
C5a Activates a Pro-Inflammatory Gene Expression Profile in Human Gaucher iPSC-Derived Macrophages

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
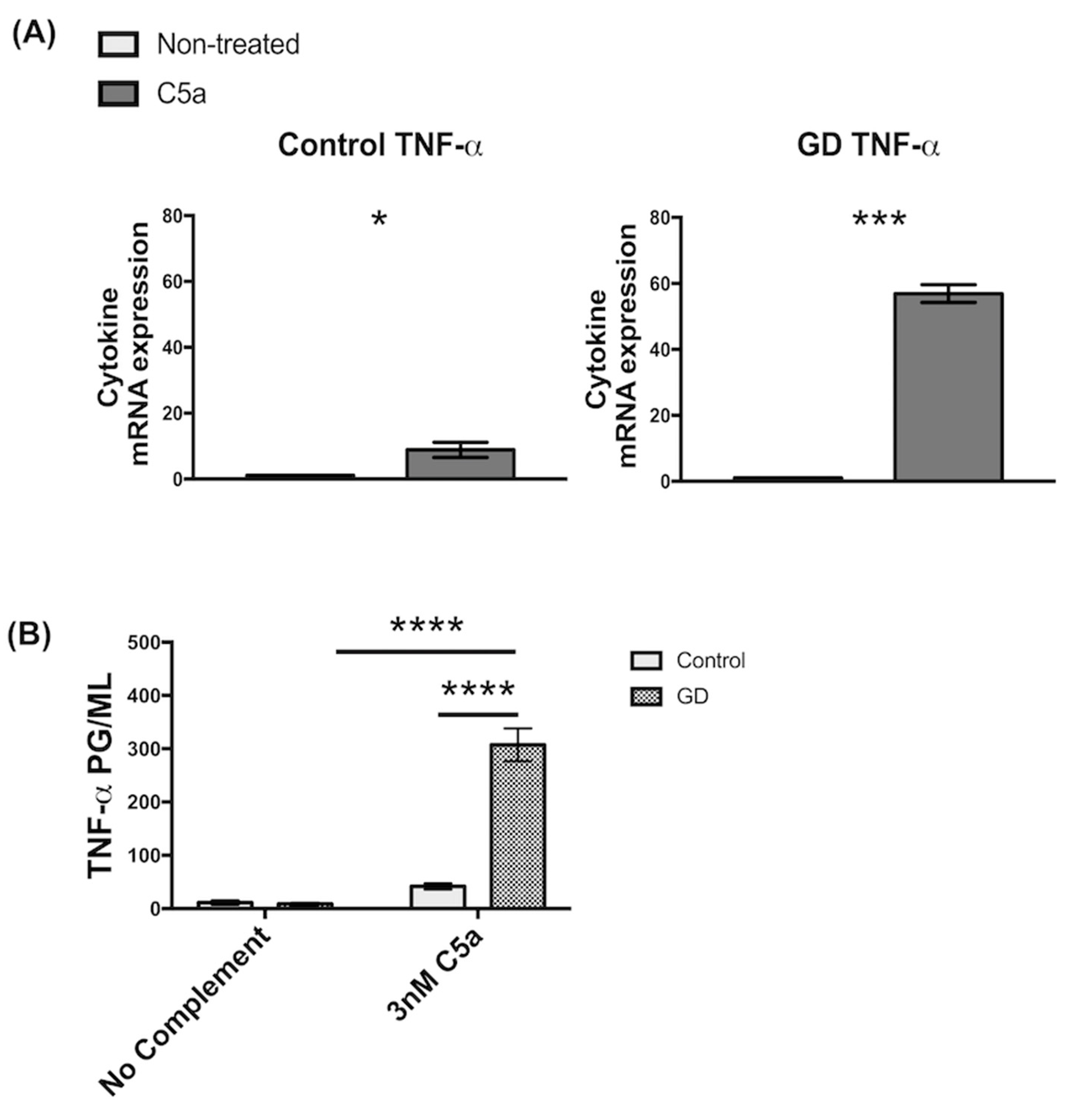
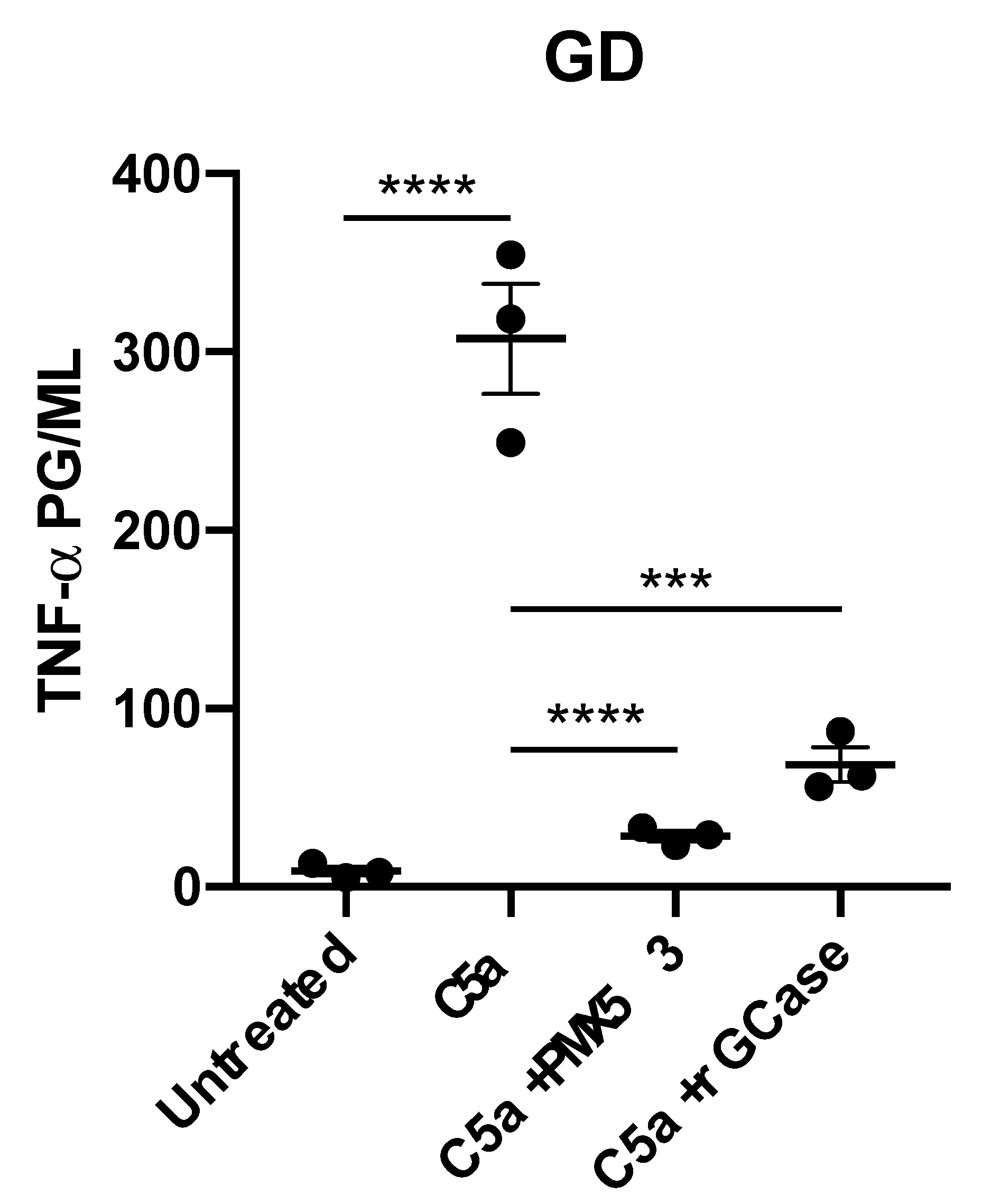
2.1. Recombinant C5a Induces an Elevated TNF-α-Mediated Inflammatory Response in GD Macrophages
2.2. Microarray Analysis of GD Macrophages
