
Tomatidine and Patchouli Alcohol as Inhibitors of SARS-CoV-2 Enzymes (3CLpro, PLpro and NSP15) by Molecular Docking and Molecular Dynamics Simulations

 ,
, 
 ,
,  ,
,  , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Molecular Docking Analysis
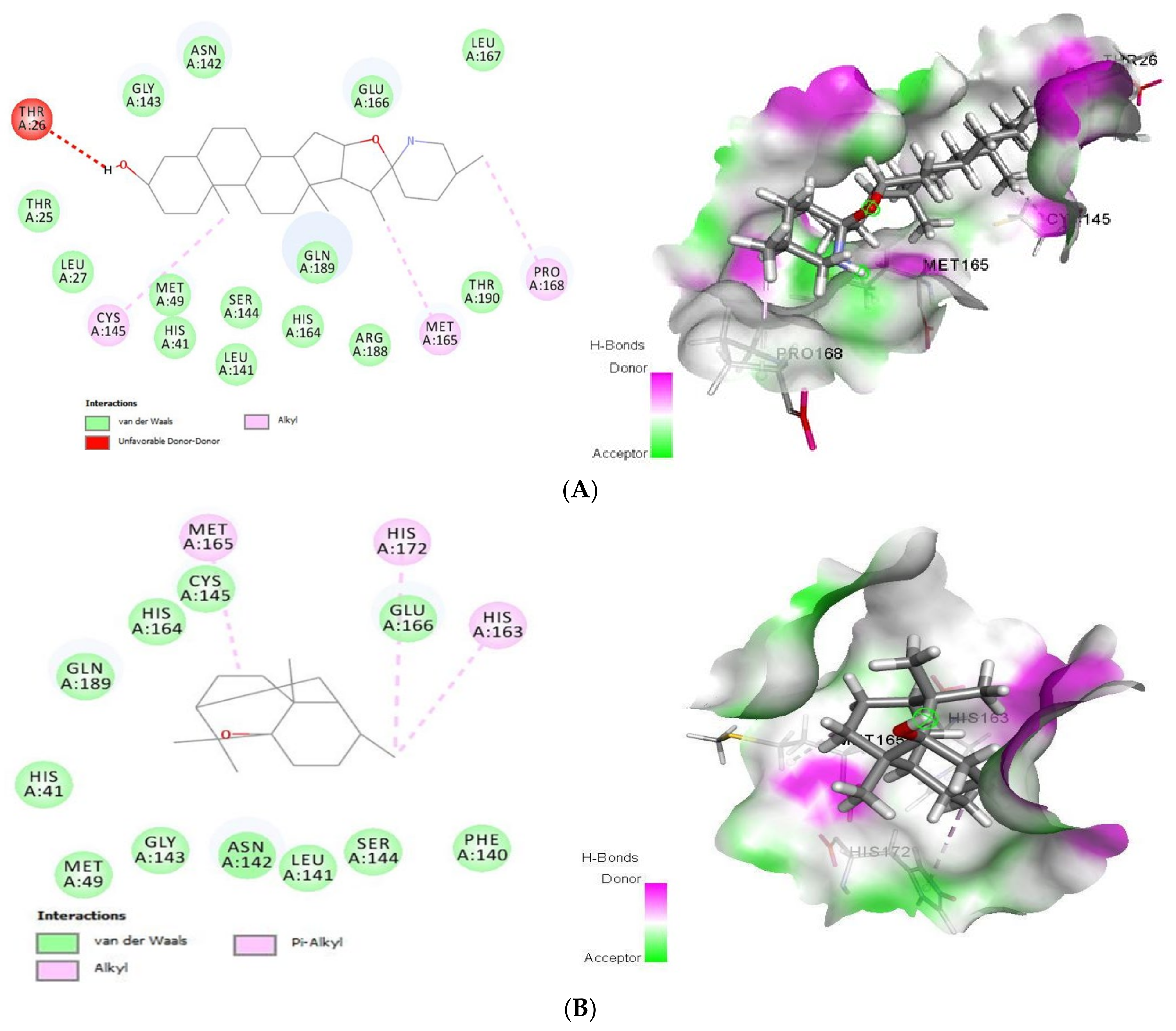
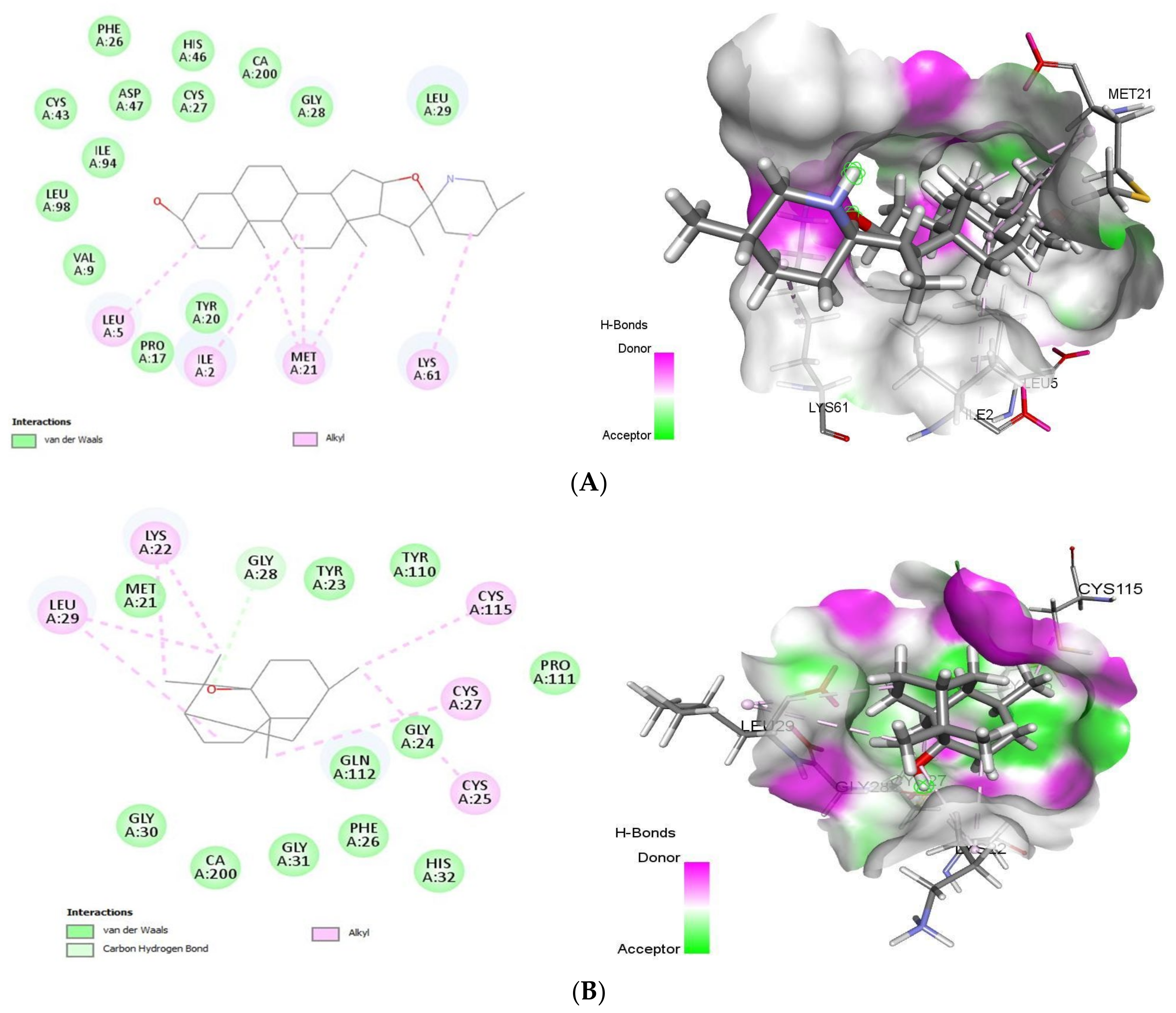
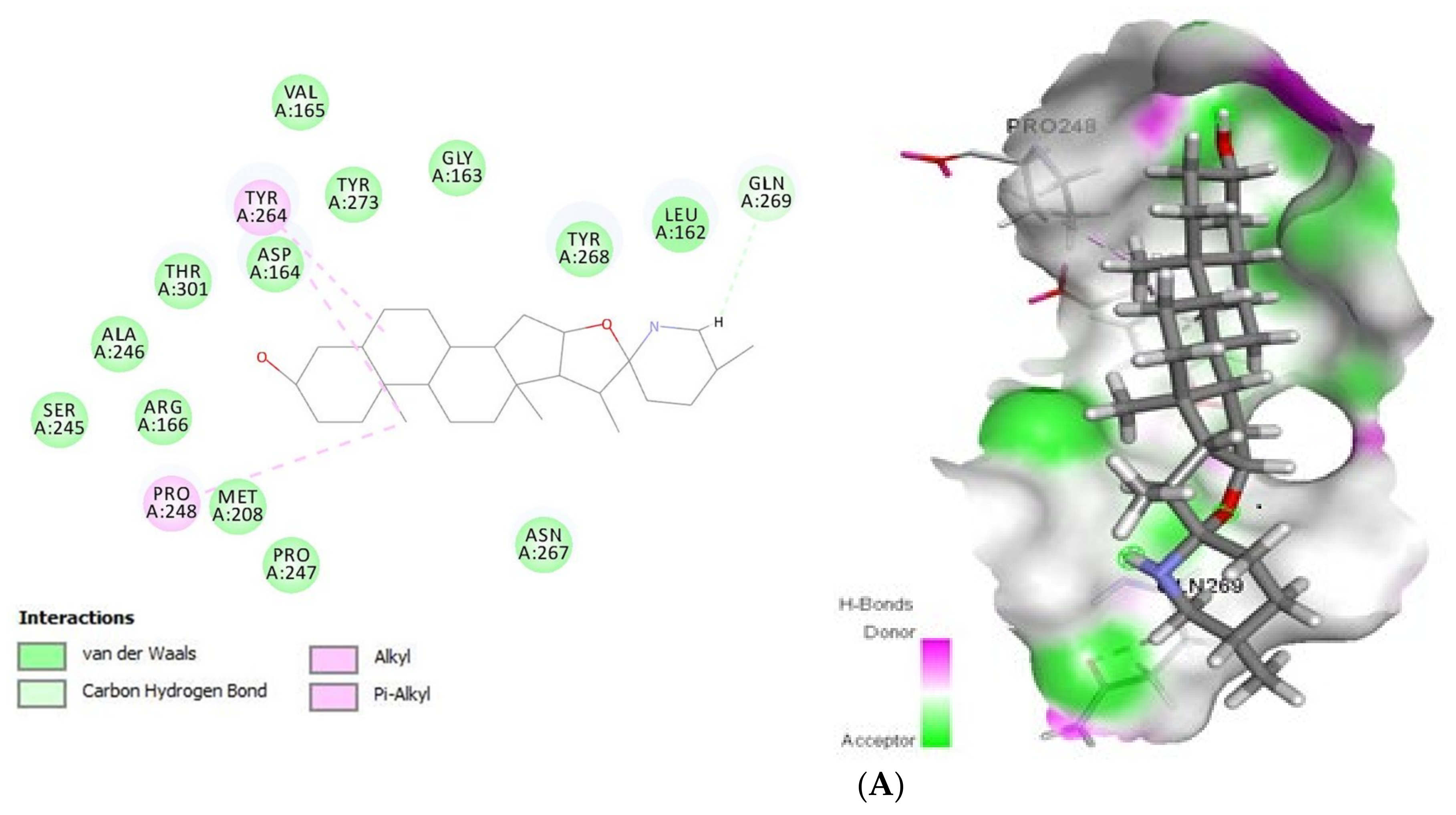
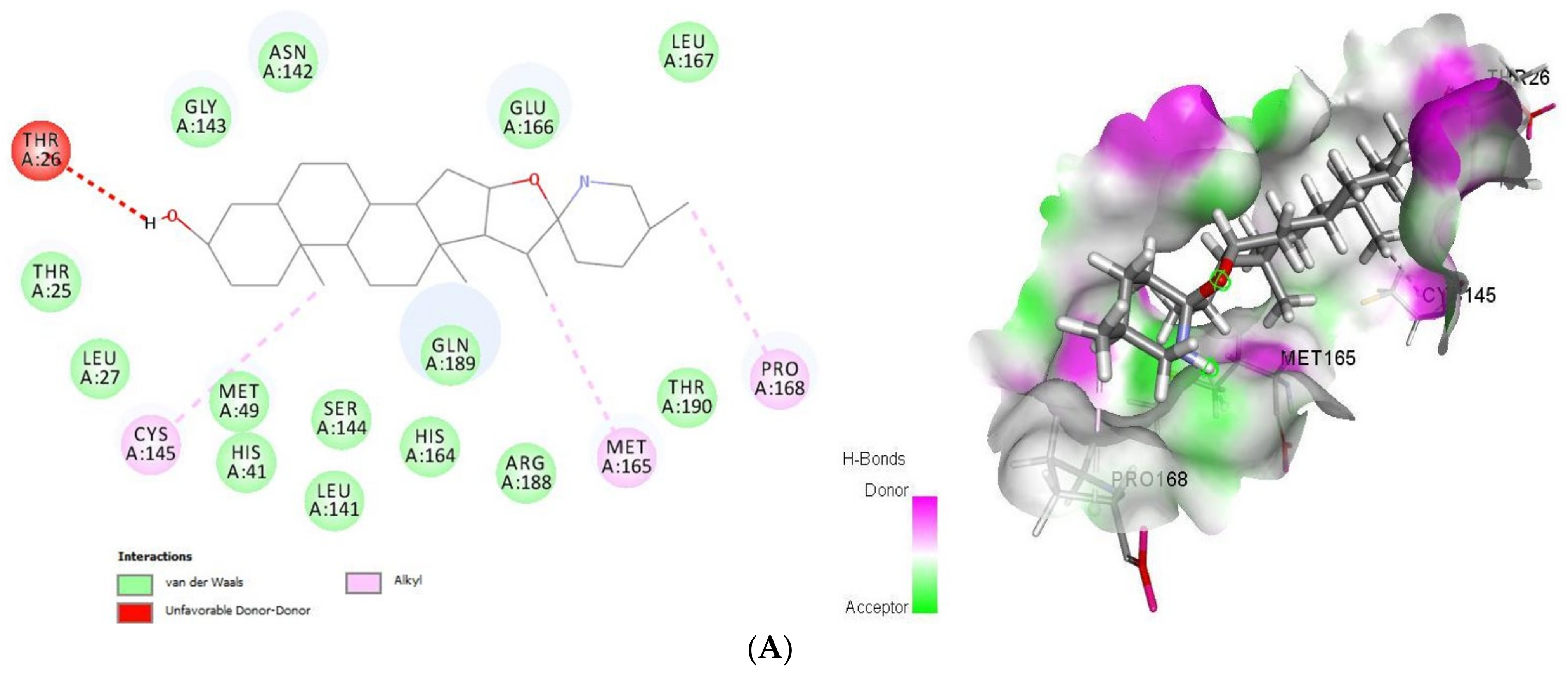
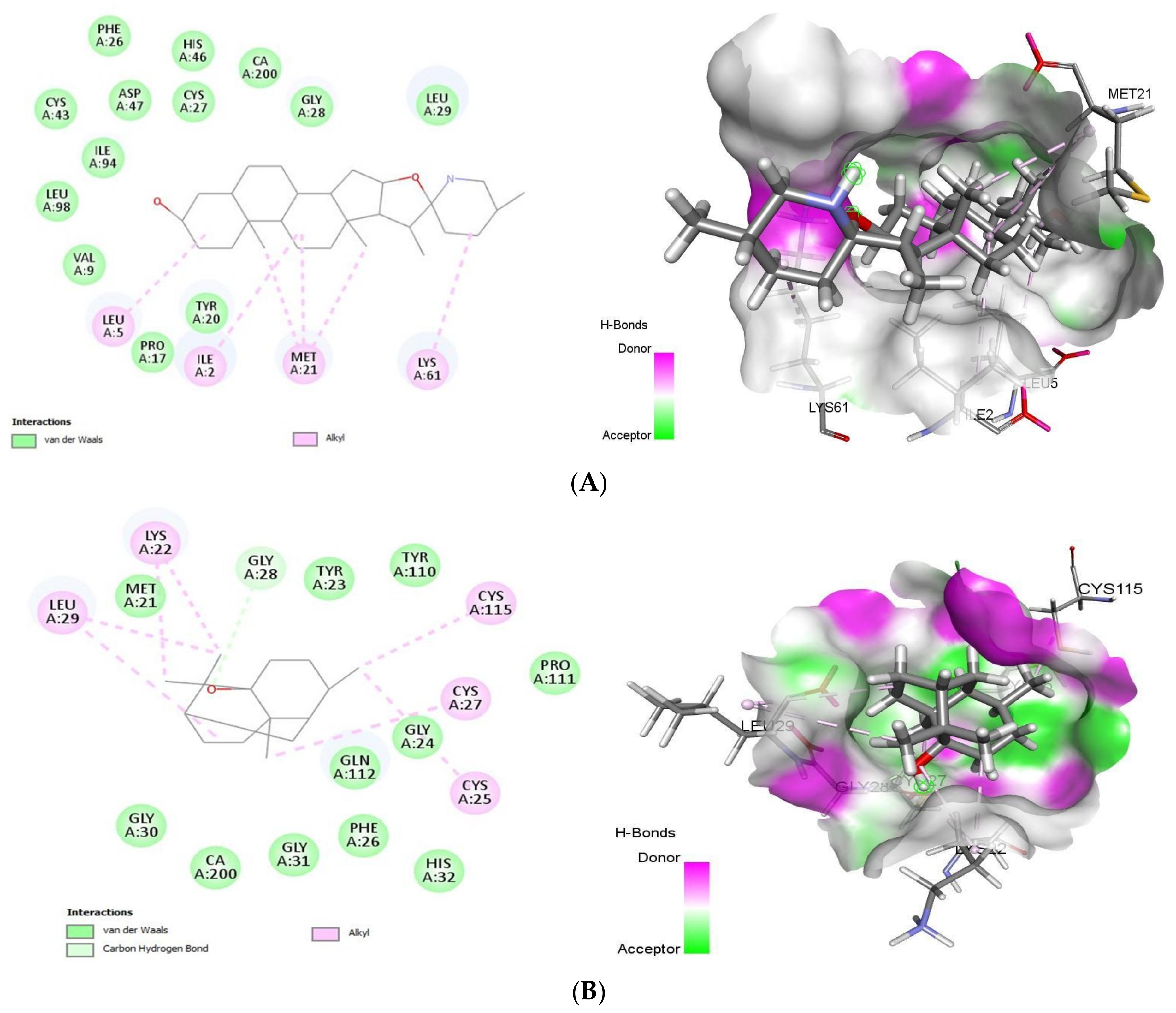
2.1.1. Docking Analysis of Tomatidine and Patchouli Alcohol with Target 3CLpro
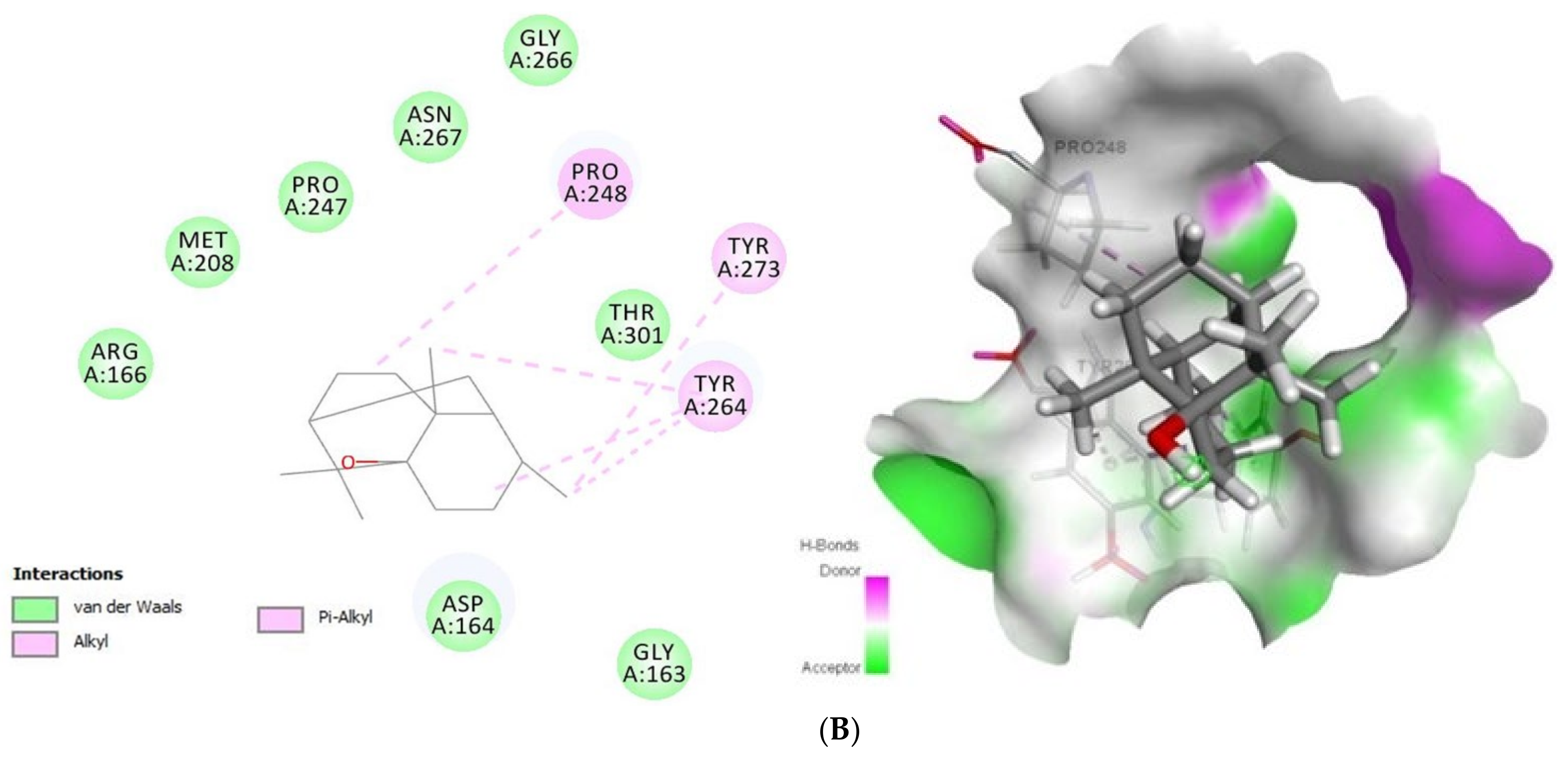
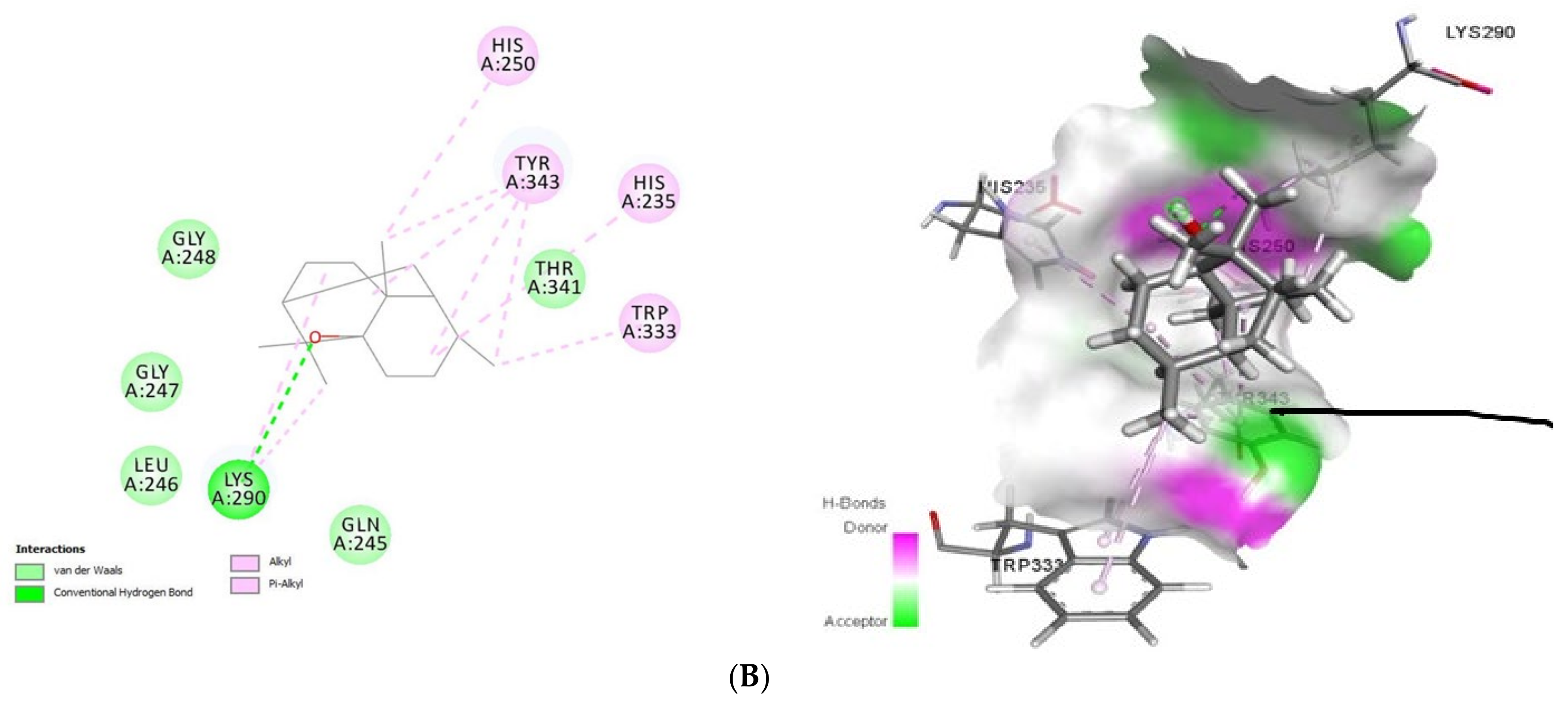
2.1.2. Docking Analysis of Tomatidine and Patchouli Alcohol with Target Plpro
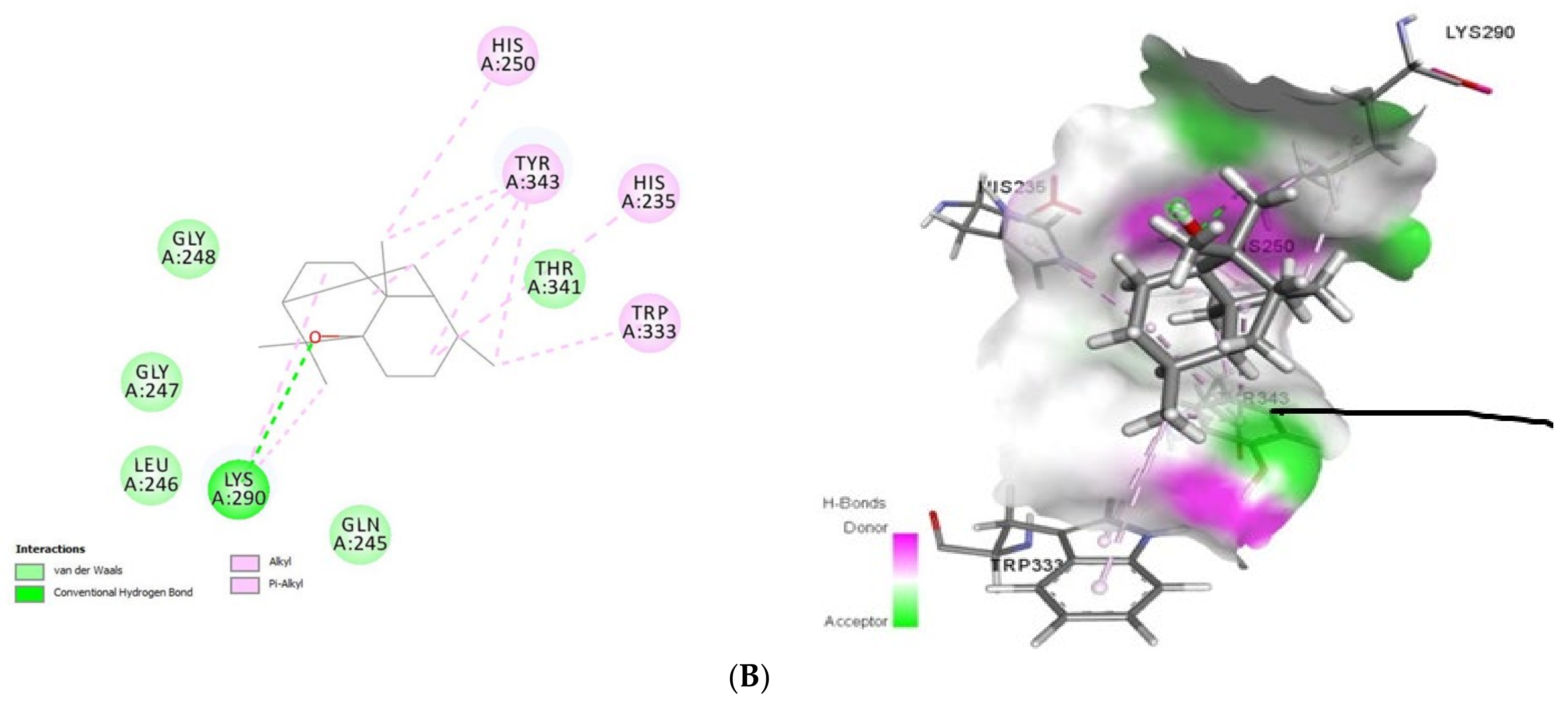
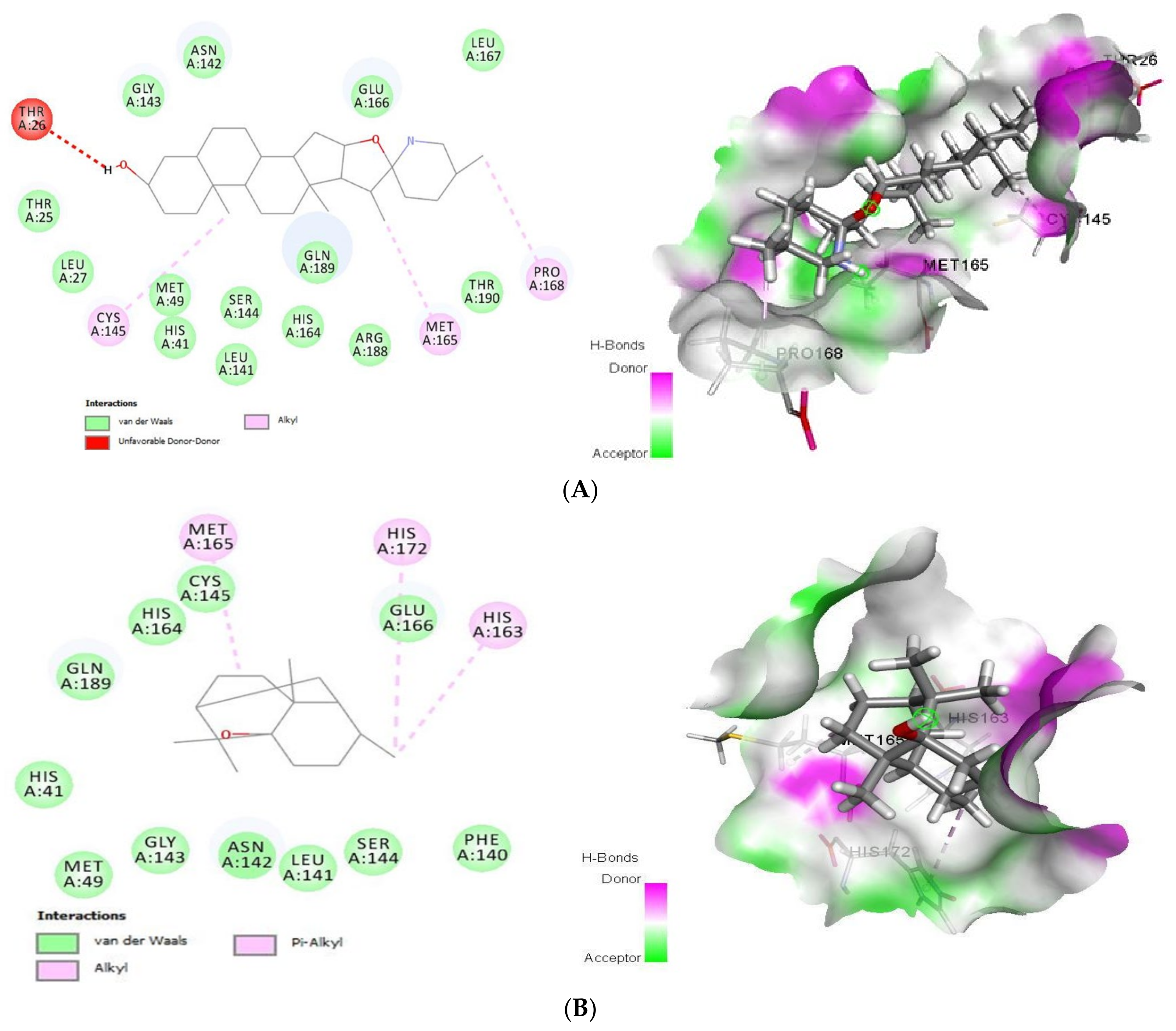
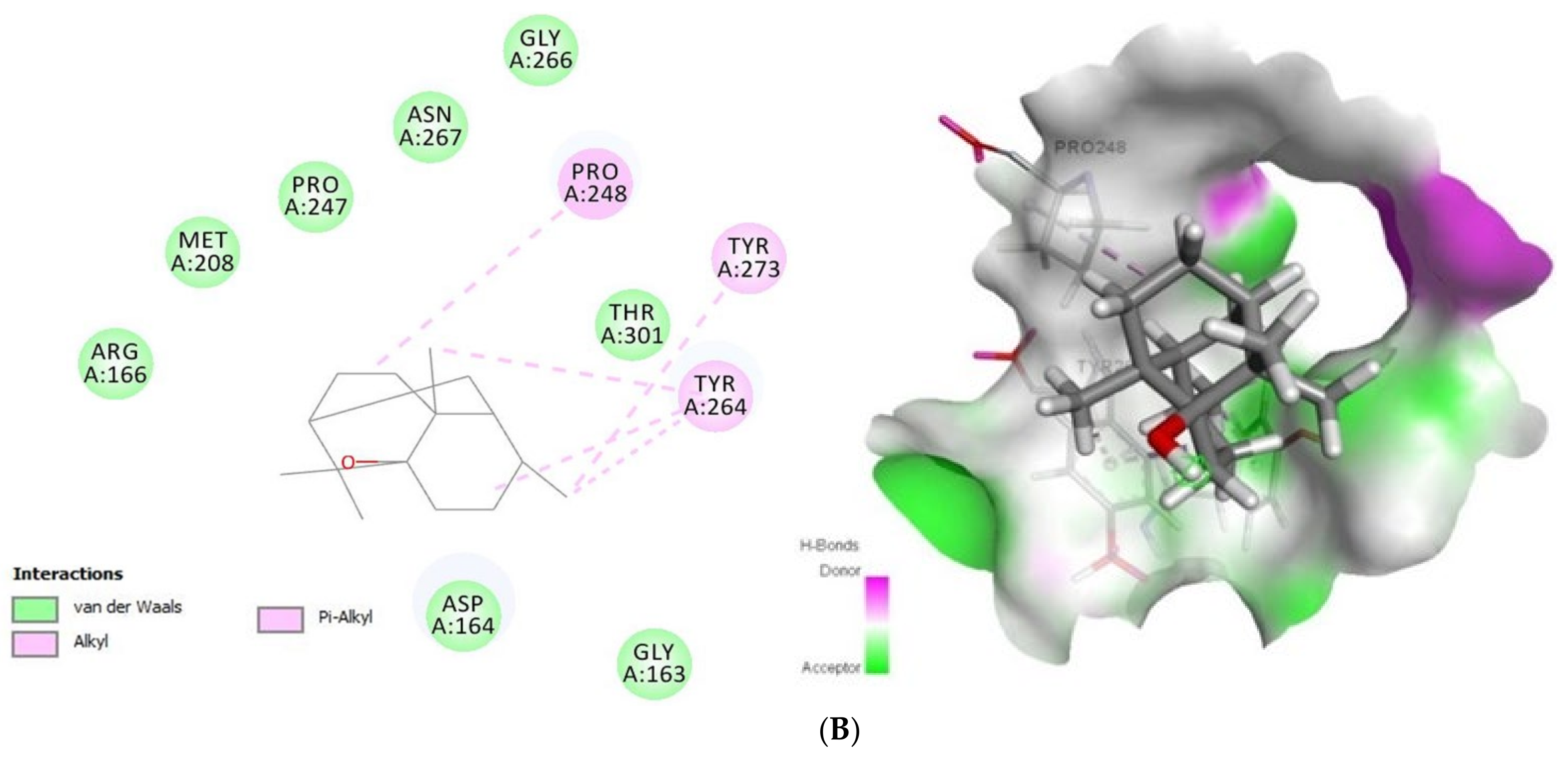
2.1.3. Docking Analysis of Tomatidine and Patchouli Alcohol with Target NSP15
2.1.4. Docking Analysis of Tomatidine and Patchouli Alcohol with Anti-Inflammatory Mediators
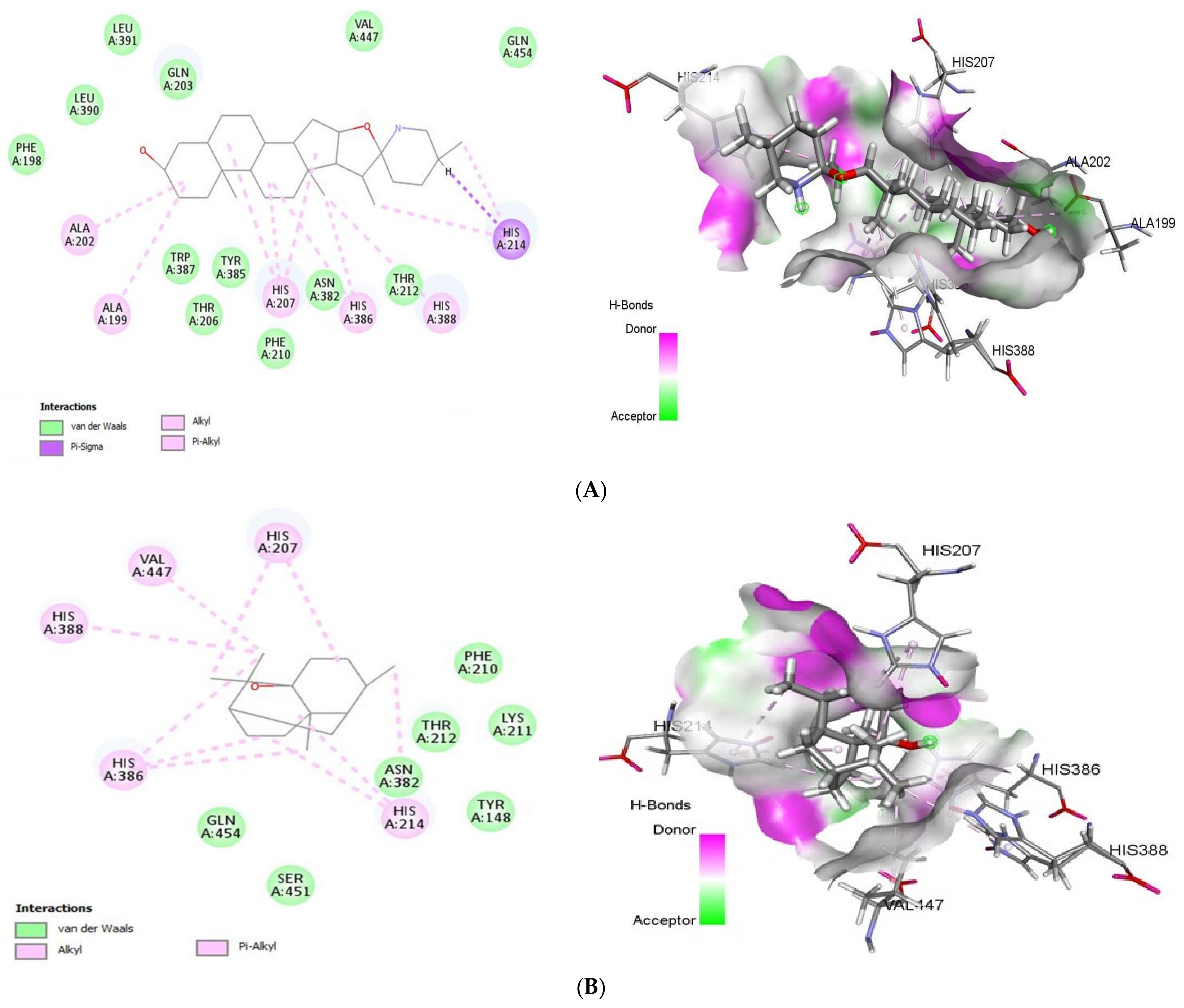
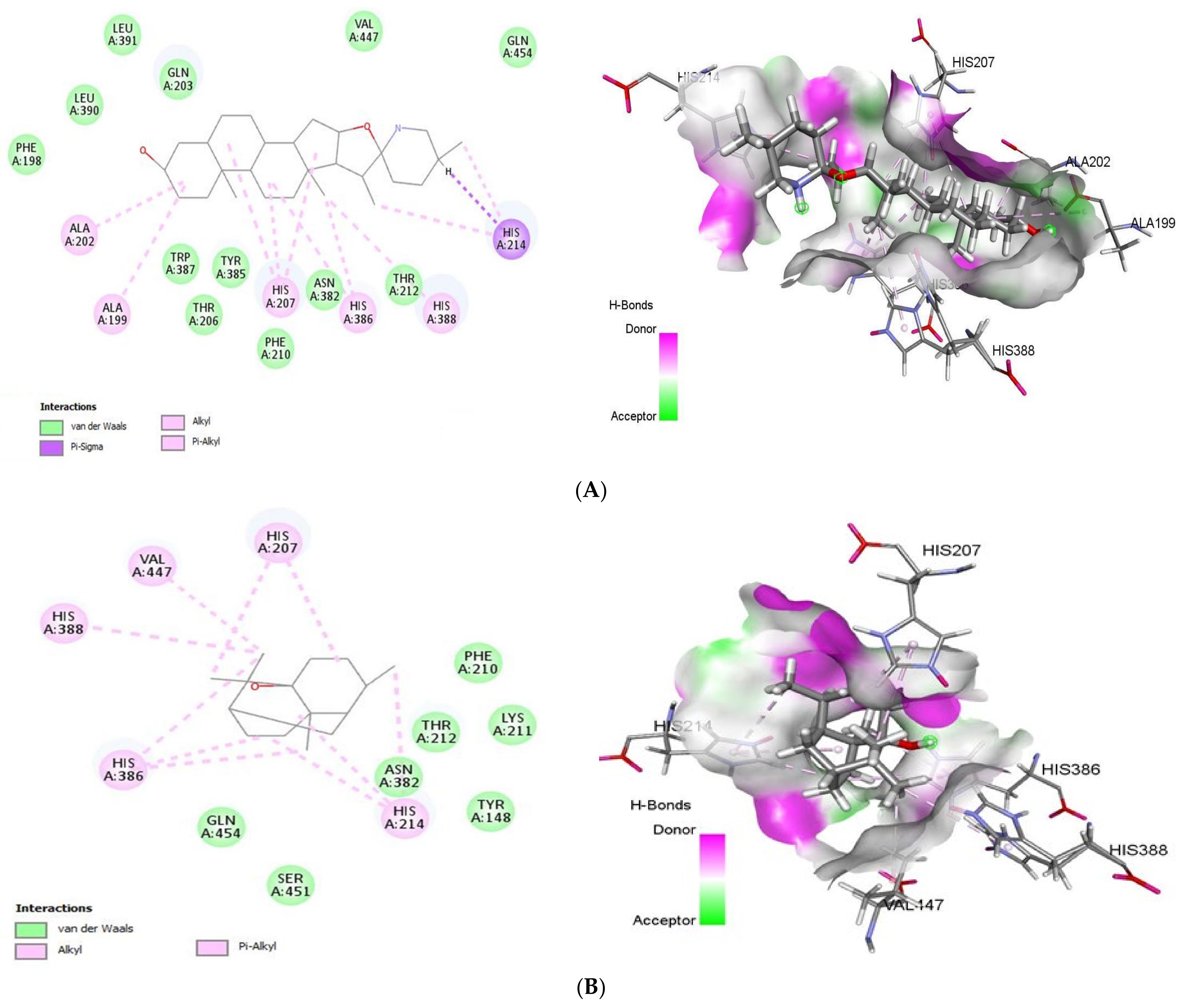
2.1.5. Docking Analysis of Tomatidine and Patchouli Alcohol with Target COX-2
2.1.6. Docking Analysis of Tomatidine and Patchouli Alcohol with Target sPLA-2
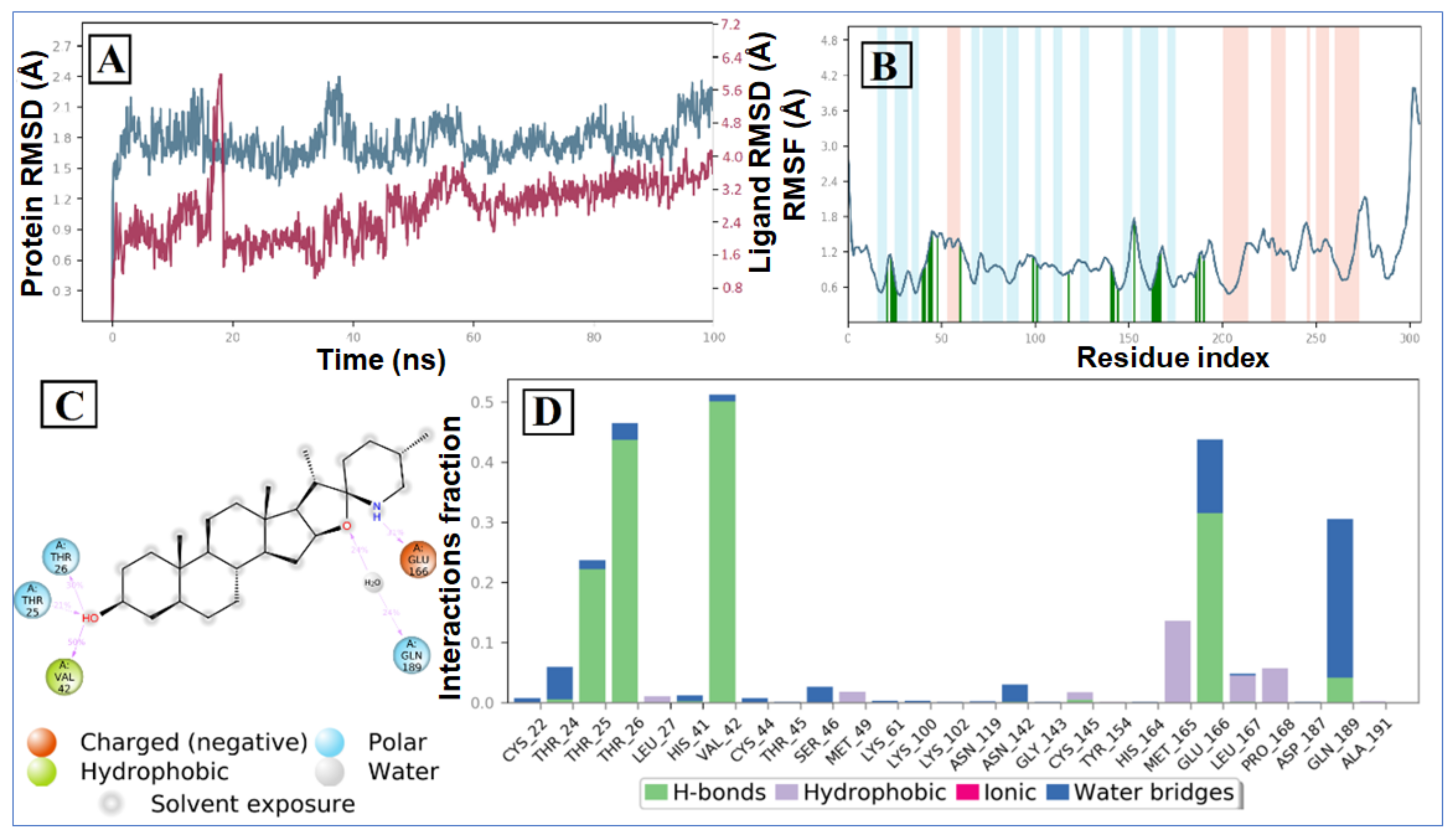
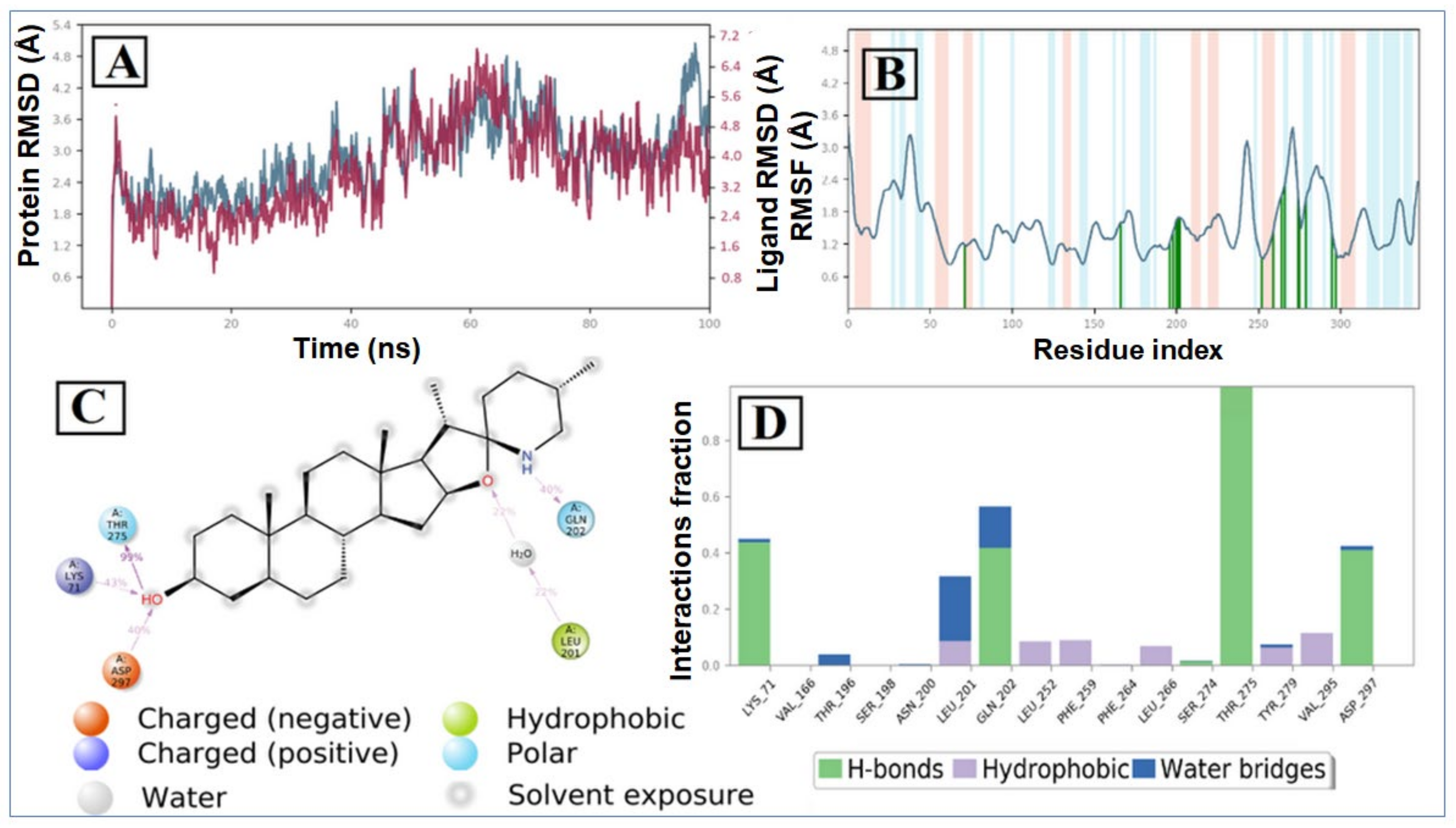
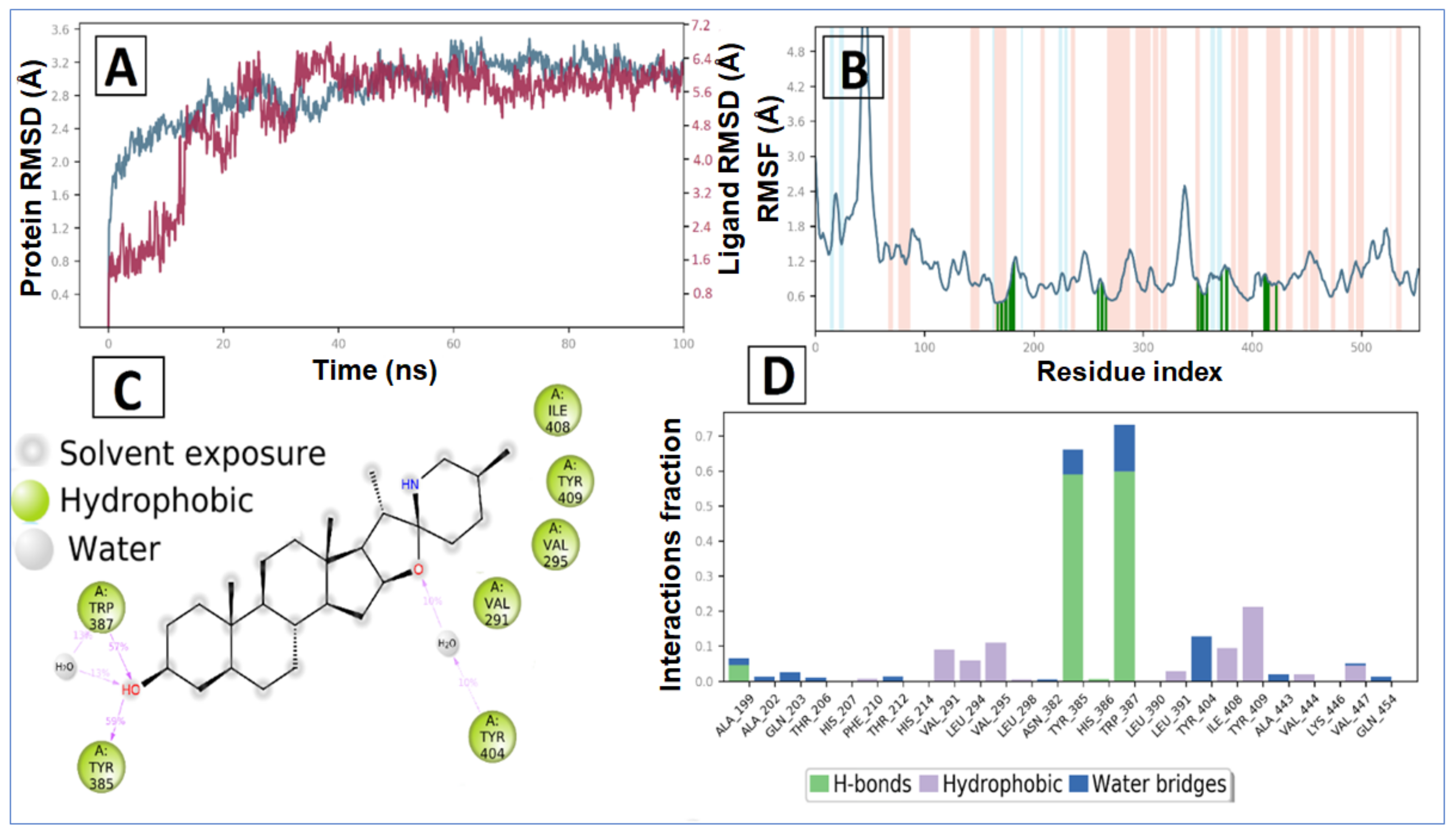
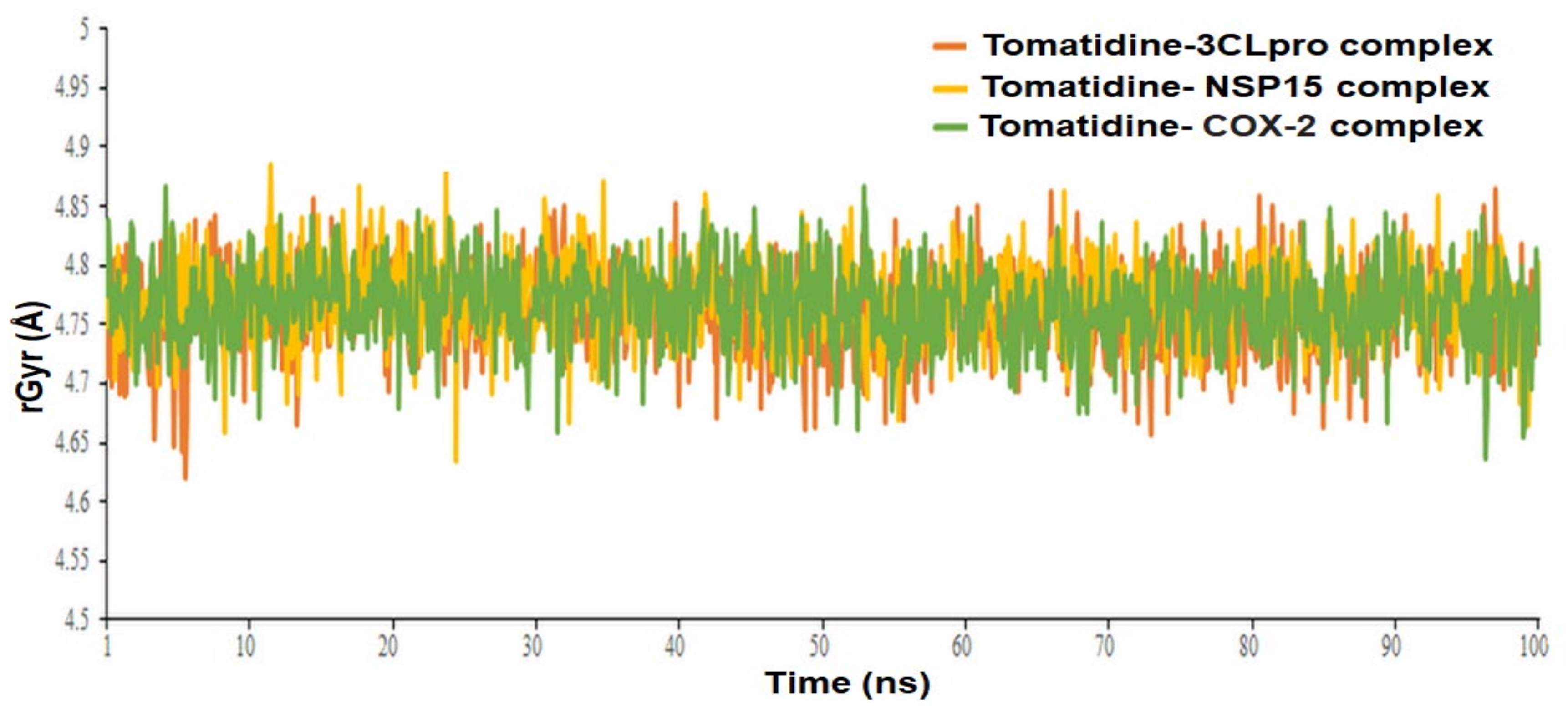
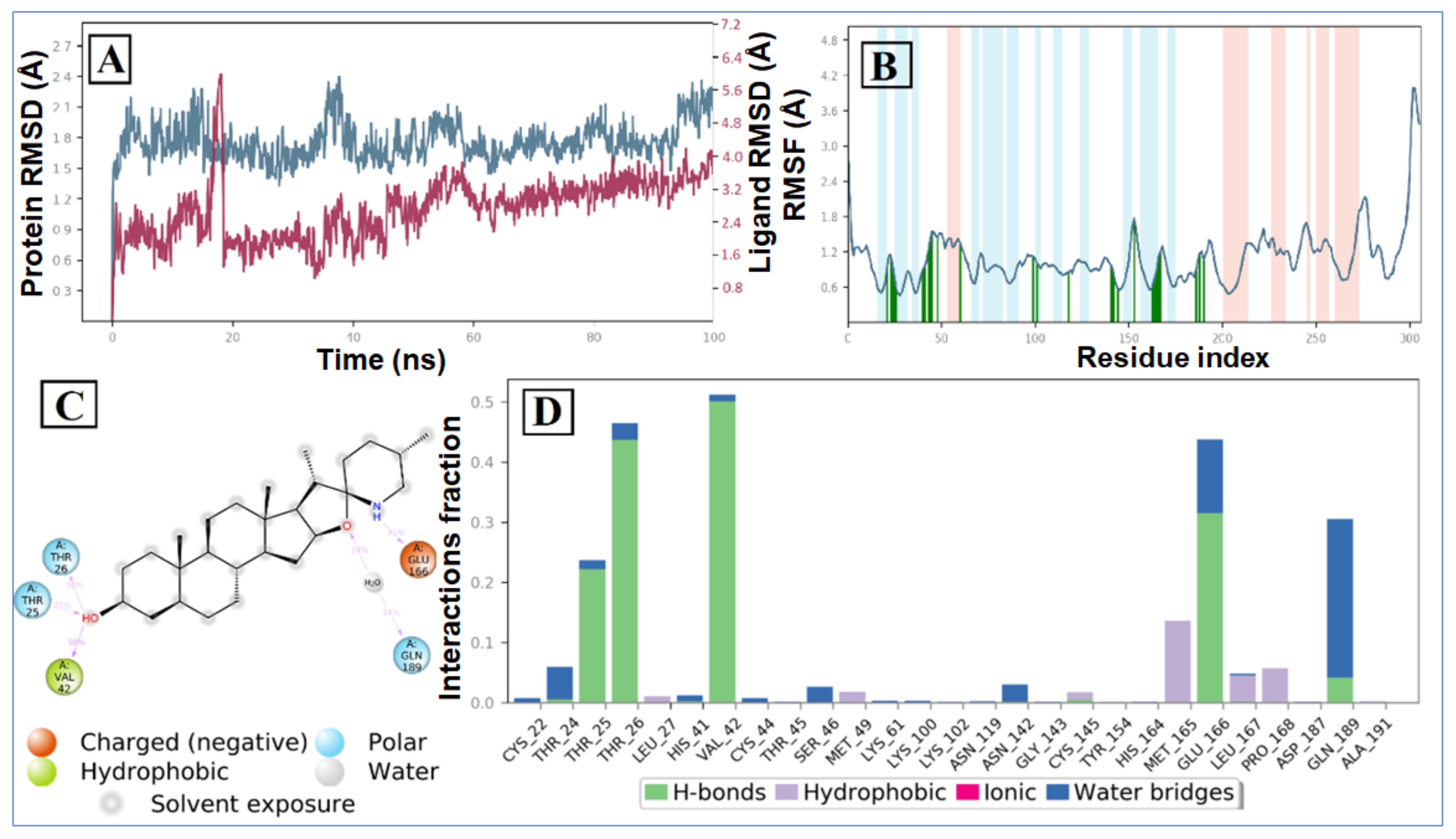
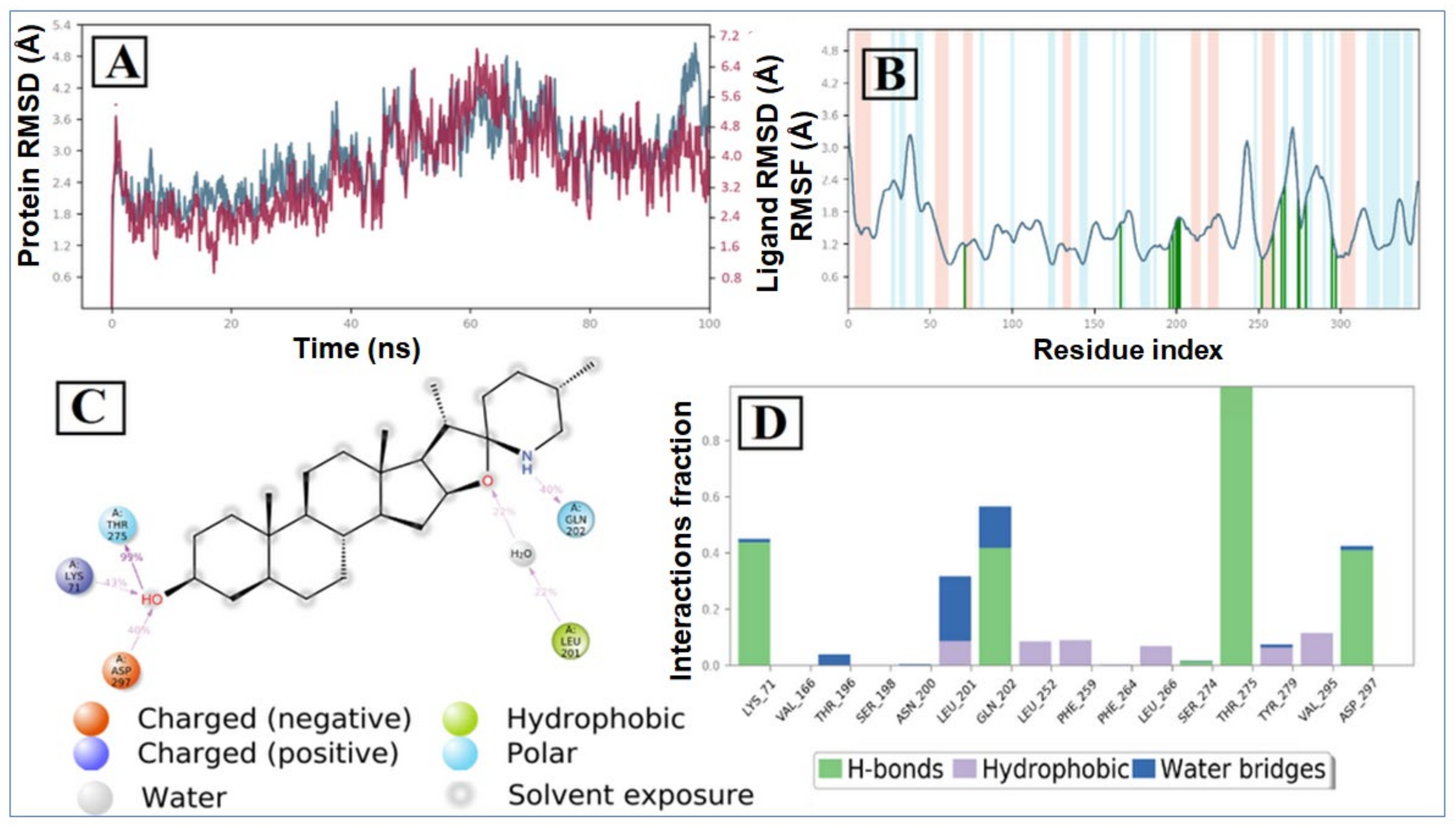
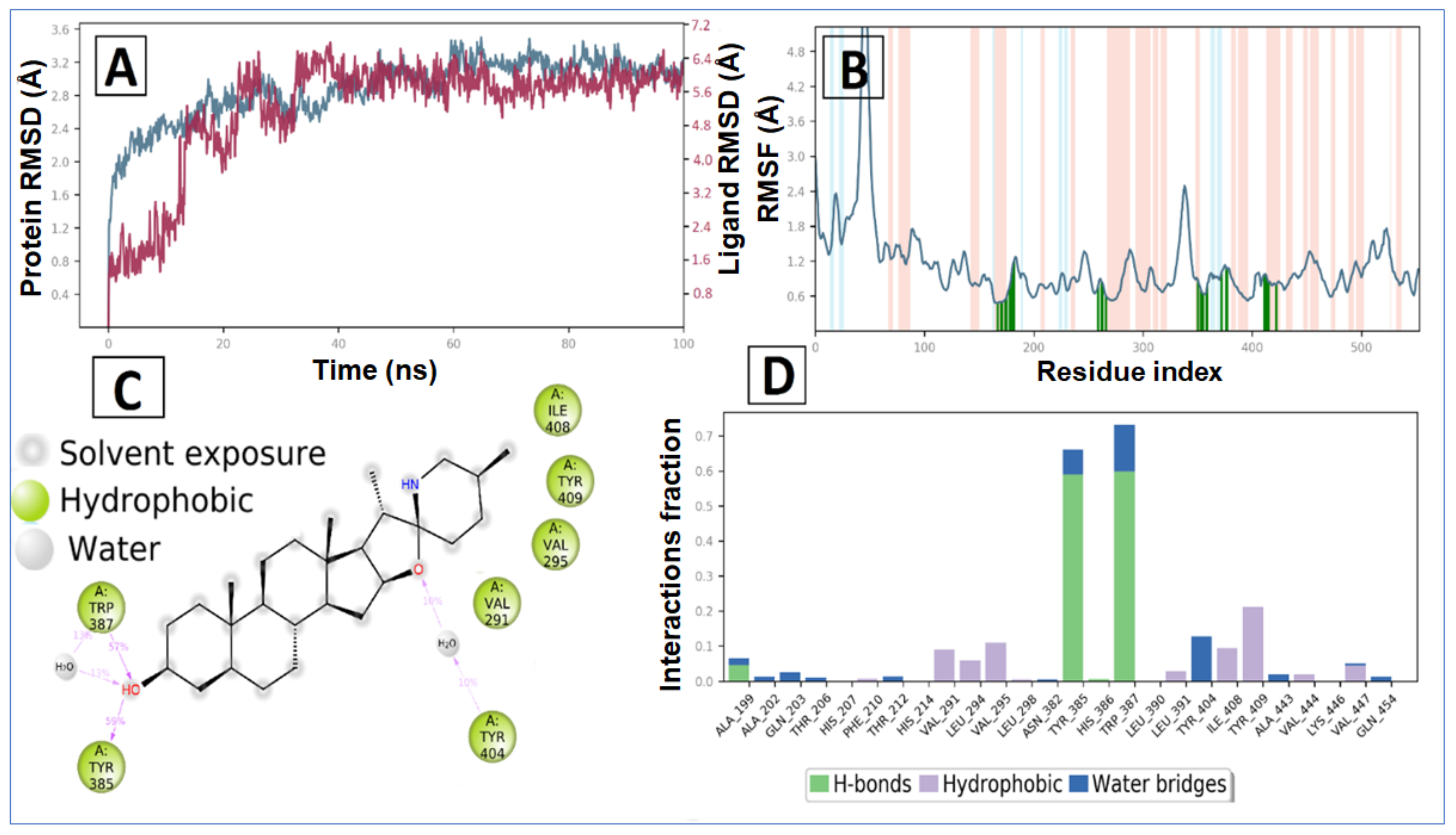
2.2. Molecular Dynamic Simulation and Post Dynamic MMGBSA Binding Free Energy Analysis
2.3. Pharmacokinetics, Drug-Likeliness and Toxicity Studies
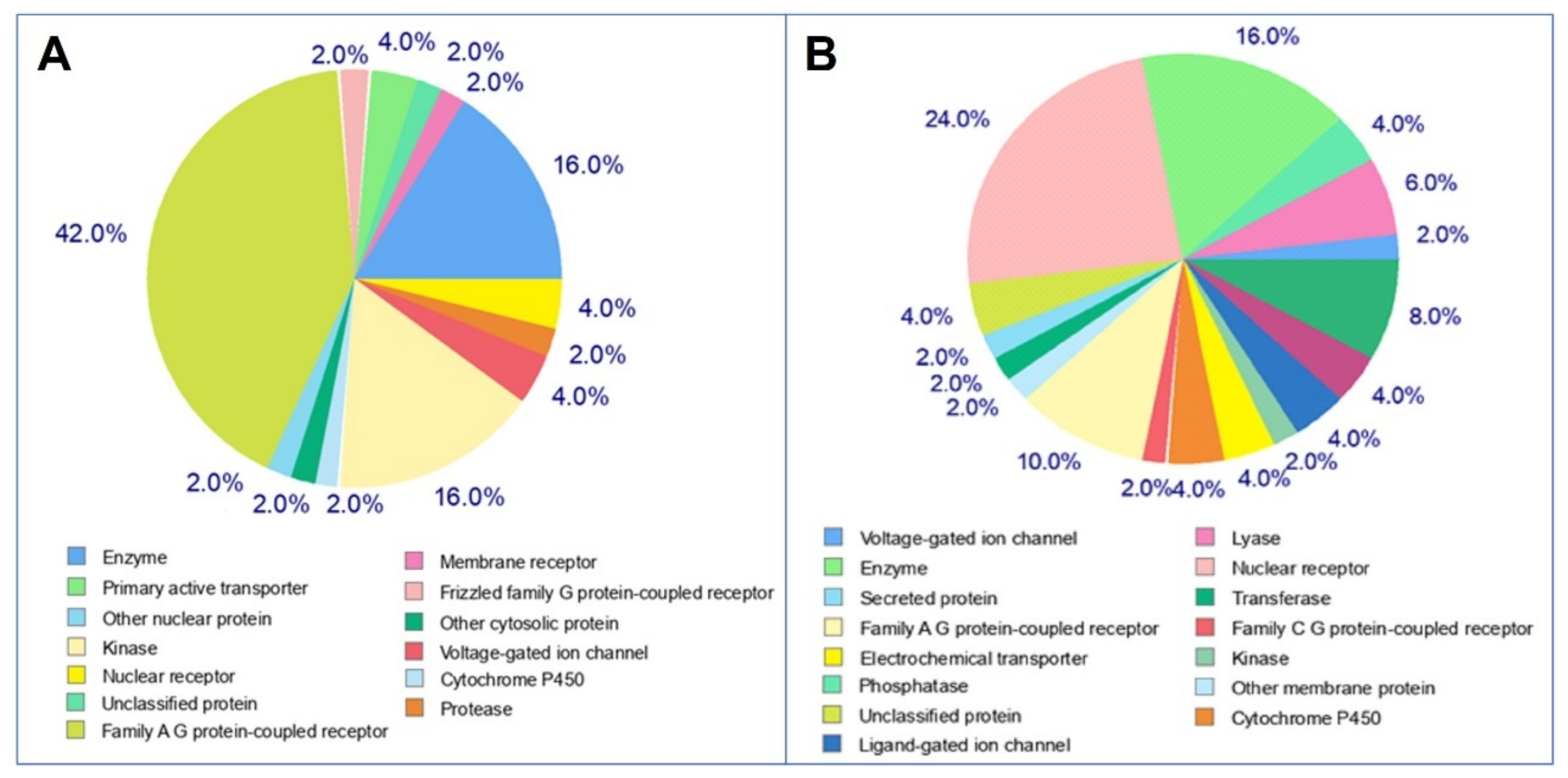
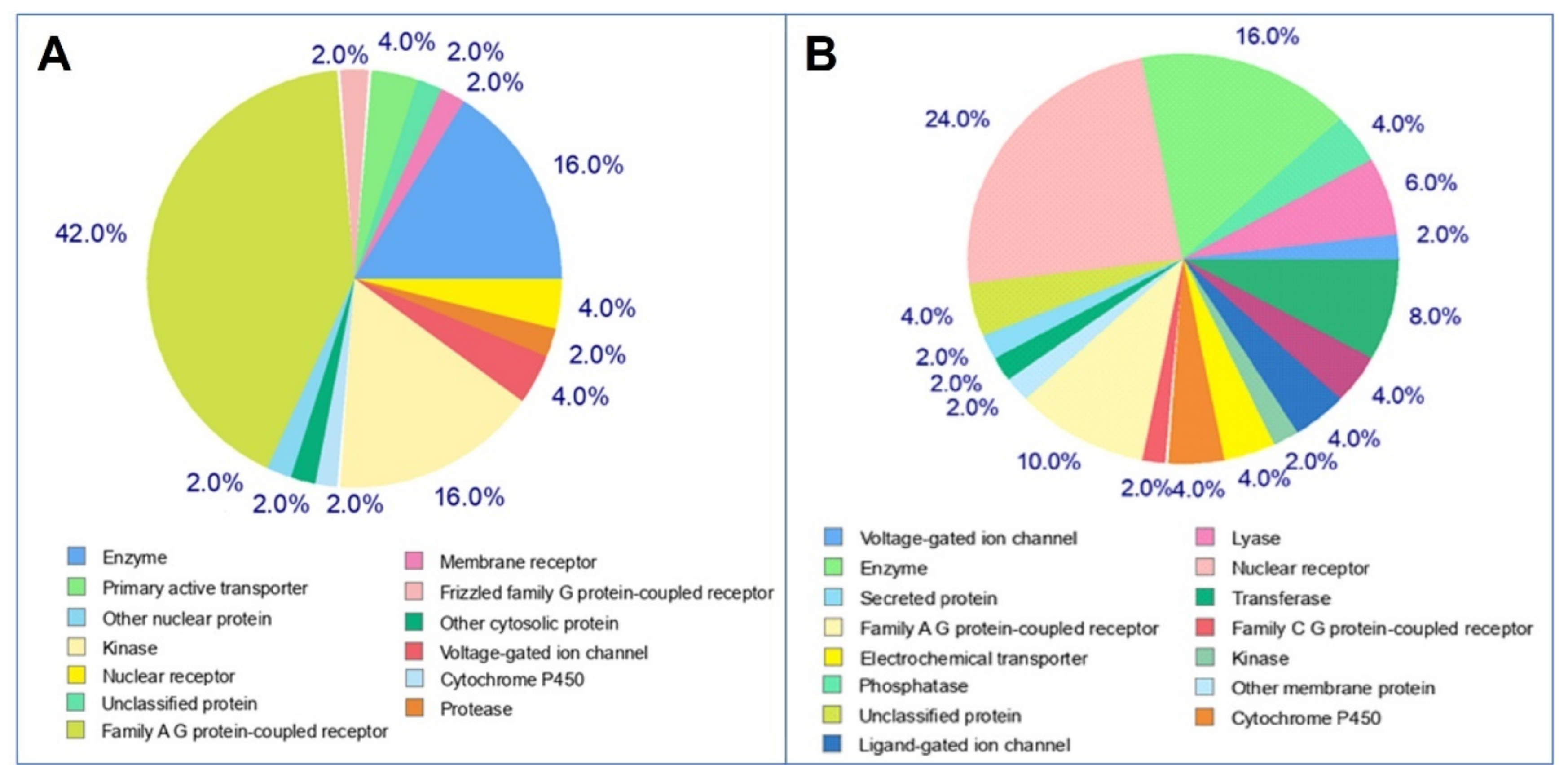
2.4. Target Prediction
3. Materials and Methods
3.1. Molecular Docking
3.1.1. Docking Using AutoDock Vina
3.1.2. Docking Using GOLD (Genetic Optimization for Ligand Docking)
3.2. Intra-Molecular Interactions in Docked Complex
3.3. Molecular Dynamic Simulation and Post Dynamic MMGBSA Binding free Energy Analysis
3.4. ADMET Prediction
3.5. Molecular Target Predictions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deshpande, R.R.; Tiwari, A.P.; Nyayanit, N.; Modak, M. In silico molecular docking analysis for repurposing therapeutics against multiple proteins from SARS-CoV-2. Eur. J. Pharmacol. 2020, 886, 173430. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, A.; Alshawaf, E.; Marafie, S.K.; Abu-Farha, M.; Abubaker, J.; Al-Mulla, F. Higher binding affinity of furin for SARS-CoV-2 spike (S) protein D614G mutant could be associated with higher SARS-CoV-2 infectivity. Perspective 2021, 103, 611–616. [Google Scholar]
