meta-Analysis. Transfus. Med. Rev. 2019, 33, 98–110. [Google Scholar] [CrossRef]
- Jazaeri, A.A.; Zsiros, E.; Amaria, R.N.; Artz, A.S.; Edwards, R.P.; Wenham, R.M.; Slomovitz, B.M.; Walther, A.; Thomas, S.S.; Chesney, J.A.; et al. Safety and Efficacy of Adoptive Cell Transfer Using Autologous Tumor Infiltrating Lymphocytes (LN-145) for Treatment of Recurrent, Metastatic, or Persistent Cervical Carcinoma. J. Clin. Oncol. 2019, 37, 2538. [Google Scholar] [CrossRef]
- Sarnaik, A.; Thomas, S.S.; Davar, D.; Kirkwood, J.M.; Kluger, H.M.; Lutzky, J.; Wilson, M.; Pavlick, A.C.; Curti, B.D.; Whitman, E.D.; et al. Safety and Efficacy of Cryopreserved Autologous Tumor Infiltrating Lymphocyte Therapy (LN-144, Lifileucel) in Advanced Metastatic Melanoma Patients Previously Treated with at Least One Prior Systemic Therapy. J. Clin. Oncol. 2019, 37, 136. [Google Scholar] [CrossRef]
- Kuwabara, T.; Matsui, Y.; Ishikawa, F.; Kondo, M. Regulation of T-Cell Signaling by Post-Translational Modifications in Autoimmune Disease. Int. J. Mol. Sci. 2018, 19, 819. [Google Scholar] [CrossRef] [PubMed]
- Raposo, B.; Merky, P.; Lundqvist, C.; Yamada, H.; Urbonaviciute, V.; Niaudet, C.; Viljanen, J.; Kihlberg, J.; Kyewski, B.; Ekwall, O.; et al. T Cells Specific for Post-Translational Modifications Escape Intrathymic Tolerance Induction. Nat. Commun. 2018, 9, 353. [Google Scholar] [CrossRef]
- Li, J.; Chai, Q.-Y.; Liu, C.H. The Ubiquitin System: A Critical Regulator of Innate Immunity and Pathogen–Host Interactions. Cell. Mol. Immunol. 2016, 13, 560–576. [Google Scholar] [CrossRef]
- Malynn, B.A.; Ma, A. Ubiquitin Makes Its Mark on Immune Regulation. Immunity 2010, 33, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The Role of Ubiquitination in Tumorigenesis and Targeted Drug Discovery. Signal Transduct. Target. Ther. 2020, 5, 11. [Google Scholar] [CrossRef]
- Fujita, Y.; Tinoco, R.; Li, Y.; Senft, D.; Ronai, Z.A. Ubiquitin Ligases in Cancer Immunotherapy—Balancing Antitumor and Autoimmunity. Trends Mol. Med. 2019, 25, 428–443. [Google Scholar] [CrossRef]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T Cells in Tumor Microenvironment: New Mechanisms, Potential Therapeutic Strategies and Future Prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Pickart, C.M. Mechanisms Underlying Ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef]
- Deshaies, R.J.; Joazeiro, C.A.P. RING Domain E3 Ubiquitin Ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Kumar, S. Mammalian HECT Ubiquitin-Protein Ligases: Biological and Pathophysiological Aspects. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2014, 1843, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Spratt, D.E.; Walden, H.; Shaw, G.S. RBR E3 Ubiquitin Ligases: New Structures, New Insights, New Questions. Biochem. J. 2014, 458, 421–437. [Google Scholar] [CrossRef]
- Ciechanover, A. N-Terminal Ubiquitination: More Protein Substrates Join In. Trends Cell Biol. 2004, 14, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Schwartzkopff, B.; Platta, H.W.; Hasan, S.; Girzalsky, W.; Erdmann, R. Cysteine-Specific Ubiquitination Protects the Peroxisomal Import Receptor Pex5p against Proteasomal Degradation. Biosci. Rep. 2015, 35, e00215. [Google Scholar] [CrossRef]
- Wang, X.; Herr, R.A.; Chua, W.-J.; Lybarger, L.; Wiertz, E.J.H.J.; Hansen, T.H. Ubiquitination of Serine, Threonine, or Lysine Residues on the Cytoplasmic Tail Can Induce ERAD of MHC-I by Viral E3 Ligase MK3. J. Cell Biol. 2007, 177, 613–624. [Google Scholar] [CrossRef]
- Bhogaraju, S.; Kalayil, S.; Liu, Y.; Bonn, F.; Colby, T.; Matic, I.; Dikic, I. Phosphoribosylation of Ubiquitin Promotes Serine Ubiquitination and Impairs Conventional Ubiquitination. Cell 2016, 167, 1636–1649.e13. [Google Scholar] [CrossRef] [PubMed]
- McClellan, A.J.; Laugesen, S.H.; Ellgaard, L. Cellular Functions and Molecular Mechanisms of Non-Lysine Ubiquitination. Open Biol. 2019, 9, 190147. [Google Scholar] [CrossRef]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin Chain Diversity at a Glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef]
- Yau, R.; Rape, M. The Increasing Complexity of the Ubiquitin Code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin Modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef]
- Deol, K.K.; Lorenz, S.; Strieter, E.R. Enzymatic Logic of Ubiquitin Chain Assembly. Front. Physiol. 2019, 10, 835. [Google Scholar] [CrossRef]
- Haglund, K.; Sigismund, S.; Polo, S.; Szymkiewicz, I.; Di Fiore, P.P.; Dikic, I. Multiple Monoubiquitination of RTKs Is Sufficient for Their Endocytosis and Degradation. Nat. Cell Biol. 2003, 5, 461–466. [Google Scholar] [CrossRef]
- Hicke, L. Protein Regulation by Monoubiquitin. Nat. Rev. Mol. Cell Biol. 2001, 2, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Zhang, Z.; van Dam, H.; Zhang, L.; Zhou, F. Regulation of TGF-β Superfamily Signaling by SMAD Mono-Ubiquitination. Cells 2014, 3, 981–993. [Google Scholar] [CrossRef]
- Tang, L.-Y.; Yamashita, M.; Coussens, N.P.; Tang, Y.; Wang, X.; Li, C.; Deng, C.-X.; Cheng, S.Y.; Zhang, Y.E. Ablation of Smurf2 Reveals an Inhibition in TGF-β Signalling through Multiple Mono-Ubiquitination of Smad3. EMBO J. 2011, 30, 4777–4789. [Google Scholar] [CrossRef] [PubMed]
- Sewduth, R.N.; Baietti, M.F.; Sablina, A.A. Cracking the Monoubiquitin Code of Genetic Diseases. Int. J. Mol. Sci. 2020, 21, 3036. [Google Scholar] [CrossRef]
- Bremm, A.; Komander, D. Emerging Roles for Lys11-Linked Polyubiquitin in Cellular Regulation. Trends Biochem. Sci. 2011, 36, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.N.; Baughman, J.M.; Phu, L.; Tea, J.S.; Yu, C.; Coons, M.; Kirkpatrick, D.S.; Bingol, B.; Corn, J.E. USP30 and Parkin Homeostatically Regulate Atypical Ubiquitin Chains on Mitochondria. Nat. Cell Biol. 2015, 17, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Fiil, B.K.; Gyrd-Hansen, M. The Met1-Linked Ubiquitin Machinery in Inflammation and Infection. Cell Death Differ. 2021, 28, 557–569. [Google Scholar] [CrossRef]
- Wu, X.; Lei, C.; Xia, T.; Zhong, X.; Yang, Q.; Shu, H.-B. Regulation of TRIF-Mediated Innate Immune Response by K27-Linked Polyubiquitination and Deubiquitination. Nat. Commun. 2019, 10, 4115. [Google Scholar] [CrossRef]
- Dang, F.; Nie, L.; Wei, W. Ubiquitin Signaling in Cell Cycle Control and Tumorigenesis. Cell Death Differ. 2021, 28, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, G.-R.; Kim, H.; Jo, Y.-J.; Hong, S.-E.; Lee, J.; Lee, H.I.; Jang, Y.-S.; Oh, S.-H.; Lee, H.J.; et al. Effective Killing of Cancer Cells and Regression of Tumor Growth by K27 Targeting Sulfiredoxin. Free Radic. Biol. Med. 2016, 101, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.E.H.; Boardman, A.P.; Wang, D.C.; Huttlin, E.L.; Everley, R.A.; Dephoure, N.; Zhou, C.; Koren, I.; Gygi, S.P.; Elledge, S.J. Quantitative Proteomic Atlas of Ubiquitination and Acetylation in the DNA Damage Response. Mol. Cell 2015, 59, 867–881. [Google Scholar] [CrossRef]
- Gersch, M.; Gladkova, C.; Schubert, A.F.; Michel, M.A.; Maslen, S.; Komander, D. Mechanism and Regulation of the Lys6-Selective Deubiquitinase USP30. Nat. Struct. Mol. Biol. 2017, 24, 920–930. [Google Scholar] [CrossRef]
