
Tryptophan: From Diet to Cardiovascular Diseases


{kind=link}
{kind=link}
Abstract
:1. Introduction
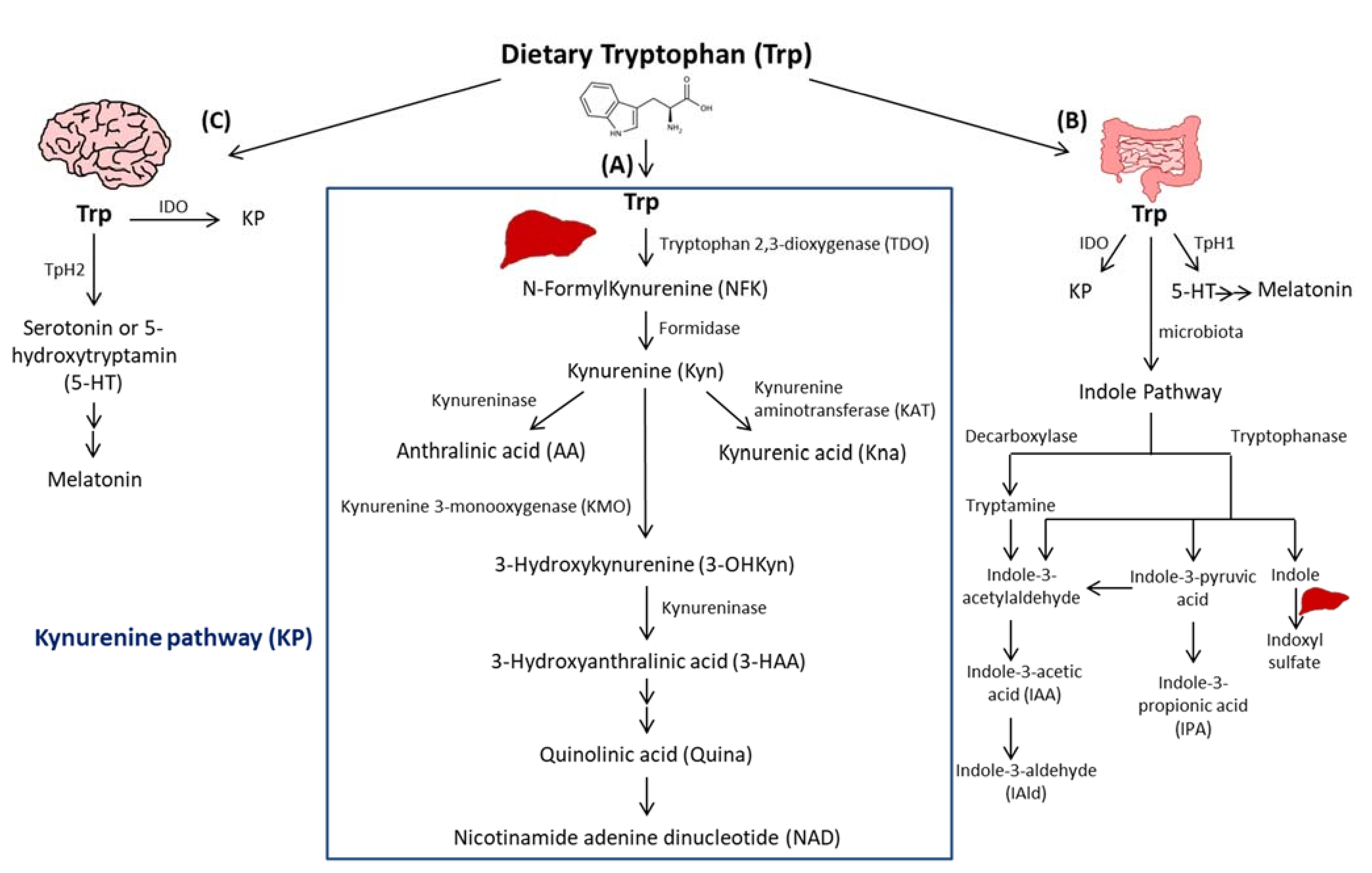
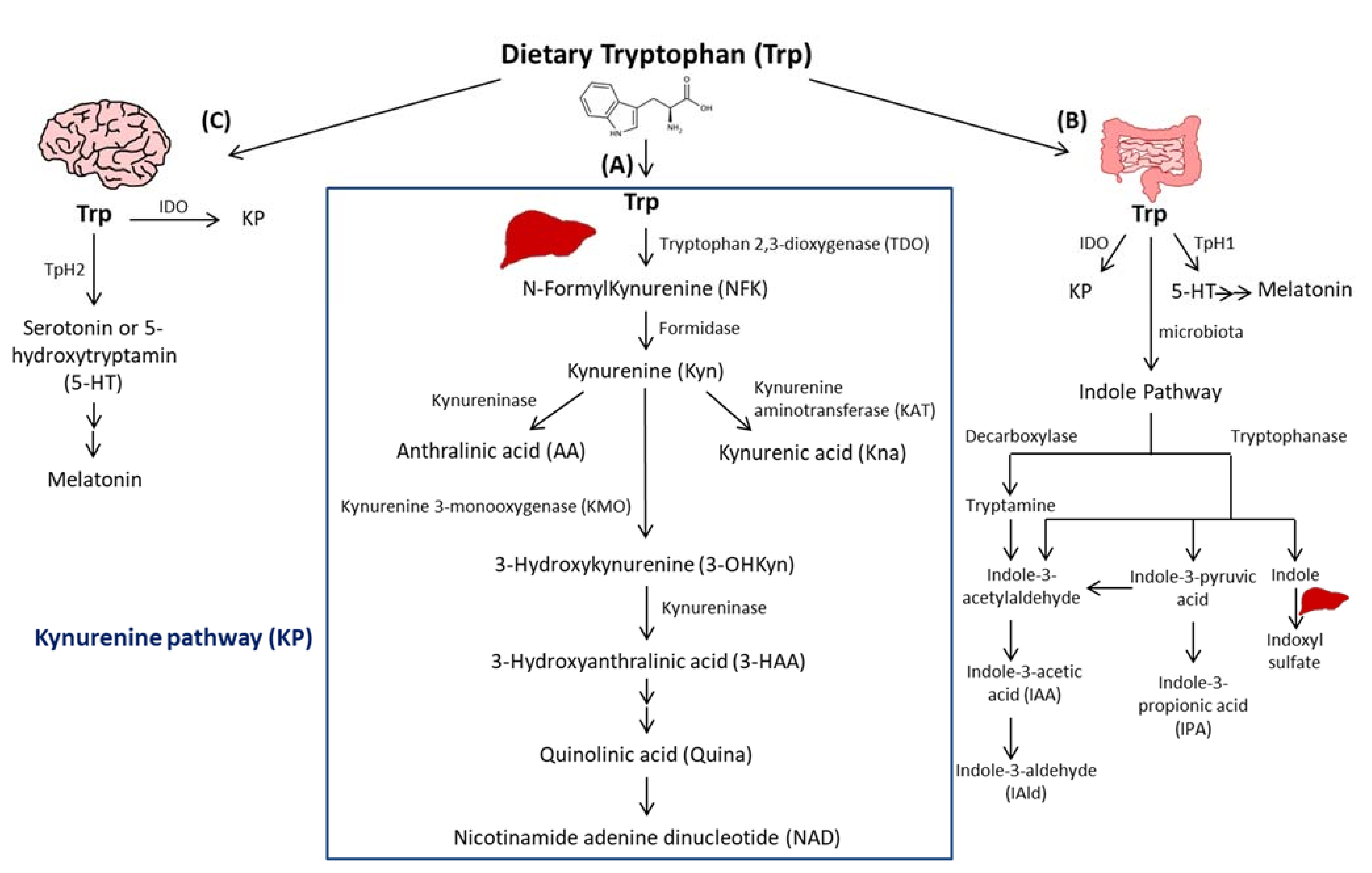
2. Trp Absorption and Catabolism
3. Indoleamine 2, 3-Dioxygenase 1
4. Trp Catabolism in Cardiometabolic Diseases
4.1. Metabolic Syndrome
4.2. Abdominal Aortic Aneurysm
4.3. Atherosclerosis
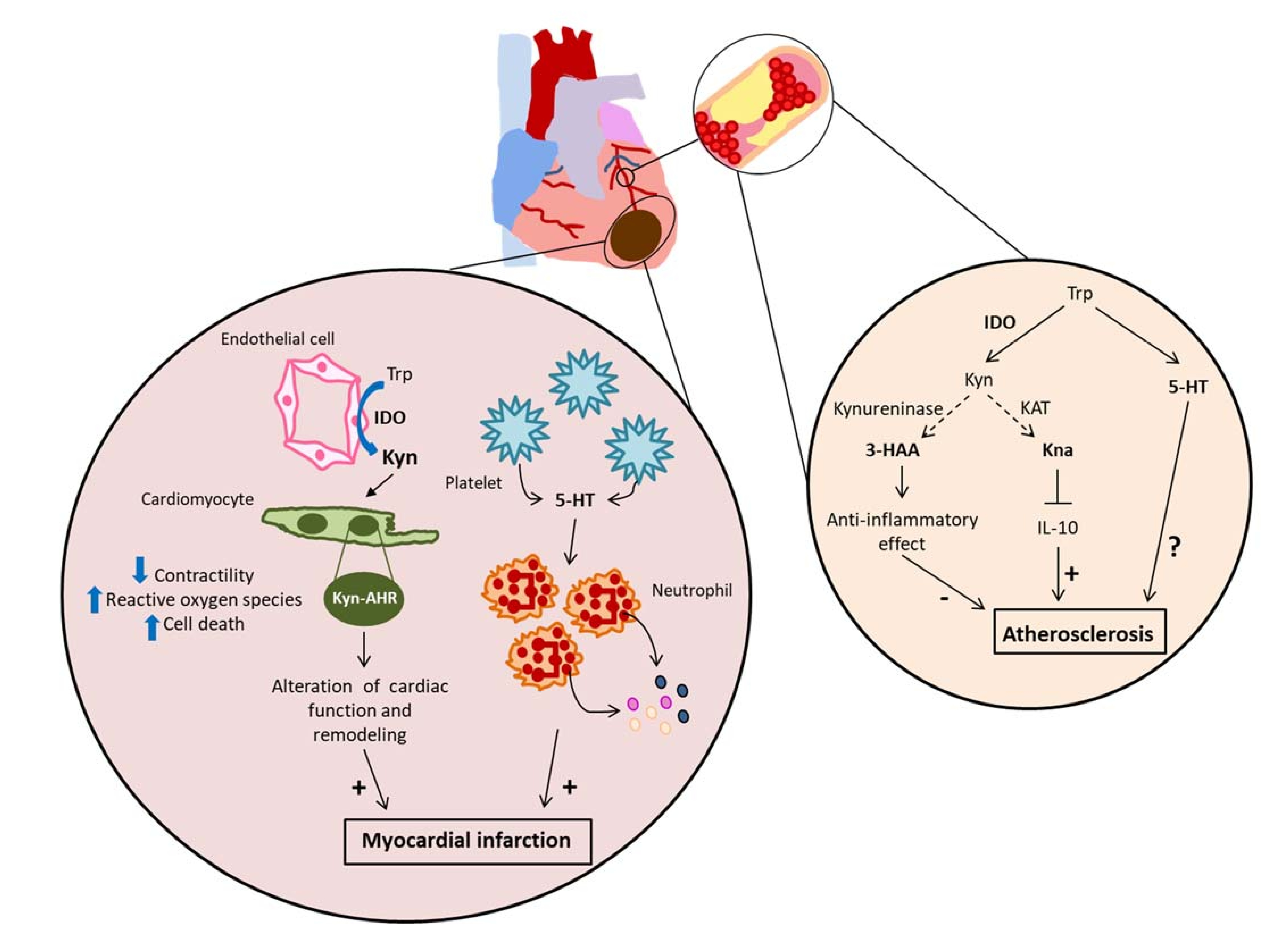
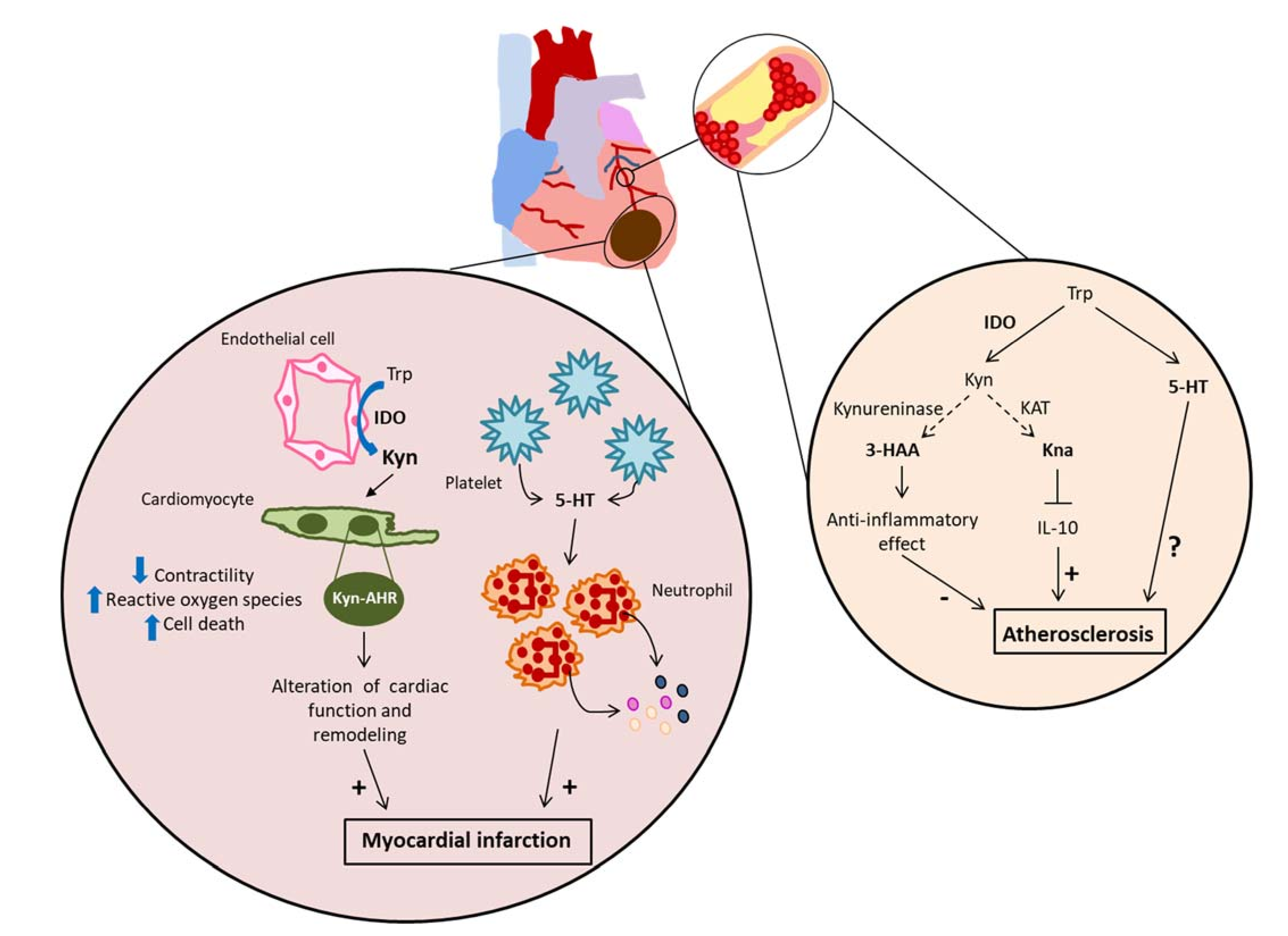
4.4. Acute Myocardial Infarction
5. Therapeutic Approaches
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef]
- Theodorou, K.; Boon, R.A. Endothelial Cell Metabolism in Atherosclerosis. Front. Cell Dev. Biol. 2018, 6, 82. [Google Scholar] [CrossRef] [Green Version]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D. Fourth Universal Definition of Myocardial Infarction (2018). J. Am. Coll. Cardiol. 2018, 72, 2231–2264. [Google Scholar] [CrossRef]
- Ibanez, B.; James, S.; Agewall, S.; Antunes, M.J.; Bucciarelli-Ducci, C.; Bueno, H.; Caforio, A.L.P.; Crea, F.; Goudevenos, J.A.; Halvorsen, S.; et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur. Heart J. 2018, 39, 119–177. [Google Scholar] [CrossRef] [Green Version]
- Ross, R. Atherosclerosis—An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, Atherosclerosis, and Coronary Artery Disease. New Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Inflammation in Atherosclerosis: From Pathophysiology to Practice. J. Am. Coll. Cardiol. 2009, 54, 2129–2138. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Zhenyukh, O.; González-Amor, M.; Díez, R.R.; Esteban, V.; Ruiz-Ortega, M.; Salaices, M.; Mas, S.; Briones, A.M.; Egido, J. Branched-chain amino acids promote endothelial dysfunction through increased reactive oxygen species generation and inflammation. J. Cell. Mol. Med. 2018, 22, 4948–4962. [Google Scholar] [CrossRef]