- Ren, L.L.; Wang, Y.M.; Wu, Z.Q.; Xiang, Z.C.; Guo, L.; Xu, T.; Wang, J.M. Identification of a novel coronavirus causing severe pneumonia in human: A descriptive study. Chin. Med. J. 2020, 133, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.K.; Vemula, S.; Donde, R.; Gouda, G.; Behera, L.; Vadde, R. In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. J. Biomol. Struct. Dyn. 2021, 39, 2617–2627. [Google Scholar] [CrossRef] [Green Version]
- Prajapat, M.; Sarma, P.; Shekhar, N.; Avti, P.; Sinha, S.; Kaur, H.; Kumar, S.; Bhattacharyya, A.; Kumar, H.; Bansal, S.; et al. Drug targets for corona virus: A systematic review. Indian J. Pharmacol. 2020, 52, 56–65. [Google Scholar] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Maurya, D.K.; Sharma, D. Evaluation of traditional ayurvedic Kadha for prevention and management of the novel Coronavirus (SARS-CoV-2) using in silico approach. J. Biomol. Struct. Dyn. 2020, 30, 1–16. [Google Scholar]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Mol. Inform. 2020, 39, 2000028. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.; Mahmud, S.; Alam, A.S.M.R.U.; Rahman, M.E.; Ahmed, F.; Rahmatullah, M. Comparative molecular investigation of the potential inhibitors against SARS-CoV-2 main protease: A molecular docking study. J. Biomol. Struct. Dyn. 2020, 6317–6323. [Google Scholar] [CrossRef]
- Lei, J.; Kusov, Y.; Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 2018, 149, 58–74. [Google Scholar] [CrossRef]
- Herowati, R.; Widodo, G.P. Molecular docking analysis: Interaction studies of natural compounds to anti-inflammatory targets quantitative structure-activity relationship. InTechOpen 2017, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Neupane, N.P.; Karn, A.K.; Mukeri, I.H.; Pathak, P.; Kumar, P.; Singh, S.; Qureshi, I.A.; Jha, T.; Verma, A. Molecular dynamics analysis of phytochemicals from Ageratinaadenophora against COVID-19 main protease (Mpro) and human angiotensin-converting enzyme 2 (ACE2). Biocatal. Agric. Biotechnol. 2021, 32, 101924. [Google Scholar]
- Baby, K.; Maity, S.; Mehta, C.H.; Suresh, A.; Nayak, U.Y.; Nayak, Y. SARS-CoV-2 entry inhibitors by dual targeting TMPRSS2 and ACE2: An in silico drug repurposing study. Eur. J. Pharmacol. 2021, 896, 173922. [Google Scholar] [CrossRef] [PubMed]
- Mseddi, K.; Alimi, F.; Noumi, E.; Veettil, V.N.; Deshpande, S.; Adnan, M.; Hamdi, A.; Elkahoui, S.; Alghamdi, A.; Kadri, A.; et al. Thymus musilii Velen. as a promising source of potent bioactive compounds with its pharmacological properties: In vitro and in silico analysis. Arab. J. Chem. 2020, 13, 6782–6801. [Google Scholar] [CrossRef]