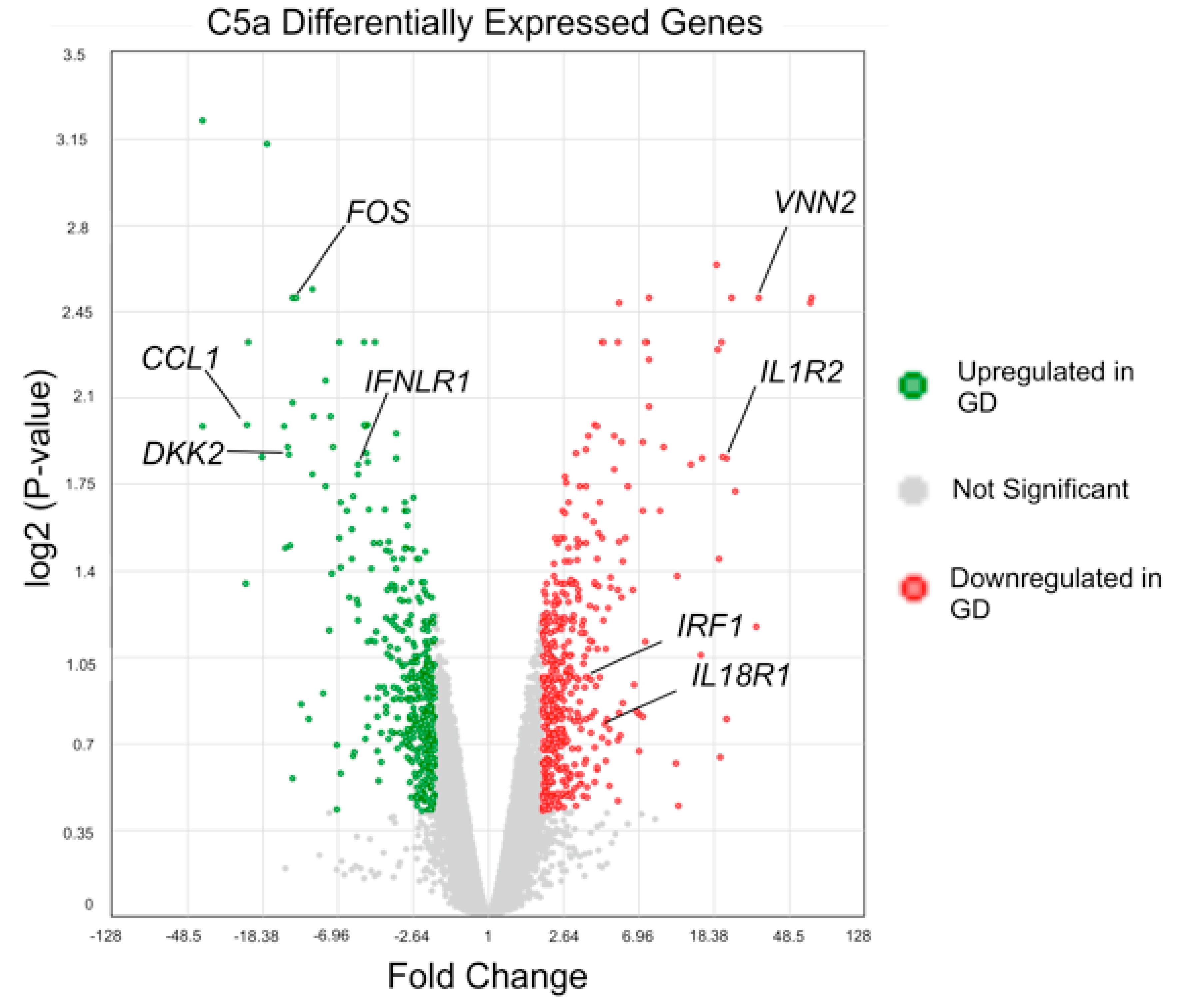
2.3. Differentially Expressed Genes between Untreated GD vs. Control Macrophages
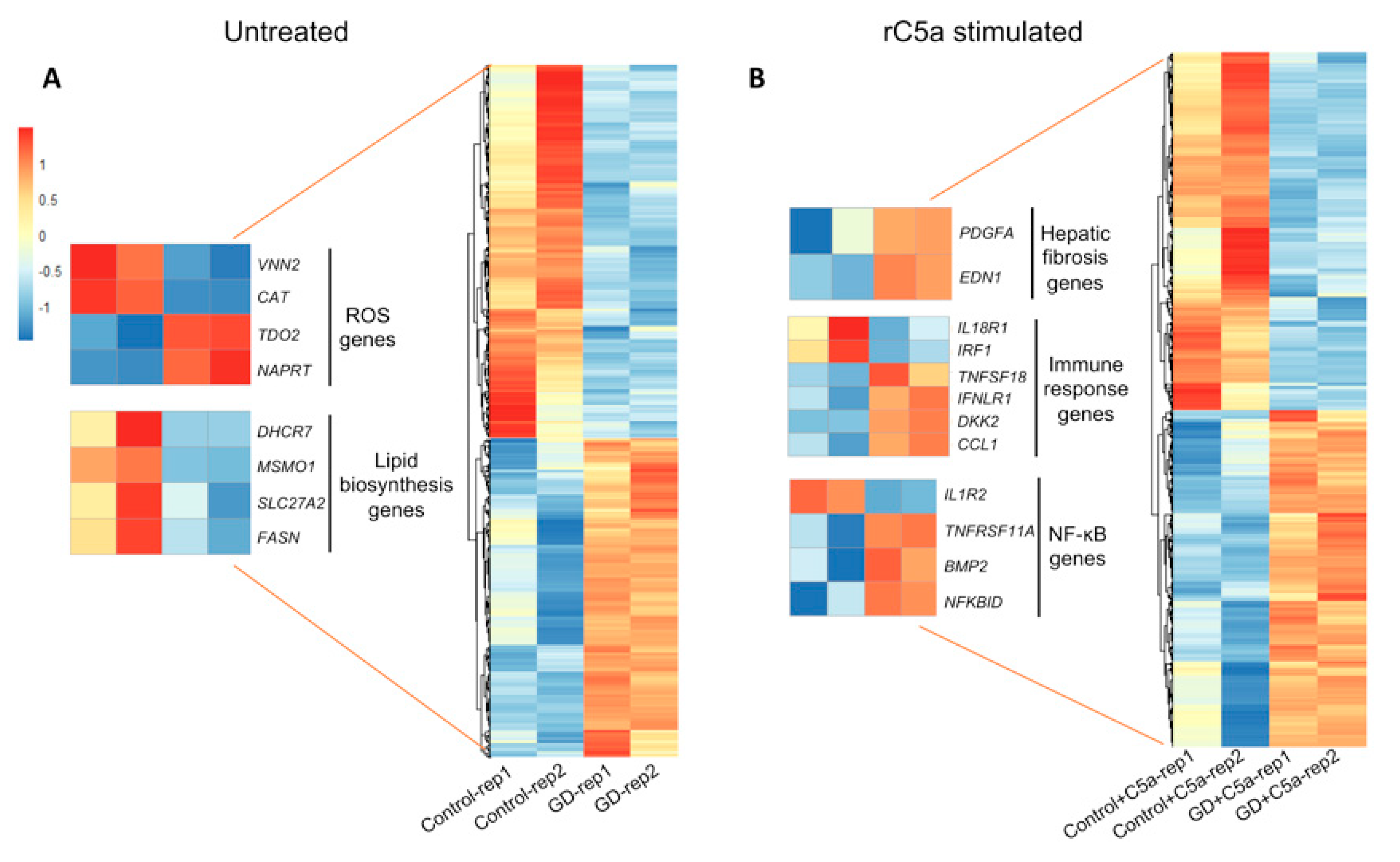
2.3.1. Redox Imbalance
2.3.2. Gene Array Analysis Indicates Dysregulated Cholesterol Metabolism in GD
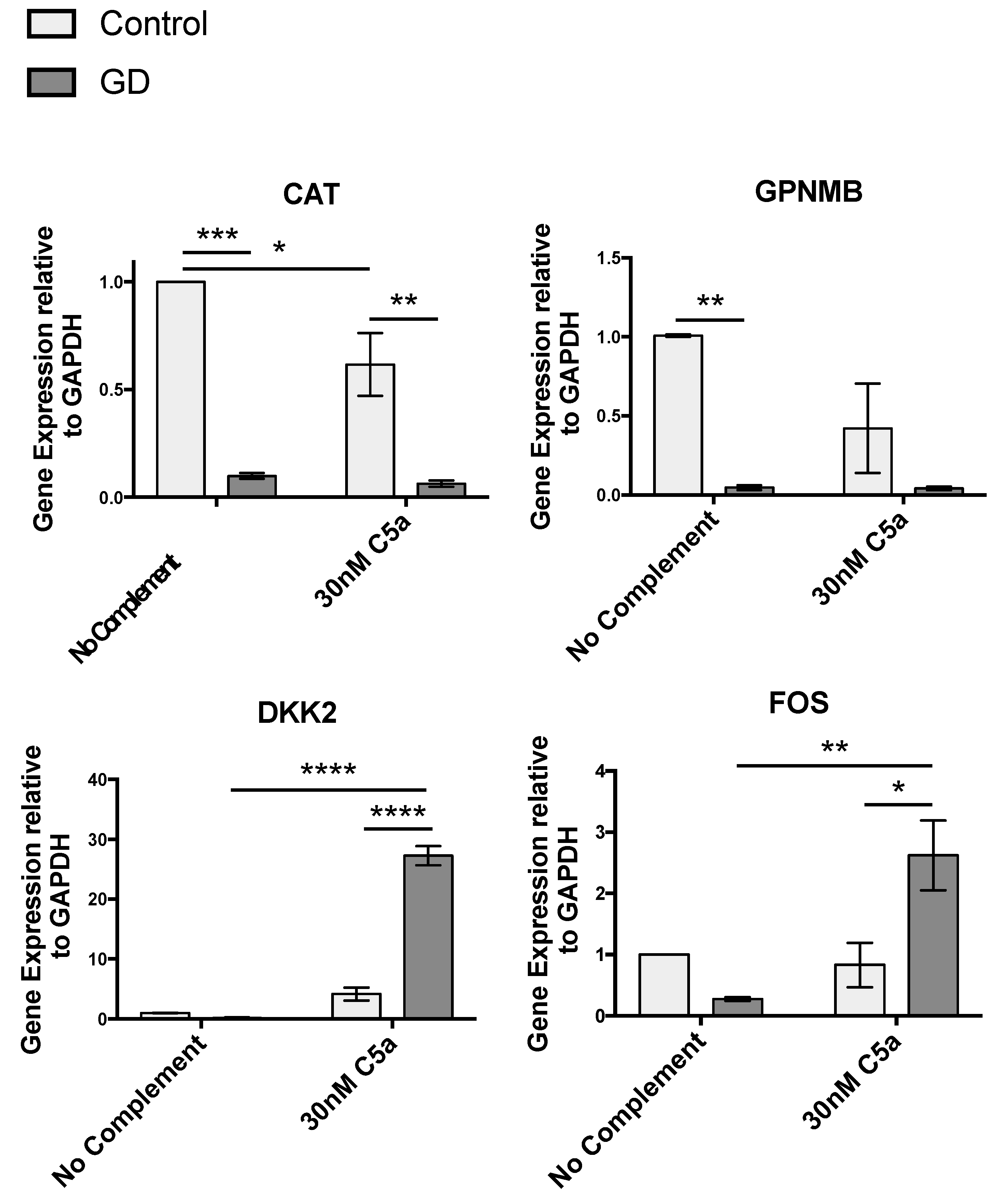
2.3.3. Gene Array Analysis Suggests Potential New Biomarkers for GD
2.4. rC5a-Stimulated GD vs. rC5a-Stimulated Control Macrophages
2.4.1. Pathway Analysis
2.4.2. C5a Treatment of GD Macrophages Upregulates Genes Involved in Hepatic Fibrosis
2.4.3. Inflammatory Gene Expression Profile Induced by rC5a in GD Macrophages
2.4.4. Wnt Signaling
3. Materials and Methods
3.1. iPSC Lines and Directed Differentiation to Macrophages
3.2. Flow Cytometry
3.3. Measurement of Cytokine Expression by qRT-PCR
3.4. Measurement of Cytokine Secretion by ELISA
3.5. Microarray Analysis
3.6. Phagocytosis Assays
3.6.1. Fcγ-Mediated Phagocytosis
3.6.2. Complement Receptor-Mediated Phagocytosis
3.7. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- National Human Genome Research Institute. About Gaucher Disease. Available online: http://www.genome.gov/25521505 (accessed on 4 April 2014).