- Nitz, K.; Lacy, M.; Atzler, D. Amino Acids and Their Metabolism in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2019, 39, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Eussen, S.J.; Ueland, P.M.; Vollset, S.E.; Nygård, O.; Midttun, Ø.; Sulo, G.; Ulvik, A.; Meyer, K.; Pedersen, E.R.; Tell, G.S. Kynurenines as predictors of acute coronary events in the Hordaland Health Study. Int. J. Cardiol. 2015, 189, 18–24. [Google Scholar] [CrossRef]
- Pedersen, E.R.; Svingen, G.F.T.; Schartum-Hansen, H.; Ueland, P.M.; Ebbing, M.; Nordrehaug, J.E.; Igland, J.; Seifert, R.; Nilsen, R.M.; Nygård, O. Urinary excretion of kynurenine and tryptophan, cardiovascular events, and mortality after elective coronary angiography. Eur. Hear. J. 2013, 34, 2689–2696. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, E.R.; Tuseth, N.; Eussen, S.J.; Ueland, P.M.; Strand, E.; Svingen, G.F.T.; Midttun, Ø.; Meyer, K.; Mellgren, G.; Ulvik, A.; et al. Associations of Plasma Kynurenines With Risk of Acute Myocardial Infarction in Patients with Stable Angina Pectoris. Arter. Thromb. Vasc. Biol. 2015, 35, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, E.R.; Midttun, Ø.; Ueland, P.M.; Schartum-Hansen, H.; Seifert, R.; Igland, J.; Nordrehaug, J.E.; Ebbing, M.; Svingen, G.; Bleie, Ø.; et al. Systemic Markers of Interferon-γ–Mediated Immune Activation and Long-Term Prognosis in Patients With Stable Coronary Artery Disease. Arter. Thromb. Vasc. Biol. 2011, 31, 698–704. [Google Scholar] [CrossRef] [Green Version]