- Alminderej, F.; Bakari, S.; Almundarij, T.I.; Snoussi, M.; Aouadi, K.; Kadri, A. Antioxidant Activities of a New Chemotype of Piper cubeba L. Fruit Essential Oil (Methyleugenol/Eugenol): In Silico Molecular Docking and ADMET Studies. Plants 2020, 9, 1534. [Google Scholar] [CrossRef] [PubMed]
- Daoud, A.; Ben Mefteh, F.; Mnafgui, K.; Turki, M.; Jmal, S.; Ben Amar, R.; Ayadi, F.; ElFeki, A.; Abid, L.; Rateb, M.E.; et al. Cardiopreventive effect of ethanolic extract of date palm pollen against isoproterenol induced myocardial infarction in rats through the inhibition of the angiotensin-converting enzyme. Exp. Toxicol. Pathol. 2017, 69, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Bakari, S.; Hajlaoui, H.; Daoud, A.; Mighri, H.; Ross-Garcia, J.M.; Gharsallah, N.; Kadri, A. Phytochemicals, antioxidant and antimicrobial potentials and LC-MS analysis of hydroalcoholic extracts of leaves and flowers of Erodium glaucophyllum collected from Tunisian Sahara. Food Sci. Technol. 2018, 38, 310–317. [Google Scholar] [CrossRef] [Green Version]
- Felhi, S.; Saoudi, M.; Daoud, A.; Hajlaoui, H.; Ncir, M.; Chaabane, R.; El Feki, A.; Gharsallah, N.; Kadri, A. Investigation of phytochemical contents, in vitro antioxidant and antibacterial behavior and in vivo anti-inflammatory potential of Ecballium elaterium methanol fruits extract. Food Sci. Technol. 2017, 37, 558–563. [Google Scholar] [CrossRef] [Green Version]
- Felhi, S.; Hajlaoui, H.; Ncir, M.; Bakari, S.; Ktari, N.; Saoudi, M.; Gharsallah, N.; Kadri, A. Nutritional, phytochemical and antioxidant evaluation and FT-IR analysis of freeze-dried extracts of Ecballium elaterium fruit juice from three localities. Food Sci. Technol. 2016, 36, 646–655. [Google Scholar] [CrossRef] [Green Version]
- Caprioli, G.; Cahill, M.; Logrippo, S.; James, K. Elucidation of the mass fragmentation pathways of tomatidine and β1- hydroxytomatine using orbitrap mass spectrometry. Nat. Prod. Commun. 2015, 10, 575–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chagnon, F.; Guay, I.; Bonin, M.A.; Mitchell, G.; Bouarab, K.; Malouin, F.; Marsault, E. Unraveling the structure-activity relationship of tomatidine, a steroid alkaloid with unique antibiotic properties against persistent forms of Staphylococcus aureus. Eur. J. Med. Chem. 2014, 80, 605–620. [Google Scholar] [CrossRef]
- Yan, K.H.; Lee, L.M.; Yan, S.H.; Yuang, H.C.; Li, C.C.; Lin, H.T.; Chen, P.S. Tomatidine inhibits invasion of human lung adenocarcinoma cell A549 by reducing matrix metalloproteinases expression. Chem. Biol. Interact. 2013, 203, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chen, S.; Van Doren, J.; Li, D.; Farichon, C.; He, Y.; Zhang, Q.; Conney, A.H.; Goodin, S.; Du, Z.; et al. α-Tomatine inhibits growth and induces apoptosis in HL-60 human myeloid leukemia cells. Mol. Med. Rep. 2015, 11, 4573–4578. [Google Scholar] [CrossRef] [Green Version]