- Fei, C.; Li, Z.; Li, C.; Chen, Y.; Chen, Z.; He, X.; Mao, L.; Wang, X.; Zeng, R.; Li, L. Smurf1-Mediated Lys29-Linked Nonproteolytic Polyubiquitination of Axin Negatively Regulates Wnt/β-Catenin Signaling. Mol. Cell. Biol. 2013, 33, 4095–4105. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zheng, Q.; Erramilli, S.K.; Pan, M.; Park, S.; Xie, Y.; Li, J.; Fei, J.; Kossiakoff, A.A.; Liu, L.; et al. K29-Linked Ubiquitin Signaling Regulates Proteotoxic Stress Response and Cell Cycle. Nat. Chem. Biol. 2021, 17, 896–905. [Google Scholar] [CrossRef]
- Yuan, W.-C.; Lee, Y.-R.; Lin, S.-Y.; Chang, L.-Y.; Tan, Y.P.; Hung, C.-C.; Kuo, J.-C.; Liu, C.-H.; Lin, M.-Y.; Xu, M.; et al. K33-Linked Polyubiquitination of Coronin 7 by Cul3-KLHL20 Ubiquitin E3 Ligase Regulates Protein Trafficking. Mol. Cell 2014, 54, 586–600. [Google Scholar] [CrossRef]
- Huang, H.; Jeon, M.-S.; Liao, L.; Yang, C.; Elly, C.; Yates, J.R.; Liu, Y.-C. K33-Linked Polyubiquitination of T Cell Receptor-Zeta Regulates Proteolysis-Independent T Cell Signaling. Immunity 2010, 33, 60–70. [Google Scholar] [CrossRef]
- Yang, M.; Chen, T.; Li, X.; Yu, Z.; Tang, S.; Wang, C.; Gu, Y.; Liu, Y.; Xu, S.; Li, W.; et al. K33-Linked Polyubiquitination of Zap70 by Nrdp1 Controls CD8(+) T Cell Activation. Nat. Immunol. 2015, 16, 1253–1262. [Google Scholar] [CrossRef]
- Yau, R.G.; Doerner, K.; Castellanos, E.R.; Haakonsen, D.L.; Werner, A.; Wang, N.; Yang, X.W.; Martinez-Martin, N.; Matsumoto, M.L.; Dixit, V.M.; et al. Assembly and Function of Heterotypic Ubiquitin Chains in Cell-Cycle and Protein Quality Control. Cell 2017, 171, 918–933.e20. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Saeki, Y.; Ishido, S.; Kanno, J.; Tanaka, K. The K48-K63 Branched Ubiquitin Chain Regulates NF-ΚB Signaling. Mol. Cell 2016, 64, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Tsuchiya, H.; Saeki, Y.; Tanaka, K. K63 Ubiquitylation Triggers Proteasomal Degradation by Seeding Branched Ubiquitin Chains. Proc. Natl. Acad. Sci. USA 2018, 115, E1401–E1408. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Huang, T.-Y.; Lin, Y.-T.; Lin, S.-Y.; Li, W.-H.; Hsiao, H.-J.; Yan, R.-L.; Tang, H.-W.; Shen, Z.-Q.; Chen, G.-C.; et al. VPS34 K29/K48 Branched Ubiquitination Governed by UBE3C and TRABID Regulates Autophagy, Proteostasis and Liver Metabolism. Nat. Commun. 2021, 12, 1322. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Clague, M.J.; Urbé, S. Breaking the Chains: Structure and Function of the Deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and Cellular Roles of Ubiquitin-Specific Deubiquitinating Enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef]
- Grou, C.P.; Pinto, M.P.; Mendes, A.V.; Domingues, P.; Azevedo, J.E. The de Novo Synthesis of Ubiquitin: Identification of Deubiquitinases Acting on Ubiquitin Precursors. Sci. Rep. 2015, 5, 12836. [Google Scholar] [CrossRef] [PubMed]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in Disease Pathogenesis and Treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- Ndubaku, C.; Tsui, V. Inhibiting the Deubiquitinating Enzymes (DUBs). J. Med. Chem. 2015, 58, 1581–1595. [Google Scholar] [CrossRef]
- Schulman, B.A.; Wade Harper, J. Ubiquitin-like Protein Activation by E1 Enzymes: The Apex for Downstream Signalling Pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331. [Google Scholar] [CrossRef]
- Michelle, C.; Vourc’h, P.; Mignon, L.; Andres, C.R. What Was the Set of Ubiquitin and Ubiquitin-Like Conjugating Enzymes in the Eukaryote Common Ancestor? J. Mol. Evol. 2009, 68, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Bengtson, M.H.; Ulbrich, A.; Matsuda, A.; Reddy, V.A.; Orth, A.; Chanda, S.K.; Batalov, S.; Joazeiro, C.A.P. Genome-Wide and Functional Annotation of Human E3 Ubiquitin Ligases Identifies MULAN, a Mitochondrial E3 That Regulates the Organelle’s Dynamics and Signaling. PLoS ONE 2008, 3, e1487. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.B.; Luna-Vargas, M.P.A.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.G.; Sixma, T.K.; Bernards, R. A Genomic and Functional Inventory of Deubiquitinating Enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef]
- Samelson, L.E.; Patel, M.D.; Weissman, A.M.; Harford, J.B.; Klausner, R.D. Antigen Activation of Murine T Cells Induces Tyrosine Phosphorylation of a Polypeptide Associated with the T Cell Antigen Receptor. Cell 1986, 46, 1083–1090. [Google Scholar] [CrossRef]
- Malissen, M.; Minard, K.; Mjolsness, S.; Kronenberg, M.; Goverman, J.; Hunkapiller, T.; Prystowsky, M.B.; Yoshikai, Y.; Fitch, F.; Mak, T.W. Mouse T Cell Antigen Receptor: Structure and Organization of Constant and Joining Gene Segments Encoding the Beta Polypeptide. Cell 1984, 37, 1101–1110. [Google Scholar] [CrossRef]
- Letourneur, F.; Klausner, R.D. Activation of T Cells by a Tyrosine Kinase Activation Domain in the Cytoplasmic Tail of CD3 Epsilon. Science 1992, 255, 79–82. [Google Scholar] [CrossRef]
- Zhang, W.; Sloan-Lancaster, J.; Kitchen, J.; Trible, R.P.; Samelson, L.E. LAT: The ZAP-70 Tyrosine Kinase Substrate That Links T Cell Receptor to Cellular Activation. Cell 1998, 92, 83–92. [Google Scholar] [CrossRef]
- Lo, W.-L.; Shah, N.H.; Ahsan, N.; Horkova, V.; Stepanek, O.; Salomon, A.R.; Kuriyan, J.; Weiss, A. Lck Promotes Zap70-Dependent LAT Phosphorylation by Bridging Zap70 to LAT. Nat. Immunol. 2018, 19, 733–741. [Google Scholar] [CrossRef]
- Essen, L.O.; Perisic, O.; Katan, M.; Wu, Y.; Roberts, M.F.; Williams, R.L. Structural Mapping of the Catalytic Mechanism for a Mammalian Phosphoinositide-Specific Phospholipase C. Biochemistry 1997, 36, 1704–1718. [Google Scholar] [CrossRef]
- Iborra, S.; Ramos, M.; Arana, D.M.; Lázaro, S.; Aguilar, F.; Santos, E.; López, D.; Fernández-Malavé, E.; Del Val, M. N-Ras Couples Antigen Receptor Signaling to Eomesodermin and to Functional CD8+ T Cell Memory but Not to Effector Differentiation. J. Exp. Med. 2013, 210, 1463–1479. [Google Scholar] [CrossRef]
- Joseph, N.; Reicher, B.; Barda-Saad, M. The Calcium Feedback Loop and T Cell Activation: How Cytoskeleton Networks Control Intracellular Calcium Flux. Biochim. Biophys. Acta 2014, 1838, 557–568. [Google Scholar] [CrossRef]
- Thome, M. CARMA1, BCL-10 and MALT1 in Lymphocyte Development and Activation. Nat. Rev. Immunol. 2004, 4, 348–359. [Google Scholar] [CrossRef]
- Bretscher, P.; Cohn, M. A Theory of Self-Nonself Discrimination: Paralysis and Induction Involve the Recognition of One and Two Determinants on an Antigen, Respectively. Science 1970, 169, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L. Coordination of T Cell Activation and Migration through Formation of the Immunological Synapse. Ann. N. Y. Acad. Sci. 2003, 987, 51–59. [Google Scholar] [CrossRef]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 Family Revisited. Annu. Rev. Immunol. 2005, 23, 515–548. [Google Scholar] [CrossRef] [PubMed]
- Helou, Y.A.; Petrashen, A.P.; Salomon, A.R. Vav1 Regulates T Cell Activation through a Feedback Mechanism and Crosstalk between the T Cell Receptor and CD28. J. Proteome Res. 2015, 14, 2963–2975. [Google Scholar] [CrossRef]
- Kane, L.P.; Weiss, A. The PI-3 Kinase/Akt Pathway and T Cell Activation: Pleiotropic Pathways Downstream of PIP3. Immunol. Rev. 2003, 192, 7–20. [Google Scholar] [CrossRef]
- Acuto, O.; Michel, F. CD28-Mediated Co-Stimulation: A Quantitative Support for TCR Signalling. Nat. Rev. Immunol. 2003, 3, 939–951. [Google Scholar] [CrossRef]