- Saadi, M.; Vodovar, N.; Paven, E.; Sadoune, M.; Barnabas, G.; Desrumeaux, G.; Sirol, M.; Mercadier, J.-J.; Launay, J.; Logeart, D. Plasma Indoleamine 2,3-dioxygenase is predictive of left ventricular remodeling after myocardial infarction. Arch. Cardiovasc. Dis. Suppl. 2020, 12, 144. [Google Scholar] [CrossRef]
- Hopkins, F.G.; Cole, S.W. A contribution to the chemistry of proteids. J. Physiol. 1901, 27, 418–428. [Google Scholar] [CrossRef]
- Richard, D.M.; Dawes, M.A.; Mathias, C.; Acheson, A.; Hill-Kapturczak, N.; Dougherty, D.M. L-Tryptophan: Basic Metabolic Functions, Behavioral Research and Therapeutic Indications. Int. J. Tryptophan Res. 2009, 2, IJTR–S2129–60. [Google Scholar] [CrossRef] [Green Version]
- Palego, L.; Betti, L.; Rossi, A.; Giannaccini, G. Tryptophan Biochemistry: Structural, Nutritional, Metabolic, and Medical Aspects in Humans. J. Amino Acids 2016, 2016, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Keszthelyi, D.; Troost, F.; Masclee, A.A.M. Understanding the role of tryptophan and serotonin metabolism in gastrointestinal function. Neurogastroenterol. Motil. 2009, 21, 1239–1249. [Google Scholar] [CrossRef]
- Taleb, S. Tryptophan Dietary Impacts Gut Barrier and Metabolic Diseases. Front. Immunol. 2019, 10, 2113. [Google Scholar] [CrossRef]
- Badawy, A.A.-B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Yu, Y.; Shen, Y.; Liu, Q.; Zhao, Z.; Sharma, R.; Reiter, R.J. Melatonin Synthesis and Function: Evolutionary History in Animals and Plants. Front. Endocrinol. 2019, 10, 249. [Google Scholar] [CrossRef]