- Chiu, F.L.; Lin, J.K. Tomatidine inhibits iNOS and COX-2 through suppression of NF-κB and JNK pathways in LPS-stimulated mouse macrophages. FEBS Lett. 2008, 582, 2407–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.Y.; Huang, W.C.; Liou, C.J.; Chen, L.C.; Shen, J.J.; Kuo, M.L. Tomatidine Attenuates Airway Hyperresponsiveness and Inflammation by Suppressing Th2 Cytokines in a Mouse Model of Asthma. Mediat. Inflamm. 2017, 2017, 5261803. [Google Scholar] [CrossRef] [Green Version]
- Troost, B.; Mulder, L.M.; Diosa-Toro, M.; Van de Pol, D.; Rodenhuis-Zybert, I.A.; Smit, J.M. Tomatidine, a natural steroidal alkaloid shows antiviral activity towards chikungunya virus in vitro. Sci. Rep. 2020, 10, 6364. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Peng, C.; Xie, X.; Zhang, S.; Cao, X. Availability, Pharmaceutics, Security, Pharmacokinetics, and Pharmacological Activities of Patchouli Alcohol. Evid. Based Complement. Alternat. Med. 2017, 2017, 4850612. [Google Scholar] [CrossRef]
- Shingh, B.G.; Kunwar, A. In Silico Investigation on the Binding of Organoselenium Compounds with Target Proteins of SARS-CoV-2 Infection Cycle. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Tamilanban, T.; Naveen Kumar, V.; Narayanan, J.; Prathusa, S.; Dhivya, N.; Manasa, K. In silico Molecular docking of Luteolin from Momordica charantia for dementia in Alzheimer’s disease. Res. J. Pharm. Technol. 2020, 13, 2381–2386. [Google Scholar] [CrossRef]
- Diosa-Toro, M.; Troost, B.; Van de Pol, D.; Heberle, A.M.; Urcuqui-Inchima, S.; Thedieck, K.; Smit, J.M. Tomatidine, a novel antiviral compound towards dengue virus. Antivir. Res. 2019, 161, 90–99. [Google Scholar] [CrossRef]
- Wang, P.; Bai, J.; Liu, X.; Wang, M.; Wang, X.; Jiang, P. Tomatidine inhibits porcine epidemic diarrhea virus replication by targeting 3CL protease. Vet. Res. 2020, 51, 136. [Google Scholar] [CrossRef] [PubMed]
- Jain, D.; Abid Ali Khan, M.; Zaim, M.; Thakur, R. Antiviral evaluations of some steroids and their glycosides: A new report. Natl. Acad. Sci. Lett. 1990, 13, 3–4. [Google Scholar]
- Thorne, H.V.; Clarke, G.F.; Skuce, R. The inactivation of herpes simplex virus by some Solanaceae glycoalkaloids. Antivir. Res. 1985, 5, 335–343. [Google Scholar] [CrossRef]
- Bier, E.; Smelkinson, M.; Krug, R.; Malur, M.; Oldstone, M.; Teijaro, J. Protection and Treatment against Influenza Infection. U.S. Patent 9095579, 4 August 2015. [Google Scholar]