- Michel, F.; Attal-Bonnefoy, G.; Mangino, G.; Mise-Omata, S.; Acuto, O. CD28 as a Molecular Amplifier Extending TCR Ligation and Signaling Capabilities. Immunity 2001, 15, 935–945. [Google Scholar] [CrossRef]
- Sharpe, A.H. Mechanisms of Costimulation. Immunol. Rev. 2009, 229, 5–11. [Google Scholar] [CrossRef]
- Croft, M. The Role of TNF Superfamily Members in T-Cell Function and Diseases. Nat. Rev. Immunol. 2009, 9, 271–285. [Google Scholar] [CrossRef]
- O’Neill, R.E.; Cao, X. Co-Stimulatory and Co-Inhibitory Pathways in Cancer Immunotherapy. Adv. Cancer Res. 2019, 143, 145–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, F.; Cao, J.; Wang, X.; Cheng, H.; Qi, K.; Wang, G.; Xu, K.; Zheng, J.; Fu, Y.-X.; et al. A Chimeric Antigen Receptor with Antigen-Independent OX40 Signaling Mediates Potent Antitumor Activity. Sci. Transl. Med. 2021, 13, eaba7308. [Google Scholar] [CrossRef]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of Large, Established Tumor Xenografts with Genetically Retargeted Human T Cells Containing CD28 and CD137 Domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Greene, J.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7-1 (CD80) and B7-2 (CD86) Bind with Similar Avidities but Distinct Kinetics to CD28 and CTLA-4 Receptors. Immunity 1994, 1, 793–801. [Google Scholar] [CrossRef]
- Linsley, P.S.; Bradshaw, J.; Greene, J.; Peach, R.; Bennett, K.L.; Mittler, R.S. Intracellular Trafficking of CTLA-4 and Focal Localization Towards Sites of TCR Engagement. Immunity 1996, 4, 535–543. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell Extrinsic Function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef]
- Guntermann, C.; Alexander, D.R. CTLA-4 Suppresses Proximal TCR Signaling in Resting Human CD4 + T Cells by Inhibiting ZAP-70 Tyr 319 Phosphorylation: A Potential Role for Tyrosine Phosphatases. J. Immunol. 2002, 168, 4420–4429. [Google Scholar] [CrossRef]
- Patsoukis, N.; Wang, Q.; Strauss, L.; Boussiotis, V.A. Revisiting the PD-1 Pathway. Sci. Adv. 2020, 6, eabd2712. [Google Scholar] [CrossRef]
- Pedoeem, A.; Azoulay-Alfaguter, I.; Strazza, M.; Silverman, G.J.; Mor, A. Programmed Death-1 Pathway in Cancer and Autoimmunity. Clin. Immunol. 2014, 153, 145–152. [Google Scholar] [CrossRef]
- Sheppard, K.-A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R.; et al. PD-1 Inhibits T-Cell Receptor Induced Phosphorylation of the ZAP70/CD3zeta Signalosome and Downstream Signaling to PKCtheta. FEBS Lett. 2004, 574, 37–41. [Google Scholar] [CrossRef]
- Gautron, A.-S.; Dominguez-Villar, M.; de Marcken, M.; Hafler, D.A. Enhanced Suppressor Function of TIM-3+ FoxP3+ Regulatory T Cells. Eur. J. Immunol. 2014, 44, 2703–2711. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-Derived Suppressor Cells as Regulators of the Immune System. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-Specific Cell Surface Protein Tim-3 Regulates Macrophage Activation and Severity of an Autoimmune Disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in Cancer Immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- McNeill, L.; Salmond, R.J.; Cooper, J.C.; Carret, C.K.; Cassady-Cain, R.L.; Roche-Molina, M.; Tandon, P.; Holmes, N.; Alexander, D.R. The Differential Regulation of Lck Kinase Phosphorylation Sites by CD45 Is Critical for T Cell Receptor Signaling Responses. Immunity 2007, 27, 425–437. [Google Scholar] [CrossRef]
- Monjas, A.; Alcover, A.; Alarcón, B. Engaged and Bystander T Cell Receptors Are Down-Modulated by Different Endocytotic Pathways. J. Biol. Chem. 2004, 279, 55376–55384. [Google Scholar] [CrossRef] [PubMed]
- Barr, V.A.; Balagopalan, L.; Barda-Saad, M.; Polishchuk, R.; Boukari, H.; Bunnell, S.C.; Bernot, K.M.; Toda, Y.; Nossal, R.; Samelson, L.E. T-Cell Antigen Receptor-Induced Signaling Complexes: Internalization Via a Cholesterol-Dependent Endocytic Pathway. Traffic 2006, 7, 1143–1162. [Google Scholar] [CrossRef]
- Liu, H.; Rhodes, M.; Wiest, D.L.; Vignali, D.A.A. On the Dynamics of TCR:CD3 Complex Cell Surface Expression and Downmodulation. Immunity 2000, 13, 665–675. [Google Scholar] [CrossRef]
- Von Essen, M.; Bonefeld, C.M.; Siersma, V.; Rasmussen, A.B.; Lauritsen, J.P.H.; Nielsen, B.L.; Geisler, C. Constitutive and Ligand-Induced TCR Degradation. J. Immunol. 2004, 173, 384–393. [Google Scholar] [CrossRef]
- Schwartz, R.H. T Cell Anergy. Annu. Rev. Immunol. 2003, 21, 305–334. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and Cellular Insights into T Cell Exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.S.; Cox, M.A.; Zajac, A.J. T-Cell Exhaustion: Characteristics, Causes and Conversion: T-Cell Exhaustion. Immunology 2010, 129, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.P.; Effros, R.B. T Cell Replicative Senescence in Human Aging. Curr. Pharm. Des. 2013, 19, 1680–1698. [Google Scholar] [CrossRef]
- Wongjitrat, C.; Sukwit, S.; Chuenchitra, T.; Seangjaruk, P.; Rojanasang, P.; Romputtan, P.; Srisurapanon, S. CTLA-4 and Its Ligands on the Surface of T- and B-Lymphocyte Subsets in Chronic Hepatitis B Virus Infection. J. Med. Assoc. Thail. Chotmaihet Thangphaet 2013, 96 (Suppl. 1), S54–S59. [Google Scholar]
- Dong, Y.; Li, X.; Zhang, L.; Zhu, Q.; Chen, C.; Bao, J.; Chen, Y. CD4+ T Cell Exhaustion Revealed by High PD-1 and LAG-3 Expression and the Loss of Helper T Cell Function in Chronic Hepatitis B. BMC Immunol. 2019, 20, 27. [Google Scholar] [CrossRef]
- Weng, N.; Akbar, A.N.; Goronzy, J. CD28−T Cells: Their Role in the Age-Associated Decline of Immune Function. Trends Immunol. 2009, 30, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Stachura, P.; Xu, H.C.; Bhatia, S.; Borkhardt, A.; Lang, P.A.; Pandyra, A.A. Senescent Tumor CD8+ T Cells: Mechanisms of Induction and Challenges to Immunotherapy. Cancers 2020, 12, 2828. [Google Scholar] [CrossRef]
- Kasakovski, D.; Xu, L.; Li, Y. T Cell Senescence and CAR-T Cell Exhaustion in Hematological Malignancies. J. Hematol. Oncol. 2018, 11, 91. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune Checkpoint Signaling and Cancer Immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T Cell Immunotherapy for Human Cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, F.; Liu, L. Prognostic Significance of PD-L1 in Solid Tumor: An Updated Meta-Analysis. Medicine 2017, 96, e6369. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive Correlates of Response to the Anti-PD-L1 Antibody MPDL3280A in Cancer Patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Andrea, A.E.; Chiron, A.; Bessoles, S.; Hacein-Bey-Abina, S. Engineering Next-Generation CAR-T Cells for Better Toxicity Management. Int. J. Mol. Sci. 2020, 21, 8620. [Google Scholar] [CrossRef] [PubMed]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 147–164. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-J.; Kuen, D.-S.; Chung, Y. Future Prospects of Immune Checkpoint Blockade in Cancer: From Response Prediction to Overcoming Resistance. Exp. Mol. Med. 2018, 50, 1–13. [Google Scholar] [CrossRef]