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef]
- Sheth, J.; Bhavsar, R.; Mistri, M.; Pancholi, D.; Bavdekar, A.; Dalal, A.; Ranganath, P.; Girisha, K.M.; Shukla, A.; Phadke, S.; et al. Gaucher disease: Single gene molecular characterization of one-hundred Indian patients reveals novel variants and the most prevalent mutation. BMC Med. Genet. 2019, 20, 31. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D. The Human Gene Mutation Database (HGMD). Hum. Genet. 2020. [Google Scholar] [CrossRef]
- Mignot, C.; Gelot, A.; De Villemeur, T.B. Gaucher disease. Handb. Clin. Neurol. 2013, 113, 1709–1715. [Google Scholar]
- Mucci, J.M.; Rozenfeld, P. Pathogenesis of Bone Alterations in Gaucher Disease: The Role of Immune System. J. Immunol. Res. 2015, 2015, 192761. [Google Scholar] [CrossRef] [Green Version]
- Burstein, Y.; Rechavi, G.; Rausen, A.R.; Frisch, B.; Spirer, Z. Association of Gaucher’s disease and lymphoid malignancy in 2 children. Scand. J. Haematol. 1985, 35, 445–447. [Google Scholar] [CrossRef]
- Hawkesford, M.P.; Bowey, A.J.; Rao, J.; Meara, N.J. Synchronous presentation of Gaucher disease and solitary plasmacytoma with progression to multiple myeloma. Scott. Med. J. 2011, 56, 236. [Google Scholar] [CrossRef] [PubMed]
- de Fost, M.; Vom Dahl, S.; Weverling, G.J.; Brill, N.; Brett, S.; Haussinger, D.; Hollak, C.E. Increased incidence of cancer in adult Gaucher disease in Western Europe. Blood Cells Mol. Dis. 2006, 36, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Shoenfeld, Y.; Gallant, L.A.; Shaklai, M.; Livni, E.; Djaldetti, M.; Pinkhas, J. Gaucher’s disease: A disease with chronic stimulation of the immune system. Arch. Pathol. Lab. Med. 1982, 106, 388–391. [Google Scholar] [PubMed]
- Rosenbloom, B.E.; Weinreb, N.J.; Zimran, A.; Kacena, K.A.; Charrow, J.; Ward, E. Gaucher disease and cancer incidence: A study from the Gaucher Registry. Blood 2005, 105, 4569–4572. [Google Scholar] [CrossRef]
- Akiyama, T.; Sato, S.; Ko, S.B.H.; Sano, O.; Sato, S.; Saito, M.; Nagai, H.; Ko, M.S.H.; Iwata, H. Synthetic mRNA-based differentiation method enables early detection of Parkinson’s phenotypes in neurons derived from Gaucher disease-induced pluripotent stem cells. Stem Cells Transl. Med. 2021, 10, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, M.P.; Jones, J.W.; Kane, M.; Awad, O.; Park, T.S.; Zambidis, E.T.; Feldman, R.A. Elevated glucosylsphingosine in Gaucher disease induced pluripotent stem cell neurons deregulates lysosomal compartment through mammalian target of rapamycin complex 1. Stem Cells Transl. Med. 2021, 10, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, B.E.; Weinreb, N.J. Gaucher disease: A comprehensive review. Crit. Rev. Oncog. 2013, 18, 163–175. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine: National Institutes of Health. Genetics Home Reference: Gaucher Disease. Available online: http://ghr.nlm.nih.gov/condition/gaucher-disease (accessed on 18 August 2020).
- Panicker, L.M.; Miller, D.; Awad, O.; Bose, V.; Lun, Y.; Park, T.S.; Zambidis, E.T.; Sgambato, J.A.; Feldman, R.A. Gaucher iPSC-derived macrophages produce elevated levels of inflammatory mediators and serve as a new platform for therapeutic development. Stem Cells 2014, 32, 2338–2349. [Google Scholar] [CrossRef] [Green Version]
- Deganuto, M.; Pittis, M.G.; Pines, A.; Dominissini, S.; Kelley, M.R.; Garcia, R.; Quadrifoglio, F.; Bembi, B.; Tell, G. Altered intracellular redox status in Gaucher disease fibroblasts and impairment of adaptive response against oxidative stress. J. Cell. Physiol. 2007, 212, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Lugowska, A.; Hetmanczyk-Sawicka, K.; Iwanicka-Nowicka, R.; Fogtman, A.; Ciesla, J.; Purzycka-Olewiecka, J.K.; Sitarska, D.; Ploski, R.; Filocamo, M.; Lualdi, S.; et al. Gene expression profile in patients with Gaucher disease indicates activation of inflammatory processes. Sci. Rep. 2019, 9, 6060. [Google Scholar] [CrossRef] [Green Version]
- Van Rossum, A.; Holsopple, M. Enzyme Replacement or Substrate Reduction? A Review of Gaucher Disease Treatment Options. Hosp. Pharm. 2016, 51, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Mistry, P.K.; Taddei, T.; vom Dahl, S.; Rosenbloom, B.E. Gaucher disease and malignancy: A model for cancer pathogenesis in an inborn error of metabolism. Crit. Rev. Oncog. 2013, 18, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Pandey, M.K.; Burrow, T.A.; Rani, R.; Martin, L.J.; Witte, D.; Setchell, K.D.; McKay, M.A.; Magnusen, A.F.; Zhang, W.; Liou, B.; et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature 2017, 543, 108–112. [Google Scholar] [CrossRef]