- Bailly, B.; Richard, C.-A.; Sharma, G.; Wang, L.; Johansen, L.; Cao, J.; Pendharkar, V.; Sharma, D.-C.; Galloux, M.; Wang, Y.; et al. Targeting human respiratory syncytial virus transcription anti- termination factor M2-1 to inhibit in vivo viral replication. Sci. Rep. 2016, 6, 25806. [Google Scholar] [CrossRef] [PubMed]
- Vergoten, G.; Bailly, C. In silico analysis of echinocandins binding to the main proteases of coronaviruses PEDV (3CLpro) and SARS-CoV-2 (Mpro). Silico Pharmacol. 2021, 9, 41. [Google Scholar] [CrossRef]
- Kiyohara, H.; Ichino, C.; Kawamura, Y.; Nagai, T.; Sato, N.; Yamada, H. Patchouli alcohol: In vitro direct anti-influenza virus sesquiterpene in Pogostemon cablin Benth. J. Nat. Med. 2012, 66, 55–61. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, Y.; Wang, S.; Liu, W.; Hao, C.; Wang, W. Inhibition effects of patchouli alcohol against influenza a virus through targeting cellular PI3K/Akt and ERK/MAPK signaling pathways. Virol. J. 2019, 16, 163. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Li, B.; Wang, X.; Jin, M.; Wang, G. Inhibitory effect and possible mechanism of action of patchouli alcohol against influenza A (H2N2) virus. Molecules 2011, 16, 6489–6501. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Twilley, D.; Esmear, T.; Oosthuizen, C.B.; Reid, A.-M.; Nel, M.; Lall, N. Anti-SARS-CoV Natural Products With the Potential to Inhibit SARS-CoV-2 (COVID-19). Front. Pharmacol. 2020, 11, 561334. [Google Scholar] [CrossRef]
- Pamuru, R.R.; Ponneri, N.; Damu, A.G.; Vadde, R. Targeting Natural Products for the Treatment of COVID-19—An Updated Review. Curr. Pharm. Des. 2020, 26, 5278–5285. [Google Scholar] [CrossRef] [PubMed]
- Prasansuklab, A.; Theerasri, A.; Rangsinth, P.; Sillapachaiyaporn, C.; Chuchawankul, S.; Tencomnao, T. Anti-COVID-19 drug candidates: A review on potential biological activities of natural products in the management of new coronavirus infection. J. Tradit. Complement. Med. 2021, 11, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, S.; Dubey, A.; Yadava, U.; Mishra, S.K.; Kang, S.G.; Dwivedi, V.D. Exploration of natural compounds with anti-SARS-CoV-2 activity via inhibition of SARS-CoV-2 Mpro. Brief Bioinform. 2021, 22, 1361–1377. [Google Scholar] [CrossRef]
- Teli, D.M.; Shah, M.B.; Chhabria, M.T. In silico Screening of Natural Compounds as Potential Inhibitors of SARS-CoV-2 Main Protease and Spike RBD: Targets for COVID-19. Front. Mol. Biosci. 2021, 7, 599079. [Google Scholar] [CrossRef]
- AlAjmi, M.F.; Azhar, A.; Hasan, S.; Alshabr, A.Z.; Hussain, A.; Rehman, M.T. Identification of Natural Compounds (Proanthocyanidin and Rhapontin) as High-Affinity Inhibitor of SARS-CoV-2 Mpro and PLpro using Computational Strategies. Arch. Med. Sci. 2021. [Google Scholar] [CrossRef]