- Larson, R.C.; Maus, M.V. Recent Advances and Discoveries in the Mechanisms and Functions of CAR T Cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, A.; Müller, S.; Favier, B.; Penna, D.; Guiraud, M.; Delmas, C.; Champagne, E.; Valitutti, S. T-Cell Activation Is Accompanied by an Ubiquitination Process Occurring at the Immunological Synapse. Immunol. Lett. 2005, 98, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Bachmaier, K.; Krawczyk, C.; Kozieradzki, I.; Kong, Y.-Y.; Sasaki, T.; Oliveira-dos-Santos, A.; Mariathasan, S.; Bouchard, D.; Wakeham, A.; Itie, A.; et al. Negative Regulation of Lymphocyte Activation and Autoimmunity by the Molecular Adaptor Cbl-b. Nature 2000, 403, 211–216. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, N.; Mueller, D.L. Casitas B-Lineage Lymphoma b Inhibits Antigen Recognition and Slows Cell Cycle Progression at Late Times during CD4 + T Cell Clonal Expansion. J. Immunol. 2008, 181, 5331–5339. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.J.; Kole, H.K.; Brown, K.; Naramura, M.; Fukuhara, S.; Hu, R.-J.; Jang, I.K.; Gutkind, J.S.; Shevach, E.; Gu, H. Cbl-b Regulates the CD28 Dependence of T-Cell Activation. Nature 2000, 403, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.; Jang, I.K.; Hodes, R.; Gu, H. Ablation of Cbl-b Provides Protection against Transplanted and Spontaneous Tumors. J. Clin. Investig. 2007, 117, 1029–1036. [Google Scholar] [CrossRef]
- Loeser, S.; Loser, K.; Bijker, M.S.; Rangachari, M.; van der Burg, S.H.; Wada, T.; Beissert, S.; Melief, C.J.M.; Penninger, J.M. Spontaneous Tumor Rejection by Cbl-b–Deficient CD8+ T Cells. J. Exp. Med. 2007, 204, 879–891. [Google Scholar] [CrossRef]
- Singh, T.P.; Vieyra-Garcia, P.A.; Wagner, K.; Penninger, J.; Wolf, P. Cbl-b Deficiency Provides Protection against UVB-Induced Skin Damage by Modulating Inflammatory Gene Signature. Cell Death Dis. 2018, 9, 835. [Google Scholar] [CrossRef] [PubMed]
- Stromnes, I.M.; Blattman, J.N.; Tan, X.; Jeevanjee, S.; Gu, H.; Greenberg, P.D. Abrogating Cbl-b in Effector CD8+ T Cells Improves the Efficacy of Adoptive Therapy of Leukemia in Mice. J. Clin. Investig. 2010, 120, 3722–3734. [Google Scholar] [CrossRef]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 Ligase Cbl-b and TAM Receptors Regulate Cancer Metastasis via Natural Killer Cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef]
- Schanz, O.; Cornez, I.; Yajnanarayana, S.P.; David, F.S.; Peer, S.; Gruber, T.; Krawitz, P.; Brossart, P.; Heine, A.; Landsberg, J.; et al. Tumor Rejection in Cblb−/− Mice Depends on IL-9 and Th9 Cells. J. Immunother. Cancer 2021, 9, e002889. [Google Scholar] [CrossRef]
- Kumar, J.; Kumar, R.; Singh, A.K.; Tsakem, E.L.; Kathania, M.; Riese, M.J.; Theiss, A.L.; Davila, M.L.; Venuprasad, K. Deletion of Cbl-b Inhibits CD8+ T-Cell Exhaustion and Promotes CAR T-Cell Function. J. Immunother. Cancer 2021, 9, e001688. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.; Wang, Z.-E.; Shen, L.; Schroeder, A.; Eckalbar, W.; Weiss, A. Cbl-b Deficiency Prevents Functional but Not Phenotypic T Cell Anergy. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef] [PubMed]
- Jeon, M.-S.; Atfield, A.; Venuprasad, K.; Krawczyk, C.; Sarao, R.; Elly, C.; Yang, C.; Arya, S.; Bachmaier, K.; Su, L.; et al. Essential Role of the E3 Ubiquitin Ligase Cbl-b in T Cell Anergy Induction. Immunity 2004, 21, 167–177. [Google Scholar] [CrossRef]
- Wohlfert, E.A.; Callahan, M.K.; Clark, R.B. Resistance to CD4+CD25+ Regulatory T Cells and TGF-β in Cbl-B−/− Mice. J. Immunol. 2004, 173, 1059–1065. [Google Scholar] [CrossRef]
- Fujiwara, M.; Anstadt, E.J.; Clark, R.B. Cbl-b Deficiency Renders T Cells Resistant to PD-L1/PD-1 Mediated Suppression. J. Immunol. 2016, 196, 55.22. [Google Scholar]
- Li, D.; Gál, I.; Vermes, C.; Alegre, M.-L.; Chong, A.S.F.; Chen, L.; Shao, Q.; Adarichev, V.; Xu, X.; Koreny, T.; et al. Cutting Edge: Cbl-b: One of the Key Molecules Tuning CD28- and CTLA-4-Mediated T Cell Costimulation. J. Immunol. 2004, 173, 7135–7139. [Google Scholar] [CrossRef]
- Gronski, M.A.; Boulter, J.M.; Moskophidis, D.; Nguyen, L.T.; Holmberg, K.; Elford, A.R.; Deenick, E.K.; Kim, H.O.; Penninger, J.M.; Odermatt, B.; et al. TCR Affinity and Negative Regulation Limit Autoimmunity. Nat. Med. 2004, 10, 1234–1239. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Thien, C.B.F.; Gruber, T.; Hinterleitner, R.; Baier, G.; Langdon, W.Y.; Penninger, J.M. Essential Role of E3 Ubiquitin Ligase Activity in Cbl-b–Regulated T Cell Functions. J. Immunol. 2011, 186, 2138–2147. [Google Scholar] [CrossRef]
- Hinterleitner, R.; Gruber, T.; Pfeifhofer-Obermair, C.; Lutz-Nicoladoni, C.; Tzankov, A.; Schuster, M.; Penninger, J.M.; Loibner, H.; Lametschwandtner, G.; Wolf, D.; et al. Adoptive Transfer of SiRNA Cblb-Silenced CD8+ T Lymphocytes Augments Tumor Vaccine Efficacy in a B16 Melanoma Model. PLoS ONE 2012, 7, e44295. [Google Scholar] [CrossRef] [PubMed]
- Thell, K.; Urban, M.; Harrauer, J.; Haslinger, I.; Kuttke, M.; Brunner, J.S.; Vogel, A.; Schabbauer, G.; Penninger, J.; Gaweco, A. 1231P—Master Checkpoint Cbl-b Inhibition: Anti-Tumour Efficacy in a Murine Colorectal Cancer Model Following SiRNA-Based Cell Therapy. Ann. Oncol. 2019, 30, v503–v504. [Google Scholar] [CrossRef]
- Nurieva, R.I.; Zheng, S.; Jin, W.; Chung, Y.; Zhang, Y.; Martinez, G.J.; Reynolds, J.M.; Wang, S.-L.; Lin, X.; Sun, S.-C.; et al. The E3 Ubiquitin Ligase GRAIL Regulates T Cell Tolerance and Regulatory T Cell Function by Mediating T Cell Receptor-CD3 Degradation. Immunity 2010, 32, 670–680. [Google Scholar] [CrossRef]
- Anandasabapathy, N.; Ford, G.S.; Bloom, D.; Holness, C.; Paragas, V.; Seroogy, C.; Skrenta, H.; Hollenhorst, M.; Fathman, C.G.; Soares, L. GRAIL: An E3 Ubiquitin Ligase That Inhibits Cytokine Gene Transcription Is Expressed in Anergic CD4+ T Cells. Immunity 2003, 18, 535–547. [Google Scholar] [CrossRef]
- Kriegel, M.A.; Rathinam, C.; Flavell, R.A. E3 Ubiquitin Ligase GRAIL Controls Primary T Cell Activation and Oral Tolerance. Proc. Natl. Acad. Sci. USA 2009, 106, 16770–16775. [Google Scholar] [CrossRef]
- Su, L.; Lineberry, N.; Huh, Y.; Soares, L.; Fathman, C.G. A Novel E3 Ubiquitin Ligase Substrate Screen Identifies Rho Guanine Dissociation Inhibitor as a Substrate of Gene Related to Anergy in Lymphocytes. J. Immunol. 2006, 177, 7559–7566. [Google Scholar] [CrossRef]
- Hu, H.; Wang, H.; Xiao, Y.; Jin, J.; Chang, J.-H.; Zou, Q.; Xie, X.; Cheng, X.; Sun, S.-C. Otud7b Facilitates T Cell Activation and Inflammatory Responses by Regulating Zap70 Ubiquitination. J. Exp. Med. 2016, 213, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Jahan, A.S.; Lestra, M.; Swee, L.K.; Fan, Y.; Lamers, M.M.; Tafesse, F.G.; Theile, C.S.; Spooner, E.; Bruzzone, R.; Ploegh, H.L.; et al. Usp12 Stabilizes the T-Cell Receptor Complex at the Cell Surface during Signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E705–E714. [Google Scholar] [CrossRef]
- Chang, M.; Jin, W.; Chang, J.-H.; Xiao, Y.; Brittain, G.C.; Yu, J.; Zhou, X.; Wang, Y.-H.; Cheng, X.; Li, P.; et al. The Ubiquitin Ligase Peli1 Negatively Regulates T Cell Activation and Prevents Autoimmunity. Nat. Immunol. 2011, 12, 1002–1009. [Google Scholar] [CrossRef]
- Scharschmidt, E.; Wegener, E.; Heissmeyer, V.; Rao, A.; Krappmann, D. Degradation of Bcl10 Induced by T-Cell Activation Negatively Regulates NF-ΚB Signaling. Mol. Cell. Biol. 2004, 24, 3860–3873. [Google Scholar] [CrossRef]