- Dubot, P.; Astudillo, L.; Therville, N.; Sabourdy, F.; Stirnemann, J.; Levade, T.; Andrieu-Abadie, N. Are Glucosylceramide-Related Sphingolipids Involved in the Increased Risk for Cancer in Gaucher Disease Patients? Review and Hypotheses. Cancers 2020, 12, 475. [Google Scholar] [CrossRef] [Green Version]
- Limgala, R.P.; Goker-Alpan, O. Effect of Substrate Reduction Therapy in Comparison to Enzyme Replacement Therapy on Immune Aspects and Bone Involvement in Gaucher Disease. Biomolecules 2020, 10, 526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regenboog, M.; van Dussen, L.; Verheij, J.; Weinreb, N.J.; Santosa, D.; Vom Dahl, S.; Haussinger, D.; Muller, M.N.; Canbay, A.; Rigoldi, M.; et al. Hepatocellular carcinoma in Gaucher disease: An international case series. J. Inherit. Metab. Dis. 2018, 41, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Taddei, T.H.; Kacena, K.A.; Yang, M.; Yang, R.; Malhotra, A.; Boxer, M.; Aleck, K.A.; Rennert, G.; Pastores, G.M.; Mistry, P.K. The underrecognized progressive nature of N370S Gaucher disease and assessment of cancer risk in 403 patients. Am. J. Hematol. 2009, 84, 208–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starosta, R.T.; Vairo, F.P.E.; Dornelles, A.D.; Basgalupp, S.P.; Siebert, M.; Pedroso, M.L.A.; Cerski, C.T.S.; Alvares-da-Silva, M.R.; Schwartz, I.V.D. Liver involvement in patients with Gaucher disease types I and III. Mol. Genet. Metab. Rep. 2020, 22, 100564. [Google Scholar] [CrossRef]
- Horiuchi, T.; Tsukamoto, H. Complement-targeted therapy: Development of C5- and C5a-targeted inhibition. Inflamm. Regen. 2016, 36, 11. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.; Sng, J.; Boddupalli, C.S.; Seckinger, A.; Chesi, M.; Fulciniti, M.; Zhang, L.; Rauniyar, N.; Lopez, M.; Neparidze, N.; et al. Antigen-mediated regulation in monoclonal gammopathies and myeloma. JCI Insight 2018, 3, e98259. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.; Branagan, A.R.; Liu, J.; Boddupalli, C.S.; Mistry, P.K.; Dhodapkar, M.V. Clonal Immunoglobulin against Lysolipids in the Origin of Myeloma. N. Engl. J. Med. 2016, 374, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Ilan, Y. Beta-Glycosphingolipids as Mediators of Both Inflammation and Immune Tolerance: A Manifestation of Randomness in Biological Systems. Front. Immunol. 2019, 10, 1143. [Google Scholar] [CrossRef] [Green Version]
- van Eijk, M.; Ferraz, M.J.; Boot, R.G.; Aerts, J. Lyso-glycosphingolipids: Presence and consequences. Essays Biochem. 2020, 64, 565–578. [Google Scholar] [PubMed]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowan, G.; Weston-Bell, N.J.; Bryant, D.; Seckinger, A.; Hose, D.; Zojer, N.; Sahota, S.S. Massive parallel IGHV gene sequencing reveals a germinal center pathway in origins of human multiple myeloma. Oncotarget 2015, 6, 13229–13240. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, P.A. Oxy-radicals and cancer. Lancet 1994, 344, 862–863. [Google Scholar] [CrossRef]
- Weitzman, S.A.; Gordon, L.I. Inflammation and cancer: Role of phagocyte-generated oxidants in carcinogenesis. Blood 1990, 76, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Imamura, R.; Nitta, H.; Taniguchi, K.; Saito, F.; Kikuchi, K.; Ogi, H.; Tanaka, T.; Katabuchi, H.; Nakayama, H.; et al. Enhancement of cancer invasion and growth via the C5a-C5a receptor system: Implications for cancer promotion by autoimmune diseases and association with cervical cancer invasion. Oncol. Lett. 2019, 17, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Medler, T.R.; Murugan, D.; Horton, W.; Kumar, S.; Cotechini, T.; Forsyth, A.M.; Leyshock, P.; Leitenberger, J.J.; Kulesz-Martin, M.; Margolin, A.A.; et al. Complement C5a Fosters Squamous Carcinogenesis and Limits T Cell Response to Chemotherapy. Cancer Cell 2018, 34, 561–578.e566. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Liu, Q.; Li, T.; Liao, Q.; Zhao, Y. Role of the complement system in the tumor microenvironment. Cancer Cell Int. 2019, 19, 300. [Google Scholar] [CrossRef]
- Mizukami, H.; Mi, Y.; Wada, R.; Kono, M.; Yamashita, T.; Liu, Y.; Werth, N.; Sandhoff, R.; Sandhoff, K.; Proia, R.L. Systemic inflammation in glucocerebrosidase-deficient mice with minimal glucosylceramide storage. J. Clin. Investig. 2002, 109, 1215–1221. [Google Scholar] [CrossRef]