- Reigstad, C.S.; Salmonson, C.E.; Rainey, J.F., III; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J. 2014, 29, 1395–1403. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Beaulieu, J.-M.; Sotnikova, T.D.; Gainetdinov, R.; Caron, M.G. Tryptophan Hydroxylase-2 Controls Brain Serotonin Synthesis. Science 2004, 305, 217. [Google Scholar] [CrossRef]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Walther, D.J.; Peter, J.U.; Bashammakh, S.; Hortnagl, H.; Voits, M.; Fink, H.; Bader, M. Synthesis of Serotonin by a Second Tryptophan Hydroxylase Isoform. Science 2003, 299, 76. [Google Scholar] [CrossRef]
- Kulikova, E.A.; Kulikov, A.V. Tryptophan hydroxylase 2 as a therapeutic target for psychiatric disorders: Focus on animal models. Expert Opin. Ther. Targets 2019, 23, 655–667. [Google Scholar] [CrossRef]
- Borghi, M.; Puccetti, M.; Pariano, M.; Renga, G.; Stincardini, C.; Ricci, M.; Giovagnoli, S.; Costantini, C.; Romani, L. Tryptophan as a Central Hub for Host/Microbial Symbiosis. Int. J. Tryptophan Res. 2020, 13, 1178646920919755. [Google Scholar] [CrossRef]
- Choi, S.-C.; Brown, J.; Gong, M.; Ge, Y.; Zadeh, M.; Li, W.; Croker, B.P.; Michailidis, G.; Garrett, T.J.; Mohamadzadeh, M.; et al. Gut microbiota dysbiosis and altered tryptophan catabolism contribute to autoimmunity in lupus-susceptible mice. Sci. Transl. Med. 2020, 12, eaax2220. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [Green Version]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.J.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry 2013, 18, 666–673. [Google Scholar] [CrossRef] [Green Version]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N. The role of endogenous aryl hydrocarbon receptor signaling in cardiovascular physiology. J. Cardiovasc. Dis. Res. 2011, 2, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.