- Merarchi, M.; Dudha, N.; Das, B.C.; Garg, M. Natural products and phytochemicals as potential anti-SARS-CoV -2 drugs. Phytother. Res. 2021, 1–13. [Google Scholar] [CrossRef]
- Hattori, S.; Higashi-Kuwata, N.; Hayashi, H.; Allu, S.R.; Raghavaiah, J.; Bulut, H.; Das, D.; Anson, B.J.; Lendy, E.K.; Takamatsu, Y.; et al. A small molecule compound with an indole moiety inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2021, 12, 668. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, R.; Singh, R.; Ghosh, A.; Dey, D.; Sharma, P.; Velayutham, R.; Roy, S.; Ghosh, D. A review on potential of natural products in the management of COVID-19. RSC Adv. 2021, 11, 16711. [Google Scholar] [CrossRef]
- Snoussi, M.; Redissi, A.; Mosbah, A.; De Feo, V.; Adnan, M.; Aouadi, K.; Alreshidi, M.; Patel, M.; Kadri, A.; Noumi, E. Emetine, a potent alkaloid for the treatment of SARS-CoV-2 targeting papain-like protease and non-structural proteins: Pharmacokinetics, molecular docking and dynamic studies. J. Biomol. Struct. Dyn. 2021. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Bartuzi, D.; Kaczor, A.A.; Targowska-Duda, K.M.; Matosiuk, D. Recent Advances and Applications of Molecular Docking to G Protein-Coupled Receptors. Molecules 2017, 22, 340. [Google Scholar] [CrossRef] [Green Version]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, G.; Willett, P.; Glen, R.C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43–53. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D. E. Shaw Research. Schrödinger Release (2020-1): Desmond Molecular dynamics System: Maestro-Desmond Interoperability Tools; D. E. Shaw Research: New York, NY, USA, 2020. [Google Scholar]
- Ahmad, I.; Jadhav, H.; Shinde, Y.; Jagtap, V.; Girase, R.; Patel, H. Optimizing Bedaquiline for cardiotoxicity by structure based virtual screening, DFT analysis and molecular dynamic simulation studies to identify selective MDR-TB inhibitors. Silico Pharmacol. 2021, 9, 23. [Google Scholar] [CrossRef]
- Ahmad, I.; Kumar, D.; Patel, H. Computational investigation of phytochemicals from Withaniasomnifera (Indian ginseng/ashwagandha) as plausible inhibitors of GluN2B-containing NMDA receptors. J. Biomol. Struct. Dyn. 2021, 10, 1–13. [Google Scholar]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all atom force field on conformational energetics and properties of organic liquids. J. Am. Chem Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Patel, H.M.; Ahmad, I.; Pawara, R.; Shaikh, M.; Surana, S. In silico search of triple mutant T790M/C797S allosteric inhibitors to conquer acquired resistance problem in non-small cell lung cancer (NSCLC): A combined approach of structure-based virtual screening and molecular dynamics simulation. J. Biomol. Struct. Dyn. 2021, 39, 1491–1505. [Google Scholar] [CrossRef]