- Zou, Q.; Jin, J.; Hu, H.; Li, H.S.; Romano, S.; Xiao, Y.; Nakaya, M.; Zhou, X.; Cheng, X.; Yang, P.; et al. USP15 Stabilizes MDM2 to Mediate Cancer-Cell Survival and Inhibit Antitumor T Cell Responses. Nat. Immunol. 2014, 15, 562–570. [Google Scholar] [CrossRef]
- Harhaj, E.W.; Dixit, V.M. Deubiquitinases in the Regulation of NF-ΚB Signaling. Cell Res. 2011, 21, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Giordano, M.; Roncagalli, R.; Bourdely, P.; Chasson, L.; Buferne, M.; Yamasaki, S.; Beyaert, R.; van Loo, G.; Auphan-Anezin, N.; Schmitt-Verhulst, A.-M.; et al. The Tumor Necrosis Factor Alpha-Induced Protein 3 (TNFAIP3, A20) Imposes a Brake on Antitumor Activity of CD8 T Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11115–11120. [Google Scholar] [CrossRef]
- Düwel, M.; Welteke, V.; Oeckinghaus, A.; Baens, M.; Kloo, B.; Ferch, U.; Darnay, B.G.; Ruland, J.; Marynen, P.; Krappmann, D. A20 Negatively Regulates T Cell Receptor Signaling to NF-ΚB by Cleaving Malt1 Ubiquitin Chains. J. Immunol. 2009, 182, 7718–7728. [Google Scholar] [CrossRef] [PubMed]
- Reiley, W.W.; Jin, W.; Lee, A.J.; Wright, A.; Wu, X.; Tewalt, E.F.; Leonard, T.O.; Norbury, C.C.; Fitzpatrick, L.; Zhang, M.; et al. Deubiquitinating Enzyme CYLD Negatively Regulates the Ubiquitin-Dependent Kinase Tak1 and Prevents Abnormal T Cell Responses. J. Exp. Med. 2007, 204, 1475–1485. [Google Scholar] [CrossRef]
- Liu, X.; Li, H.; Zhong, B.; Blonska, M.; Gorjestani, S.; Yan, M.; Tian, Q.; Zhang, D.-E.; Lin, X.; Dong, C. USP18 Inhibits NF-ΚB and NFAT Activation during Th17 Differentiation by Deubiquitinating the TAK1-TAB1 Complex. J. Exp. Med. 2013, 210, 1575–1590. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Jin, H.; Liu, Y.-C. Regulation of T Cell Function by the Ubiquitin-Specific Protease USP9X via Modulating the Carma1-Bcl10-Malt1 Complex. Proc. Natl. Acad. Sci. USA 2013, 110, 9433–9438. [Google Scholar] [CrossRef]
- Fu, Y.; Wang, P.; Zhao, J.; Tan, Y.; Sheng, J.; He, S.; Du, X.; Huang, Y.; Yang, Y.; Li, J.; et al. USP12 Promotes CD4+ T Cell Responses through Deubiquitinating and Stabilizing BCL10. Cell Death Differ. 2021, 1–14. [Google Scholar] [CrossRef]
- Hou, D.; Cenciarelli, C.; Jensen, J.P.; Nguygen, H.B.; Weissman, A.M. Activation-Dependent Ubiquitination of a T Cell Antigen Receptor Subunit on Multiple Intracellular Lysines. J. Biol. Chem. 1994, 269, 14244–14247. [Google Scholar] [CrossRef]
- Cenciarelli, C.; Hou, D.; Hsu, K.C.; Rellahan, B.L.; Wiest, D.L.; Smith, H.T.; Fried, V.A.; Weissman, A.M. Activation-Induced Ubiquitination of the T Cell Antigen Receptor. Science 1992, 257, 795–797. [Google Scholar] [CrossRef] [PubMed]
- Haglund, K.; Dikic, I. The Role of Ubiquitylation in Receptor Endocytosis and Endosomal Sorting. J. Cell Sci. 2012, 125, 265–275. [Google Scholar] [CrossRef]
- Naramura, M.; Jang, I.-K.; Kole, H.; Huang, F.; Haines, D.; Gu, H. C-Cbl and Cbl-b Regulate T Cell Responsiveness by Promoting Ligand-Induced TCR down-Modulation. Nat. Immunol. 2002, 3, 1192–1199. [Google Scholar] [CrossRef]
- Krawczyk, C.; Bachmaier, K.; Sasaki, T.; Jones, R.G.; Snapper, S.B.; Bouchard, D.; Kozieradzki, I.; Ohashi, P.S.; Alt, F.W.; Penninger, J.M. Cbl-b Is a Negative Regulator of Receptor Clustering and Raft Aggregation in T Cells. Immunity 2000, 13, 463–473. [Google Scholar] [CrossRef]
- Li, W.; Qiu, S.; Chen, J.; Jiang, S.; Chen, W.; Jiang, J.; Wang, F.; Si, W.; Shu, Y.; Wei, P.; et al. Chimeric Antigen Receptor Designed to Prevent Ubiquitination and Downregulation Showed Durable Antitumor Efficacy. Immunity 2020, 53, 456–470.e6. [Google Scholar] [CrossRef]
- Gauthier, J.; Yakoub-Agha, I. Chimeric Antigen-Receptor T-Cell Therapy for Hematological Malignancies and Solid Tumors: Clinical Data to Date, Current Limitations and Perspectives. Curr. Res. Transl. Med. 2017, 65, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Alcázar, I.; Cortés, I.; Zaballos, A.; Hernandez, C.; Fruman, D.A.; Barber, D.F.; Carrera, A.C. P85β Phosphoinositide 3-Kinase Regulates CD28 Coreceptor Function. Blood 2009, 113, 3198–3208. [Google Scholar] [CrossRef]
- Fang, D.; Liu, Y.-C. Proteolysis-Independent Regulation of PI3K by Cbl-b–Mediated Ubiquitination in T Cells. Nat. Immunol. 2001, 2, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Qiao, G.; Li, Z.; Molinero, L.; Alegre, M.-L.; Ying, H.; Sun, Z.; Penninger, J.M.; Zhang, J. T-Cell Receptor-Induced NF-KappaB Activation Is Negatively Regulated by E3 Ubiquitin Ligase Cbl-b. Mol. Cell. Biol. 2008, 28, 2470–2480. [Google Scholar] [CrossRef]
- Guo, H.; Qiao, G.; Ying, H.; Li, Z.; Zhao, Y.; Liang, Y.; Yang, L.; Lipkowitz, S.; Penninger, J.M.; Langdon, W.Y.; et al. E3 Ubiquitin Ligase Cbl-b Regulates Pten via Nedd4 in T Cells Independently of Its Ubiquitin Ligase Activity. Cell Rep. 2012, 1, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Wang, H.Y.; Fang, N.; Altman, Y.; Elly, C.; Liu, Y.C. Cbl-b, a RING-Type E3 Ubiquitin Ligase, Targets Phosphatidylinositol 3-Kinase for Ubiquitination in T Cells. J. Biol. Chem. 2001, 276, 4872–4878. [Google Scholar] [CrossRef]
- Xie, J.-J.; Liang, J.-Q.; Diao, L.-H.; Altman, A.; Li, Y. TNFR-Associated Factor 6 Regulates TCR Signaling via Interaction with and Modification of LAT Adapter. J. Immunol. 2013, 190, 4027–4036. [Google Scholar] [CrossRef] [PubMed]
- Appleman, L.J.; Chernova, I.; Li, L.; Boussiotis, V.A. CD28 Costimulation Mediates Transcription of SKP2 and CKS1, the Substrate Recognition Components of SCFSkp2 Ubiquitin Ligase That Leads P27kip1 to Degradation. Cell Cycle 2006, 5, 2123–2129. [Google Scholar] [CrossRef][Green Version]
- Zhang, J.; Bárdos, T.; Li, D.; Gál, I.; Vermes, C.; Xu, J.; Mikecz, K.; Finnegan, A.; Lipkowitz, S.; Glant, T.T. Cutting Edge: Regulation of T Cell Activation Threshold by CD28 Costimulation through Targeting Cbl-b for Ubiquitination. J. Immunol. Baltim. Md 1950 2002, 169, 2236–2240. [Google Scholar] [CrossRef]
- Xiao, Y.; Qiao, G.; Tang, J.; Tang, R.; Guo, H.; Warwar, S.; Langdon, W.Y.; Tao, L.; Zhang, J. Protein Tyrosine Phosphatase SHP-1 Modulates T Cell Responses by Controlling Cbl-b Degradation. J. Immunol. 2015, 195, 4218–4227. [Google Scholar] [CrossRef] [PubMed]
- Gruber, T.; Hermann-Kleiter, N.; Hinterleitner, R.; Fresser, F.; Schneider, R.; Gastl, G.; Penninger, J.M.; Baier, G. PKC-Theta Modulates the Strength of T Cell Responses by Targeting Cbl-b for Ubiquitination and Degradation. Sci. Signal. 2009, 2, ra30. [Google Scholar] [CrossRef]
- Yang, B.; Gay, D.L.; MacLeod, M.K.L.; Cao, X.; Hala, T.; Sweezer, E.M.; Kappler, J.; Marrack, P.; Oliver, P.M. Nedd4 Augments the Adaptive Immune Response by Promoting Ubiquitin-Mediated Degradation of Cbl-b in Activated T Cells. Nat. Immunol. 2008, 9, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; George, C.; Jolly, C.A. CD28 Activation Does Not Down-Regulate Cbl-b Expression in Aged Rat T-Lymphocytes. Mech. Ageing Dev. 2004, 125, 595–602. [Google Scholar] [CrossRef]
- Tran, C.W.; Saibil, S.D.; Le Bihan, T.; Hamilton, S.R.; Lang, K.S.; You, H.; Lin, A.E.; Garza, K.M.; Elford, A.R.; Tai, K.; et al. Glycogen Synthase Kinase-3 Modulates Cbl-b and Constrains T Cell Activation. J. Immunol. 2017, 199, 4056–4065. [Google Scholar] [CrossRef]