-P.; Michel, M.-L.; Costa, G.D.A.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.; et al. Symbiotic Bacterial Metabolites Regulate Gastrointestinal Barrier Function via the Xenobiotic Sensor PXR and Toll-like Receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef] [Green Version]
- Laurans, L.; Venteclef, N.; Haddad, Y.; Chajadine, M.; Alzaid, F.; Metghalchi, S.; Sovran, B.; Denis, R.; Dairou, J.; Cardellini, M.; et al. Genetic deficiency of indoleamine 2,3-dioxygenase promotes gut microbiota-mediated metabolic health. Nat. Med. 2018, 24, 1113–1120. [Google Scholar] [CrossRef]
- Lamas, B.; Hernandez-Galan, L.; Galipeau, H.J.; Constante, M.; Clarizio, A.; Jury, J.; Breyner, N.M.; Caminero, A.; Rueda, G.; Hayes, C.L.; et al. Aryl hydrocarbon receptor ligand production by the gut microbiota is decreased in celiac disease leading to intestinal inflammation. Sci. Transl. Med. 2020, 12, eaba0624. [Google Scholar] [CrossRef]
- Kazemian, N.; Mahmoudi, M.; Halperin, F.; Wu, J.C.; Pakpour, S. Gut microbiota and cardiovascular disease: Opportunities and challenges. Microbiome 2020, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Cainzos-Achirica, M.; Glassner, K.; Zawahir, H.S.; Dey, A.K.; Agrawal, T.; Quigley, E.M.; Abraham, B.P.; Acquah, I.; Yahya, T.; Mehta, N.N.; et al. Inflammatory Bowel Disease and Atherosclerotic Cardiovascular Disease. J. Am. Coll. Cardiol. 2020, 76, 2895–2905. [Google Scholar] [CrossRef]
- Konopelski, P.; Ufnal, M. Indoles—Gut Bacteria Metabolites of Tryptophan with Pharmacotherapeutic Potential. Curr. Drug Metab. 2018, 19, 883–890. [Google Scholar] [CrossRef]