- Stricklett, P.K.; Hughes, A.K.; Kohan, D.E. Inhibition of p38 mitogen-activated protein kinase ameliorates cytokine up-regulated shigatoxin-1 toxicity in human brain microvascular endothelial cells. J. Infect. Dis. 2005, 191, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Karasu, E.; Demmelmaier, J.; Kellermann, S.; Holzmann, K.; Kohl, J.; Schmidt, C.Q.; Kalbitz, M.; Gebhard, F.; Huber-Lang, M.S.; Halbgebauer, R. Complement C5a Induces Pro-inflammatory Microvesicle Shedding in Severely Injured Patients. Front. Immunol. 2020, 11, 1789. [Google Scholar] [CrossRef] [PubMed]
- Vardi, A.; Ben-Dor, S.; Cho, S.M.; Kalinke, U.; Spanier, J.; Futerman, A.H. Mice defective in interferon signaling help distinguish between primary and secondary pathological pathways in a mouse model of neuronal forms of Gaucher disease. J. Neuroinflamm. 2020, 17, 265. [Google Scholar] [CrossRef]
- Kartha, R.V.; Terluk, M.R.; Brown, R.; Travis, A.; Mishra, U.R.; Rudser, K.; Lau, H.; Jarnes, J.R.; Cloyd, J.C.; Weinreb, N.J. Patients with Gaucher disease display systemic oxidative stress dependent on therapy status. Mol. Genet. Metab. Rep. 2020, 25, 100667. [Google Scholar] [CrossRef] [PubMed]
- de la Mata, M.; Cotan, D.; Oropesa-Avila, M.; Garrido-Maraver, J.; Cordero, M.D.; Villanueva Paz, M.; Delgado Pavon, A.; Alcocer-Gomez, E.; de Lavera, I.; Ybot-Gonzalez, P.; et al. Pharmacological Chaperones and Coenzyme Q10 Treatment Improves Mutant beta-Glucocerebrosidase Activity and Mitochondrial Function in Neuronopathic Forms of Gaucher Disease. Sci. Rep. 2015, 5, 10903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [CrossRef] [Green Version]
- Kirkham, P. Oxidative stress and macrophage function: A failure to resolve the inflammatory response. Biochem. Soc. Trans. 2007, 35, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, R.H.; Wight, D.G.; Lomas, D.J.; Fisher, N.C.; Schofield, J.P.; Elias, E.; Cox, T.M. Massive hepatic fibrosis in Gaucher’s disease: Clinico-pathological and radiological features. QJM 2000, 93, 237–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohte, A.E.; van Dussen, L.; Akkerman, E.M.; Nederveen, A.J.; Sinkus, R.; Jansen, P.L.; Stoker, J.; Hollak, C.E. Liver fibrosis in type I Gaucher disease: Magnetic resonance imaging, transient elastography and parameters of iron storage. PLoS ONE 2013, 8, e57507. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Pascual, F.; Busnadiego, O.; Gonzalez-Santamaria, J. The profibrotic role of endothelin-1: Is the door still open for the treatment of fibrotic diseases? Life Sci. 2014, 118, 156–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thieringer, F.; Maass, T.; Czochra, P.; Klopcic, B.; Conrad, I.; Friebe, D.; Schirmacher, P.; Lohse, A.W.; Blessing, M.; Galle, P.R.; et al. Spontaneous hepatic fibrosis in transgenic mice overexpressing PDGF-A. Gene 2008, 423, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chou, X.; Guan, F.; Fang, Z.; Lu, S.; Lei, J.; Li, Y.; Liu, W. Enhanced Wnt Signalling in Hepatocytes is Associated with Schistosoma japonicum Infection and Contributes to Liver Fibrosis. Sci. Rep. 2017, 7, 230. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell. Mol. Immunol. 2021, 18, 45–56. [Google Scholar] [CrossRef]
- Lu, J.W.; Liao, C.Y.; Yang, W.Y.; Lin, Y.M.; Jin, S.L.; Wang, H.D.; Yuh, C.H. Overexpression of endothelin 1 triggers hepatocarcinogenesis in zebrafish and promotes cell proliferation and migration through the AKT pathway. PLoS ONE 2014, 9, e85318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parameswaran, N.; Patial, S. Tumor necrosis factor-alpha signaling in macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef]
- Goel, R.R.; Wang, X.; O’Neil, L.J.; Nakabo, S.; Hasneen, K.; Gupta, S.; Wigerblad, G.; Blanco, L.P.; Kopp, J.B.; Morasso, M.I.; et al. Interferon lambda promotes immune dysregulation and tissue inflammation in TLR7-induced lupus. Proc. Natl. Acad. Sci. USA 2020, 117, 5409–5419. [Google Scholar] [CrossRef]
- Zaiss, D.M.W.; Gause, W.C.; Osborne, L.C.; Artis, D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 2015, 42, 216–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platen, C.; Dreschers, S.; Wappler, J.; Ludwig, A.; Dusterhoft, S.; Reiss, L.K.; Orlikowsky, T.W. Amphiregulin Regulates Phagocytosis-Induced Cell Death in Monocytes via EGFR and the Bcl-2 Protein Family. Mediat. Inflamm. 2019, 2019, 1603131. [Google Scholar] [CrossRef]