- Lin, J.T.; Lineberry, N.B.; Kattah, M.G.; Su, L.L.; Utz, P.J.; Fathman, C.G.; Wu, L. Naive CD4 t Cell Proliferation Is Controlled by Mammalian Target of Rapamycin Regulation of GRAIL Expression. J. Immunol. 2009, 182, 5919–5928. [Google Scholar] [CrossRef]
- Soares, L.; Seroogy, C.; Skrenta, H.; Anandasabapathy, N.; Lovelace, P.; Chung, C.D.; Engleman, E.; Fathman, C.G. Two Isoforms of Otubain 1 Regulate T Cell Anergy via GRAIL. Nat. Immunol. 2004, 5, 45–54. [Google Scholar] [CrossRef]
- Coornaert, B.; Baens, M.; Heyninck, K.; Bekaert, T.; Haegman, M.; Staal, J.; Sun, L.; Chen, Z.J.; Marynen, P.; Beyaert, R. T Cell Antigen Receptor Stimulation Induces MALT1 Paracaspase–Mediated Cleavage of the NF-ΚB Inhibitor A20. Nat. Immunol. 2008, 9, 263–271. [Google Scholar] [CrossRef]
- Zapata, J.M.; Perez-Chacon, G.; Carr-Baena, P.; Martinez-Forero, I.; Azpilikueta, A.; Otano, I.; Melero, I. CD137 (4-1BB) Signalosome: Complexity Is a Matter of TRAFs. Front. Immunol. 2018, 9, 2618. [Google Scholar] [CrossRef] [PubMed]
- Song, H.Y.; Rothe, M.; Goeddel, D.V. The Tumor Necrosis Factor-Inducible Zinc Finger Protein A20 Interacts with TRAF1/TRAF2 and Inhibits NF-KappaB Activation. Proc. Natl. Acad. Sci. USA 1996, 93, 6721–6725. [Google Scholar] [CrossRef]
- Oikawa, D.; Hatanaka, N.; Suzuki, T.; Tokunaga, F. Cellular and Mathematical Analyses of LUBAC Involvement in T Cell Receptor-Mediated NF-ΚB Activation Pathway. Front. Immunol. 2020, 11, 601926. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [PubMed]
- van der Stegen, S.J.C.; Hamieh, M.; Sadelain, M. The Pharmacology of Second-Generation Chimeric Antigen Receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Forero, I.; Azpilikueta, A.; Bolaños-Mateo, E.; Nistal-Villan, E.; Palazon, A.; Teijeira, A.; Perez-Chacon, G.; Morales-Kastresana, A.; Murillo, O.; Jure-Kunkel, M.; et al. T Cell Costimulation with Anti-CD137 Monoclonal Antibodies Is Mediated by K63–Polyubiquitin-Dependent Signals from Endosomes. J. Immunol. 2013, 190, 6694–6706. [Google Scholar] [CrossRef]
- Azpilikueta, A.; Bolaños, E.; Lang, V.; Labiano, S.; Aznar, M.A.; Etxeberria, I.; Teijeira, A.; Rodriguez-Ruiz, M.E.; Perez-Gracia, J.L.; Jure-Kunkel, M.; et al. Deubiquitinases A20 and CYLD Modulate Costimulatory Signaling via CD137 (4–1BB). OncoImmunology 2018, 7, e1368605. [Google Scholar] [CrossRef] [PubMed]
- Mahne, A.E.; Mauze, S.; Joyce-Shaikh, B.; Xia, J.; Bowman, E.P.; Beebe, A.M.; Cua, D.J.; Jain, R. Dual Roles for Regulatory T-Cell Depletion and Costimulatory Signaling in Agonistic GITR Targeting for Tumor Immunotherapy. Cancer Res. 2017, 77, 1108–1118. [Google Scholar] [CrossRef]
- Guo, Y.; Yang, L.; Lei, S.; Tan, W.; Long, J. NEDD4 Negatively Regulates GITR via Ubiquitination in Immune Microenvironment of Melanoma. OncoTargets Ther. 2019, Volume 12, 10629–10637. [Google Scholar] [CrossRef]
- Deenick, E.K.; Ma, C.S. The Regulation and Role of T Follicular Helper Cells in Immunity. Immunology 2011, 134, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Posey, A.D.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T Cell Persistence through ICOS and 4-1BB Costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef] [PubMed]
- Alves Costa Silva, C.; Facchinetti, F.; Routy, B.; Derosa, L. New Pathways in Immune Stimulation: Targeting OX40. ESMO Open 2020, 5, e000573. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-H.; Kang, S.G.; Huang, Z.; Wu, C.-J.; Jin, H.Y.; Maine, C.J.; Liu, Y.; Shepherd, J.; Sabouri-Ghomi, M.; Gonzalez-Martin, A.; et al. A MiR-155–Peli1–c-Rel Pathway Controls the Generation and Function of T Follicular Helper Cells. J. Exp. Med. 2016, 213, 1901–1919. [Google Scholar] [CrossRef]
- Huang, X.; Hao, S.; Liu, J.; Huang, Y.; Liu, M.; Xiao, C.; Wang, Y.; Pei, S.; Yu, T.; Xu, J.; et al. The Ubiquitin Ligase Peli1 Inhibits ICOS and Thereby Tfh-Mediated Immunity. Cell. Mol. Immunol. 2021, 18, 969–978. [Google Scholar] [CrossRef]
- Lineberry, N.B.; Su, L.L.; Lin, J.T.; Coffey, G.P.; Seroogy, C.M.; Fathman, C.G. Cutting Edge: The Transmembrane E3 Ligase GRAIL Ubiquitinates the Costimulatory Molecule CD40 Ligand during the Induction of T Cell Anergy. J. Immunol. 2008, 181, 1622–1626. [Google Scholar] [CrossRef] [PubMed]
- Athanasopoulos, V.; Barker, A.; Yu, D.; Tan, A.H.-M.; Srivastava, M.; Contreras, N.; Wang, J.; Lam, K.-P.; Brown, S.H.J.; Goodnow, C.C.; et al. The ROQUIN Family of Proteins Localizes to Stress Granules via the ROQ Domain and Binds Target MRNAs. FEBS J. 2010, 277, 2109–2127. [Google Scholar] [CrossRef]
- Glasmacher, E.; Hoefig, K.P.; Vogel, K.U.; Rath, N.; Du, L.; Wolf, C.; Kremmer, E.; Wang, X.; Heissmeyer, V. Roquin Binds Inducible Costimulator MRNA and Effectors of MRNA Decay to Induce MicroRNA-Independent Post-Transcriptional Repression. Nat. Immunol. 2010, 11, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Rehage, N.; Davydova, E.; Conrad, C.; Behrens, G.; Maiser, A.; Stehklein, J.E.; Brenner, S.; Klein, J.; Jeridi, A.; Hoffmann, A.; et al. Binding of NUFIP2 to Roquin Promotes Recognition and Regulation of ICOS MRNA. Nat. Commun. 2018, 9, 299. [Google Scholar] [CrossRef]
- Vogel, K.U.; Edelmann, S.L.; Jeltsch, K.M.; Bertossi, A.; Heger, K.; Heinz, G.A.; Zöller, J.; Warth, S.C.; Hoefig, K.P.; Lohs, C.; et al. Roquin Paralogs 1 and 2 Redundantly Repress the Icos and Ox40 Costimulator MRNAs and Control Follicular Helper T Cell Differentiation. Immunity 2013, 38, 655–668. [Google Scholar] [CrossRef]
- Linterman, M.A.; Rigby, R.J.; Wong, R.; Silva, D.; Withers, D.; Anderson, G.; Verma, N.K.; Brink, R.; Hutloff, A.; Goodnow, C.C.; et al. Roquin Differentiates the Specialized Functions of Duplicated T Cell Costimulatory Receptor Genes CD28 and ICOS. Immunity 2009, 30, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Hoff, H.; Kolar, P.; Ambach, A.; Radbruch, A.; Brunner-Weinzierl, M.C. CTLA-4 (CD152) Inhibits T Cell Function by Activating the Ubiquitin Ligase Itch. Mol. Immunol. 2010, 47, 1875–1881. [Google Scholar] [CrossRef]
- Paolino, M.; Penninger, J.M. E3 Ubiquitin Ligases in T-Cell Tolerance. Eur. J. Immunol. 2009, 39, 2337–2344. [Google Scholar] [CrossRef]
- Paolino, M.; Penninger, J.M. Cbl-b in T-Cell Activation. Semin. Immunopathol. 2010, 32, 137–148. [Google Scholar] [CrossRef]
- Sobhani, N.; Tardiel-Cyril, D.R.; Davtyan, A.; Generali, D.; Roudi, R.; Li, Y. CTLA-4 in Regulatory T Cells for Cancer Immunotherapy. Cancers 2021, 13, 1440. [Google Scholar] [CrossRef] [PubMed]
- Venuprasad, K.; Huang, H.; Harada, Y.; Elly, C.; Subramaniam, M.; Spelsberg, T.; Su, J.; Liu, Y.-C. The E3 Ubiquitin Ligase Itch Regulates Expression of Transcription Factor Foxp3 and Airway Inflammation by Enhancing the Function of Transcription Factor TIEG1. Nat. Immunol. 2008, 9, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Wohlfert, E.A.; Gorelik, L.; Mittler, R.; Flavell, R.A.; Clark, R.B. Cutting Edge: Deficiency in the E3 Ubiquitin Ligase Cbl-b Results in a Multifunctional Defect in T Cell TGF-β Sensitivity In Vitro and In Vivo. J. Immunol. 2006, 176, 1316–1320. [Google Scholar] [CrossRef]
- Harada, Y.; Harada, Y.; Elly, C.; Ying, G.; Paik, J.-H.; DePinho, R.A.; Liu, Y.-C. Transcription Factors Foxo3a and Foxo1 Couple the E3 Ligase Cbl-b to the Induction of Foxp3 Expression in Induced Regulatory T Cells. J. Exp. Med. 2010, 207, 1381–1391. [Google Scholar] [CrossRef]