- Chen, W.-J.; Lai, Y.-J.; Lee, J.-L.; Wu, S.-T.; Hsu, Y.-J. CREB/ATF3 signaling mediates indoxyl sulfate-induced vascular smooth muscle cell proliferation and neointimal formation in uremia. Atherosclerosis 2020, 315, 43–54. [Google Scholar] [CrossRef]
- Wlodarska, M.; Luo, C.; Kolde, R.; D’Hennezel, E.; Annand, J.W.; Heim, C.E.; Krastel, P.; Schmitt, E.K.; Omar, A.S.; Creasey, E.A.; et al. Indoleacrylic Acid Produced by Commensal Peptostreptococcus Species Suppresses Inflammation. Cell Host Microbe 2017, 22, 25–37.e6. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, K.; Hayaishi, O. Enzymic formation of d-kynurenine from d-tryptophan. Arch. Biochem. Biophys. 1967, 120, 397–403. [Google Scholar] [CrossRef]
- Yamamoto, S.; Hayaishi, O. Tryptophan pyrrolase of rabbit intestine. D- and L-tryptophan-cleaving enzyme or enzymes. J. Biol. Chem. 1967, 242, 5260–5266. [Google Scholar]
- Dai, X.; Zhu, B.T. Indoleamine 2,3-Dioxygenase Tissue Distribution and Cellular Localization in Mice: Implications for Its Biological Functions. J. Histochem. Cytochem. 2009, 58, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Yeung, A.W.; Terentis, A.; King, N.J.C.; Thomas, S.R. Role of indoleamine 2,3-dioxygenase in health and disease. Clin. Sci. 2015, 129, 601–672. [Google Scholar] [CrossRef]
- Pfefferkorn, E.R. Interferon gamma blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cells to degrade tryptophan. Proc. Natl. Acad. Sci. USA 1984, 81, 908–912. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Zhou, M.; Attwood, J.T.; Bondarev, I.; Conway, S.J.; Marshall, B.; Brown, C.; Mellor, A.L. Prevention of Allogeneic Fetal Rejection by Tryptophan Catabolism. Science 1998, 281, 1191–1193. [Google Scholar] [CrossRef]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T Cell Proliferation by Macrophage Tryptophan Catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef]