- Ahmad, I.; Shaikh, M.; Surana, S.; Ghosh, A.; Patel, H. p38α MAP kinase inhibitors to overcome EGFR tertiary C797S point mutation associated with osimertinib in non-small cell lung cancer (NSCLC): Emergence of fourth-generation EGFR inhibitor. J. Biomol. Struct. Dyn. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Martyna, G.J. Remarks on “Constant-temperature molecular dynamics with momentum conservation”. Phys. Rev. E 1994, 50, 3234–3236. [Google Scholar] [CrossRef] [PubMed]
- Kalibaeva, G.; Ferrario, M.; Ciccotti, G. Constant pressure-constant temperature molecular dynamics: A correct constrained NPT ensemble using the molecular virial. Mol. Phys. 2003, 101, 765–778. [Google Scholar] [CrossRef]
- Patel, H.M.; Shaikh, M.; Ahmad, I.; Lokwani, D.; Surana, S.J. BREED based de novo hybridization approach: Generating novel T790M/C797S-EGFR tyrosine kinase inhibitors to overcome the problem of mutation and resistance in non-small cell lung cancer (NSCLC). J. Biomol. Struct. Dyn. 2021, 39, 2838–2856. [Google Scholar] [CrossRef] [PubMed]
- Kadri, A.; Aouadi, K. In vitro antimicrobial and α-glucosidase inhibitory potential of enantiopure cycloalkylglycine derivatives: Insights into their in silico pharmacokinetic. druglikeness and medicinal chemistry properties. J. Appl. Pharm. Sci. 2020, 10, 107–115. [Google Scholar]
- Othman, I.M.M.; Gad-Elkareem, M.A.M.; Anouar, E.H.; Aouadi, K.; Kadri, A.; Snoussi, M. Design, synthesis ADMET and molecular docking of new imidazo[4.5-b]pyridine-5-thione derivatives as potential tyrosyl-tRNA synthetase inhibitors. Bioorg. Chem. 2020, 102, 104105. [Google Scholar] [CrossRef] [PubMed]
- Ghannay, S.; Kadri, A.; Aouadi, K. Synthesis, in vitro antimicrobial assessment, and computational investigation of pharmacokinetic and bioactivity properties of novel trifluoromethylated compounds using in silico ADME and toxicity prediction tools. Monatshefte Chem. 2020, 151, 267–280. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
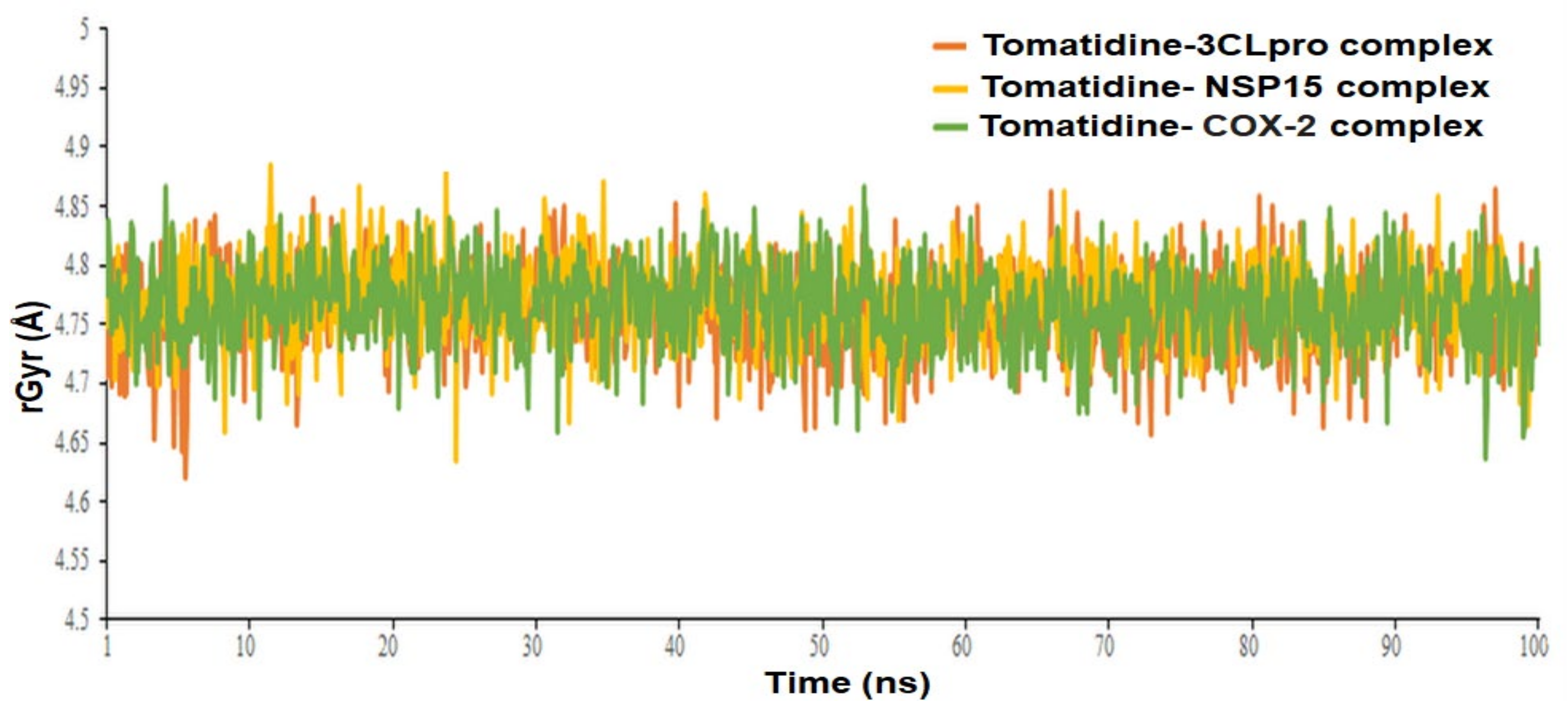
| Complex Name | Radius of Gyration, rGyr (Å) | MMGBSA, ΔG Bind (kcal/mol) | ||
|---|---|---|---|---|
| Mean | Range | Mean | Range | |
| Tomatidine-3CLpro complex | 4.76 ± 0.036 | 4.62 to 4.86 | −47.4633 ± 9.28 | −61.2963 to −31.8438 |
| Tomatidine-NSP15 complex | 4.77± 0.034 | 4.63 to 4.88 | −51.8064 ± 8.91 | −67.7417 to −35.2832 |
| Tomatidine-human COX-2 complex | 4.77± 0.034 | 4.63 to 4.86 | −54.8918 ±7.55 | −62.1318 to −38.2816 |
| Physicochemical Properties | Drug-Likeness | Bioavailability (Radar Plot) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| T | PA | T | PA | T | PA | ||||