- Hoyne, G.F.; Flening, E.; Yabas, M.; Teh, C.; Altin, J.A.; Randall, K.; Thien, C.B.F.; Langdon, W.Y.; Goodnow, C.C. Visualizing the Role of Cbl-b in Control of Islet-Reactive CD4 T Cells and Susceptibility to Type 1 Diabetes. J. Immunol. 2011, 186, 2024–2032. [Google Scholar] [CrossRef] [PubMed]
- Schartner, J.M.; Singh, A.M.; Dahlberg, P.E.; Nettenstrom, L.; Seroogy, C.M. Recurrent Superantigen Exposure in Vivo Leads to Highly Suppressive CD4+CD25+ and CD4+CD25- T Cells with Anergic and Suppressive Genetic Signatures. Clin. Exp. Immunol. 2009, 155, 348–356. [Google Scholar] [CrossRef]
- MacKenzie, D.A.; Schartner, J.; Lin, J.; Timmel, A.; Jennens-Clough, M.; Fathman, C.G.; Seroogy, C.M. GRAIL Is Up-Regulated in CD4+ CD25+ T Regulatory Cells and Is Sufficient for Conversion of T Cells to a Regulatory Phenotype. J. Biol. Chem. 2007, 282, 9696–9702. [Google Scholar] [CrossRef]
- Karwacz, K.; Bricogne, C.; MacDonald, D.; Arce, F.; Bennett, C.L.; Collins, M.; Escors, D. PD-L1 Co-Stimulation Contributes to Ligand-Induced T Cell Receptor down-Modulation on CD8+ T Cells. EMBO Mol. Med. 2011, 3, 581–592. [Google Scholar] [CrossRef]
- Lyle, C.; Richards, S.; Yasuda, K.; Napoleon, M.A.; Walker, J.; Arinze, N.; Belghasem, M.; Vellard, I.; Yin, W.; Ravid, J.D.; et al. C-Cbl Targets PD-1 in Immune Cells for Proteasomal Degradation and Modulates Colorectal Tumor Growth. Sci. Rep. 2019, 9, 20257. [Google Scholar] [CrossRef]
- Meng, X.; Liu, X.; Guo, X.; Jiang, S.; Chen, T.; Hu, Z.; Liu, H.; Bai, Y.; Xue, M.; Hu, R.; et al. FBXO38 Mediates PD-1 Ubiquitination and Regulates Anti-Tumour Immunity of T Cells. Nature 2018, 564, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Fang, W.; Zhan, J.; Hong, S.; Tang, Y.; Kang, S.; Zhang, Y.; He, X.; Zhou, T.; Qin, T.; et al. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J. Thorac. Oncol. 2015, 10, 910–923. [Google Scholar] [CrossRef]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 Pathway Contributes to Immune Escape in EGFR-Driven Lung Tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Azuma, K.; Ota, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; Kage, M.; et al. Association of PD-L1 Overexpression with Activating EGFR Mutations in Surgically Resected Nonsmall-Cell Lung Cancer. Ann. Oncol. 2014, 25, 1935–1940. [Google Scholar] [CrossRef] [PubMed]
- Okita, R.; Maeda, A.; Shimizu, K.; Nojima, Y.; Saisho, S.; Nakata, M. PD-L1 Overexpression Is Partially Regulated by EGFR/HER2 Signaling and Associated with Poor Prognosis in Patients with Non-Small-Cell Lung Cancer. Cancer Immunol. Immunother. CII 2017, 66, 865–876. [Google Scholar] [CrossRef]
- Zhang, N.; Zeng, Y.; Du, W.; Zhu, J.; Shen, D.; Liu, Z.; Huang, J.-A. The EGFR Pathway Is Involved in the Regulation of PD-L1 Expression via the IL-6/JAK/STAT3 Signaling Pathway in EGFR-Mutated Non-Small Cell Lung Cancer. Int. J. Oncol. 2016, 49, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Voli, F.; Valli, E.; Lerra, L.; Kimpton, K.; Saletta, F.; Giorgi, F.M.; Mercatelli, D.; Rouaen, J.R.C.; Shen, S.; Murray, J.E.; et al. Intratumoral Copper Modulates PD-L1 Expression and Influences Tumor Immune Evasion. Cancer Res. 2020, 80, 4129–4144. [Google Scholar] [CrossRef]
- Li, C.-W.; Lim, S.-O.; Xia, W.; Lee, H.-H.; Chan, L.-C.; Kuo, C.-W.; Khoo, K.-H.; Chang, S.-S.; Cha, J.-H.; Kim, T.; et al. Glycosylation and Stabilization of Programmed Death Ligand-1 Suppresses T-Cell Activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef]
- Li, H.; Li, C.-W.; Li, X.; Ding, Q.; Guo, L.; Liu, S.; Liu, C.; Lai, C.-C.; Hsu, J.-M.; Dong, Q.; et al. MET Inhibitors Promote Liver Tumor Evasion of the Immune Response by Stabilizing PDL1. Gastroenterology 2019, 156, 1849–1861.e13. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, C.; Liu, X.; He, Z.; Shan, B.; Zeng, Q.; Zhao, Q.; Zhu, H.; Liao, H.; Cen, X.; et al. ARIH1 Signaling Promotes Anti-Tumor Immunity by Targeting PD-L1 for Proteasomal Degradation. Nat. Commun. 2021, 12, 2346. [Google Scholar] [CrossRef]
- Qian, G.; Guo, J.; Vallega, K.A.; Hu, C.; Chen, Z.; Deng, Y.; Wang, Q.; Fan, S.; Ramalingam, S.S.; Owonikoko, T.K.; et al. Membrane-Associated RING-CH 8 Functions as a Novel PD-L1 E3 Ligase to Mediate PD-L1 Degradation Induced by EGFR Inhibitors. Mol. Cancer Res. 2021, 19, 10. [Google Scholar] [CrossRef]
- Wang, S.; Xu, L.; Che, X.; Li, C.; Xu, L.; Hou, K.; Fan, Y.; Wen, T.; Qu, X.; Liu, Y. E3 Ubiquitin Ligases Cbl-b and c-Cbl Downregulate PD-L1 in EGFR Wild-Type Non-Small Cell Lung Cancer. FEBS Lett. 2018, 592, 621–630. [Google Scholar] [CrossRef]
- Pennock, S.; Wang, Z. A Tale of Two Cbls: Interplay of c-Cbl and Cbl-b in Epidermal Growth Factor Receptor Downregulation. Mol. Cell. Biol. 2008, 28, 3020–3037. [Google Scholar] [CrossRef] [PubMed]
- Umebayashi, K.; Stenmark, H.; Yoshimori, T. Ubc4/5 and c-Cbl Continue to Ubiquitinate EGF Receptor after Internalization to Facilitate Polyubiquitination and Degradation. Mol. Biol. Cell 2008, 19, 3454–3462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D–CDK4 Kinase Destabilizes PD-L1 via Cullin 3–SPOP to Control Cancer Immune Surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef]
- Gao, L.; Xia, L.; Ji, W.; Zhang, Y.; Xia, W.; Lu, S. Knockdown of CDK5 Down-Regulates PD-L1 via the Ubiquitination-Proteasome Pathway and Improves Antitumor Immunity in Lung Adenocarcinoma. Transl. Oncol. 2021, 14, 101148. [Google Scholar] [CrossRef]
- Mezzadra, R.; Sun, C.; Jae, L.T.; Gomez-Eerland, R.; de Vries, E.; Wu, W.; Logtenberg, M.E.W.; Slagter, M.; Rozeman, E.A.; Hofland, I.; et al. Identification of CMTM6 and CMTM4 as PD-L1 Protein Regulators. Nature 2017, 549, 106–110. [Google Scholar] [CrossRef]
- Burr, M.L.; Sparbier, C.E.; Chan, Y.-C.; Williamson, J.C.; Woods, K.; Beavis, P.A.; Lam, E.Y.N.; Henderson, M.A.; Bell, C.C.; Stolzenburg, S.; et al. CMTM6 Maintains the Expression of PD-L1 and Regulates Anti-Tumour Immunity. Nature 2017, 549, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Lan, J.; Li, C.; Shi, H.; Brosseau, J.-P.; Wang, H.; Lu, H.; Fang, C.; Zhang, Y.; Liang, L.; et al. Inhibiting PD-L1 Palmitoylation Enhances T-Cell Immune Responses against Tumours. Nat. Biomed. Eng. 2019, 3, 306–317. [Google Scholar] [CrossRef]
- Lim, S.-O.; Li, C.-W.; Xia, W.; Cha, J.-H.; Chan, L.-C.; Wu, Y.; Chang, S.-S.; Lin, W.-C.; Hsu, J.-M.; Hsu, Y.-H.; et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, X.; Zhang, N.; Yin, M.; Dong, J.; Zeng, Q.; Mao, G.; Song, D.; Liu, L.; Deng, H. Berberine Diminishes Cancer Cell PD-L1 Expression and Facilitates Antitumor Immunity via Inhibiting the Deubiquitination Activity of CSN5. Acta Pharm. Sin. B 2020, 10, 2299–2312. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Q.; Mu, N.; Sun, X.; Wang, Y.; Fan, S.; Su, L.; Liu, X. The Deubiquitinase USP22 Regulates PD-L1 Degradation in Human Cancer Cells. Cell Commun. Signal. 2020, 18, 112. [Google Scholar] [CrossRef]
- Zhu, D.; Xu, R.; Huang, X.; Tang, Z.; Tian, Y.; Zhang, J.; Zheng, X. Deubiquitinating Enzyme OTUB1 Promotes Cancer Cell Immunosuppression via Preventing ER-Associated Degradation of Immune Checkpoint Protein PD-L1. Cell Death Differ. 2021, 28, 1773–1789. [Google Scholar] [CrossRef]