- Spence, J.S.; He, R.; Hoffmann, H.H.; Das, T.; Thinon, E.; Rice, C.M.; Peng, T.; Chandran, K.; Hang, H.C. IFITM3 directly engages and shuttles incoming virus particles to lysosomes. Nat. Chem. Biol. 2019, 15, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Robinson, M.E.; Ma, N.; Artadji, D.; Ahmed, M.A.; Xiao, G.; Sadras, T.; Deb, G.; Winchester, J.; Cosgun, K.N.; et al. IFITM3 functions as a PIP3 scaffold to amplify PI3K signalling in B cells. Nature 2020, 588, 491–497. [Google Scholar] [CrossRef]
- Kitatani, K.; Wada, M.; Perry, D.; Usui, T.; Sun, Y.; Obeid, L.M.; Yaegashi, N.; Grabowski, G.A.; Hannun, Y.A. Activation of p38 Mitogen-Activated Protein Kinase in Gaucher’s Disease. PLoS ONE 2015, 10, e0136633. [Google Scholar] [CrossRef]
- Aflaki, E.; Moaven, N.; Borger, D.K.; Lopez, G.; Westbroek, W.; Chae, J.J.; Marugan, J.; Patnaik, S.; Maniwang, E.; Gonzalez, A.N.; et al. Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell 2016, 15, 77–88. [Google Scholar] [CrossRef]
- Zhu, J.; Jiang, L.; Liu, Y.; Qian, W.; Liu, J.; Zhou, J.; Gao, R.; Xiao, H.; Wang, J. MAPK and NF-kappaB pathways are involved in bisphenol A-induced TNF-alpha and IL-6 production in BV2 microglial cells. Inflammation 2015, 38, 637–648. [Google Scholar] [CrossRef]
- Bernhardi, R.V. Neurodegenerative Diseases–MAPK Signalling Pathways in Neuroinflammation. In Encyclopedia of Neuroscience; Binder, M.D., Hirokawa, N., Windhorst, U., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 2614–2620. [Google Scholar]
- Flood, P.M.; Qian, L.; Peterson, L.J.; Zhang, F.; Shi, J.S.; Gao, H.M.; Hong, J.S. Transcriptional Factor NF-kappaB as a Target for Therapy in Parkinson’s Disease. Parkinson Dis. 2011, 2011, 216298. [Google Scholar]
- Du, Q.; Geller, D.A. Cross-Regulation Between Wnt and NF-kappaB Signaling Pathways. Onco Ther. 2010, 1, 155–181. [Google Scholar]
- Dasgupta, N.; Xu, Y.H.; Li, R.; Peng, Y.; Pandey, M.K.; Tinch, S.L.; Liou, B.; Inskeep, V.; Zhang, W.; Setchell, K.D.; et al. Neuronopathic Gaucher disease: Dysregulated mRNAs and miRNAs in brain pathogenesis and effects of pharmacologic chaperone treatment in a mouse model. Hum. Mol. Genet. 2015, 24, 7031–7048. [Google Scholar] [CrossRef] [PubMed]
- Awad, O.; Panicker, L.M.; Deranieh, R.M.; Srikanth, M.P.; Brown, R.A.; Voit, A.; Peesay, T.; Park, T.S.; Zambidis, E.T.; Feldman, R.A. Altered Differentiation Potential of Gaucher’s Disease iPSC Neuronal Progenitors due to Wnt/beta-Catenin Downregulation. Stem Cell Rep. 2017, 9, 1853–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, R.; Bellesso, S.; Lualdi, S.; Manzoli, R.; Pistorio, V.; Filocamo, M.; Moro, E. A transcriptional and post-transcriptional dysregulation of Dishevelled 1 and 2 underlies the Wnt signaling impairment in type I Gaucher disease experimental models. Hum. Mol. Genet. 2020, 29, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Panicker, L.M.; Miller, D.; Park, T.S.; Patel, B.; Azevedo, J.L.; Awad, O.; Masood, M.A.; Veenstra, T.D.; Goldin, E.; Stubblefield, B.K.; et al. Induced pluripotent stem cell model recapitulates pathologic hallmarks of Gaucher disease. Proc. Natl. Acad. Sci. USA 2012, 109, 18054–18059. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.M.; Zhang, X. Measuring opsonic phagocytosis via Fcgamma receptors and complement receptors on macrophages. Curr. Protoc. Immunol. 2011, 95, 14–27. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serfecz, J.C.; Saadin, A.; Santiago, C.P.; Zhang, Y.; Bentzen, S.M.; Vogel, S.N.; Feldman, R.A. C5a Activates a Pro-Inflammatory Gene Expression Profile in Human Gaucher iPSC-Derived Macrophages. Int. J. Mol. Sci. 2021, 22, 9912. https://doi.org/10.3390/ijms22189912
Serfecz JC, Saadin A, Santiago CP, Zhang Y, Bentzen SM, Vogel SN, Feldman RA. C5a Activates a Pro-Inflammatory Gene Expression Profile in Human Gaucher iPSC-Derived Macrophages. International Journal of Molecular Sciences. 2021; 22(18):9912. https://doi.org/10.3390/ijms22189912
Chicago/Turabian StyleSerfecz, Jacquelyn C., Afsoon Saadin, Clayton P. Santiago, Yuji Zhang, Søren M. Bentzen, Stefanie N. Vogel, and Ricardo A. Feldman. 2021. "C5a Activates a Pro-Inflammatory Gene Expression Profile in Human Gaucher iPSC-Derived Macrophages" International Journal of Molecular Sciences 22, no. 18: 9912. https://doi.org/10.3390/ijms22189912
APA StyleSerfecz, J. C., Saadin, A., Santiago, C. P., Zhang, Y., Bentzen, S. M., Vogel, S. N., & Feldman, R. A. (2021). C5a Activates a Pro-Inflammatory Gene Expression Profile in Human Gaucher iPSC-Derived Macrophages. International Journal of Molecular Sciences, 22(18), 9912. https://doi.org/10.3390/ijms22189912