| Molecular weight (g/mol) Num. heavy atoms Num. arom. heavy atoms Fraction Csp3 Num. rotatable bonds Num. H-bond acceptors Num. H-bond donors Molar Refractivity TPSA (Å2) | 415.65 30 0 1.00 0 3 2 127.70 41.49 | 222.37 16 0 1.00 0 1 1 68.56 20.23 | Lipinski Ghose Veber Egan Muegge Bioavailability Score Consensus log Po/w | Yes No Yes Yes No 0.55 4.90 | Yes Yes Yes Yes No 0.55 3.57 |  | 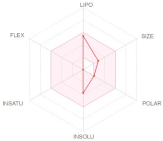 | ||
| LIPO: Lipophilicity FLEX: Flexibility SIZE: Size | INSAT: Insaturation INSOLU: Insolubility POLAR: Polarity | ||||||||
| Pharmacokinetics | |||||||||
| T | PA | T | PA | T | PA | ||||
| GI absorption | High | High | CYP1A2 inhibitor | No | No | CYP2D6 inhibitor | No | No | |
| BBB permeant | Yes | Yes | CYP2C19 inhibitor | No | No | CYP3A4 inhibitor | No | No | |
| P-gp substrate | Yes | No | CYP2C9 inhibitor | No | Yes | log Kp (skin permeation) cm/s | −4.34 | −4.87 | |
| Toxicity | |||||||||
| AMES | hERG I inhibitor | Hepatotoxicity | Skin Sensitisation | ||||||
| T | PA | T | PA | T | PA | T | PA | ||
| No | No | No | No | No | No | No | No | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zrieq, R.; Ahmad, I.; Snoussi, M.; Noumi, E.; Iriti, M.; Algahtani, F.D.; Patel, H.; Saeed, M.; Tasleem, M.; Sulaiman, S.; et al. Tomatidine and Patchouli Alcohol as Inhibitors of SARS-CoV-2 Enzymes (3CLpro, PLpro and NSP15) by Molecular Docking and Molecular Dynamics Simulations. Int. J. Mol. Sci. 2021, 22, 10693. https://doi.org/10.3390/ijms221910693
Zrieq R, Ahmad I, Snoussi M, Noumi E, Iriti M, Algahtani FD, Patel H, Saeed M, Tasleem M, Sulaiman S, et al. Tomatidine and Patchouli Alcohol as Inhibitors of SARS-CoV-2 Enzymes (3CLpro, PLpro and NSP15) by Molecular Docking and Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2021; 22(19):10693. https://doi.org/10.3390/ijms221910693
Chicago/Turabian StyleZrieq, Rafat, Iqrar Ahmad, Mejdi Snoussi, Emira Noumi, Marcello Iriti, Fahad D. Algahtani, Harun Patel, Mohd Saeed, Munazzah Tasleem, Shadi Sulaiman, and et al. 2021. "Tomatidine and Patchouli Alcohol as Inhibitors of SARS-CoV-2 Enzymes (3CLpro, PLpro and NSP15) by Molecular Docking and Molecular Dynamics Simulations" International Journal of Molecular Sciences 22, no. 19: 10693. https://doi.org/10.3390/ijms221910693
APA StyleZrieq, R., Ahmad, I., Snoussi, M., Noumi, E., Iriti, M., Algahtani, F. D., Patel, H., Saeed, M., Tasleem, M., Sulaiman, S., Aouadi, K., & Kadri, A. (2021). Tomatidine and Patchouli Alcohol as Inhibitors of SARS-CoV-2 Enzymes (3CLpro, PLpro and NSP15) by Molecular Docking and Molecular Dynamics Simulations. International Journal of Molecular Sciences, 22(19), 10693. https://doi.org/10.3390/ijms221910693