- Jingjing, W.; Wenzheng, G.; Donghua, W.; Guangyu, H.; Aiping, Z.; Wenjuan, W. Deubiquitination and Stabilization of Programmed Cell Death Ligand 1 by Ubiquitin-Specific Peptidase 9, X-Linked in Oral Squamous Cell Carcinoma. Cancer Med. 2018, 7, 4004–4011. [Google Scholar] [CrossRef]
- Wang, Z.; Kang, W.; Li, O.; Qi, F.; Wang, J.; You, Y.; He, P.; Suo, Z.; Zheng, Y.; Liu, H.-M. Abrogation of USP7 Is an Alternative Strategy to Downregulate PD-L1 and Sensitize Gastric Cancer Cells to T Cells Killing. Acta Pharm. Sin. B 2021, 11, 694–707. [Google Scholar] [CrossRef]
- Metzger, M.B.; Pruneda, J.N.; Klevit, R.E.; Weissman, A.M. RING-Type E3 Ligases: Master Manipulators of E2 Ubiquitin-Conjugating Enzymes and Ubiquitination. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2014, 1843, 47–60. [Google Scholar] [CrossRef]
- Li, X.; Song, Y. Proteolysis-Targeting Chimera (PROTAC) for Targeted Protein Degradation and Cancer Therapy. J. Hematol. Oncol. 2020, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Henning, N.J.; Boike, L.; Spradlin, J.N.; Ward, C.C.; Belcher, B.; Brittain, S.M.; Hesse, M.; Dovala, D.; McGregor, L.M.; McKenna, J.M.; et al. Deubiquitinase-Targeting Chimeras for Targeted Protein Stabilization. bioRxiv 2021. [Google Scholar] [CrossRef]
- Veggiani, G.; Gerpe, M.C.R.; Sidhu, S.S.; Zhang, W. Emerging Drug Development Technologies Targeting Ubiquitination for Cancer Therapeutics. Pharmacol. Ther. 2019, 199, 139–154. [Google Scholar] [CrossRef]
- Neklesa, T.K.; Crews, C.M. Greasy Tags for Protein Removal. Nature 2012, 487, 308–309. [Google Scholar] [CrossRef]
- Wu, H.Q.; Baker, D.; Ovaa, H. Small Molecules That Target the Ubiquitin System. Biochem. Soc. Trans. 2020, 48, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Wang, X. From Discovery to Bedside: Targeting the Ubiquitin System. Cell Chem. Biol. 2019, 26, 156–177. [Google Scholar] [CrossRef]
- Chen, S.; Liu, Y.; Zhou, H. Advances in the Development Ubiquitin-Specific Peptidase (USP) Inhibitors. Int. J. Mol. Sci. 2021, 22, 4546. [Google Scholar] [CrossRef]
- LaPlante, G.; Zhang, W. Targeting the Ubiquitin-Proteasome System for Cancer Therapeutics by Small-Molecule Inhibitors. Cancers 2021, 13, 3079. [Google Scholar] [CrossRef] [PubMed]
- Schauer, N.J.; Magin, R.S.; Liu, X.; Doherty, L.M.; Buhrlage, S.J. Advances in Discovering Deubiquitinating Enzyme (DUB) Inhibitors. J. Med. Chem. 2020, 63, 2731–2750. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Muniyappan, S.; Lee, S.-B.; Lee, B.-H. Small-Molecule Inhibitors Targeting Proteasome-Associated Deubiquitinases. Int. J. Mol. Sci. 2021, 22, 6213. [Google Scholar] [CrossRef]
- Mofers, A.; Pellegrini, P.; Linder, S.; D’Arcy, P. Proteasome-Associated Deubiquitinases and Cancer. Cancer Metastasis Rev. 2017, 36, 635–653. [Google Scholar] [CrossRef] [PubMed]
- Gruber, T.; Hinterleitner, R.; Pfeifhofer-Obermair, C.; Wolf, D.; Baier, G. Engineering Effective T-Cell Based Antitumor Immunity. OncoImmunology 2013, 2, e22893. [Google Scholar] [CrossRef][Green Version]
- Zhou, S.-K.; Chen, W.-H.; Shi, Z.-D.; Wang, S.-P.; Li, L.; Wen, X.-F.; Wang, Y.-M. Silencing the Expression of Cbl-b Enhances the Immune Activation of T Lymphocytes against RM-1 Prostate Cancer Cells in Vitro. J. Chin. Med. Assoc. 2014, 77, 630–636. [Google Scholar] [CrossRef]
- Sachet, M.; Lametschwandtner, G.; Hayden, H.; Hassler, M.; Loibner, H.; Triozzi, P.; Friedl, J. Treatment of a Cancer Patient by an Adoptive Cell Therapy Protocol Combining DC Vaccination with Cbl-b Ex Vivo Silencing. J. Immunother. Cancer 2015, 3, P172. [Google Scholar] [CrossRef]
- Triozzi, P.; Kooshki, M.; Alistar, A.; Bitting, R.; Neal, A.; Lametschwandtner, G.; Loibner, H. Phase I Clinical Trial of Adoptive Cellular Immunotherapy with APN401 in Patients with Solid Tumors. J. Immunother. Cancer 2015, 3, P175. [Google Scholar] [CrossRef]
- Wirnsberger, G.; Zwolanek, F.; Asaoka, T.; Kozieradzki, I.; Tortola, L.; Wimmer, R.A.; Kavirayani, A.; Fresser, F.; Baier, G.; Langdon, W.Y.; et al. Inhibition of CBL-B Protects from Lethal C. Albicans Sepsis. Nat. Med. 2016, 22, 915–923. [Google Scholar] [CrossRef]
- Gabrielsen, M.; Buetow, L.; Nakasone, M.A.; Ahmed, S.F.; Sibbet, G.J.; Smith, B.O.; Zhang, W.; Sidhu, S.S.; Huang, D.T. A General Strategy for Discovery of Inhibitors and Activators of RING and U-Box E3 Ligases with Ubiquitin Variants. Mol. Cell 2017, 68, 456–470.e10. [Google Scholar] [CrossRef]
- Cossu, F.; Milani, M.; Mastrangelo, E.; Lecis, D. Targeting the BIR Domains of Inhibitor of Apoptosis (IAP) Proteins in Cancer Treatment. Comput. Struct. Biotechnol. J. 2019, 17, 142–150. [Google Scholar] [CrossRef]
- Fang, D.D.; Tang, Q.; Kong, Y.; Wang, Q.; Gu, J.; Fang, X.; Zou, P.; Rong, T.; Wang, J.; Yang, D.; et al. MDM2 Inhibitor APG-115 Synergizes with PD-1 Blockade through Enhancing Antitumor Immunity in the Tumor Microenvironment. J. Immunother. Cancer 2019, 7, 327. [Google Scholar] [CrossRef] [PubMed]
- Sahin, I.; Zhang, S.; Navaraj, A.; Zhou, L.; Dizon, D.; Safran, H.; El-Deiry, W.S. AMG-232 Sensitizes High MDM2-Expressing Tumor Cells to T-Cell-Mediated Killing. Cell Death Discov. 2020, 6, 57. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Zhu, X.; Xu, P.; Li, Y. Pharmacological Inhibition of USP7 Promotes Antitumor Immunity and Contributes to Colon Cancer Therapy. OncoTargets Ther. 2019, 12, 609–617. [Google Scholar] [CrossRef]
- Mullard, A. Targeted Protein Degraders Crowd into the Clinic. Nat. Rev. Drug Discov. 2021, 20, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Cotton, A.D.; Nguyen, D.P.; Gramespacher, J.A.; Seiple, I.B.; Wells, J.A. Development of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1. J. Am. Chem. Soc. 2021, 143, 593–598. [Google Scholar] [CrossRef]
- Jan, M.; Scarfò, I.; Larson, R.C.; Walker, A.; Schmidts, A.; Guirguis, A.A.; Gasser, J.A.; Słabicki, M.; Bouffard, A.A.; Castano, A.P.; et al. Reversible ON- and OFF-Switch Chimeric Antigen Receptors Controlled by Lenalidomide. Sci. Transl. Med. 2021, 13, eabb6295. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Kang, C.H.; Choi, S.U.; Kim, Y.; Hwang, J.Y.; Jeong, H.G.; Park, C.H. A Chemical Switch System to Modulate Chimeric Antigen Receptor T Cell Activity through Proteolysis-Targeting Chimaera Technology. ACS Synth. Biol. 2020, 9, 987–992. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gavali, S.; Liu, J.; Li, X.; Paolino, M. Ubiquitination in T-Cell Activation and Checkpoint Inhibition: New Avenues for Targeted Cancer Immunotherapy. Int. J. Mol. Sci. 2021, 22, 10800. https://doi.org/10.3390/ijms221910800
Gavali S, Liu J, Li X, Paolino M. Ubiquitination in T-Cell Activation and Checkpoint Inhibition: New Avenues for Targeted Cancer Immunotherapy. International Journal of Molecular Sciences. 2021; 22(19):10800. https://doi.org/10.3390/ijms221910800
Chicago/Turabian StyleGavali, Shubhangi, Jianing Liu, Xinyi Li, and Magdalena Paolino. 2021. "Ubiquitination in T-Cell Activation and Checkpoint Inhibition: New Avenues for Targeted Cancer Immunotherapy" International Journal of Molecular Sciences 22, no. 19: 10800. https://doi.org/10.3390/ijms221910800
APA StyleGavali, S., Liu, J., Li, X., & Paolino, M. (2021). Ubiquitination in T-Cell Activation and Checkpoint Inhibition: New Avenues for Targeted Cancer Immunotherapy. International Journal of Molecular Sciences, 22(19), 10800. https://doi.org/10.3390/ijms221910800