- Mellor, A.L.; Munn, D.H. Ido expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef]
- Chon, S.Y.; Hassanain, H.H.; Gupta, S.L. Cooperative Role of Interferon Regulatory Factor 1 and p91 (STAT1) Response Elements in Interferon-γ-inducible Expression of Human Indoleamine 2,3-Dioxygenase Gene. J. Biol. Chem. 1996, 271, 17247–17252. [Google Scholar] [CrossRef] [Green Version]
- Schwarcz, R.; Köhler, C. Differential vulnerability of central neurons of the rat to quinolinic acid. Neurosci. Lett. 1983, 38, 85–90. [Google Scholar] [CrossRef]
- Gheorghe, C.E.; Martin, J.; Manriquez, F.V.; Dinan, T.G.; Cryan, J.F.; Clarke, G. Focus on the essentials: Tryptophan metabolism and the microbiome-gut-brain axis. Curr. Opin. Pharmacol. 2019, 48, 137–145. [Google Scholar] [CrossRef]
- Ting, K.K.; Brew, B.J.; Guillemin, G.J. Effect of quinolinic acid on human astrocytes morphology and functions: Implications in Alzheimer’s disease. J. Neuroinflammation 2009, 6, 36. [Google Scholar] [CrossRef] [Green Version]
- Zwilling, D.; Huang, S.-Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Guidetti, P.; Wu, H.-Q.; Lee, J.; Truong, J.; Andrews-Zwilling, Y.; Hsieh, E.W.; et al. Kynurenine 3-Monooxygenase Inhibition in Blood Ameliorates Neurodegeneration. Cell 2011, 145, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic Acid Is a Potent Endogenous Aryl Hydrocarbon Receptor Ligand that Synergistically Induces Interleukin-6 in the Presence of Inflammatory Signaling. Toxicol. Sci. 2010, 115, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, L.V.; Neyens, D.; Ramsay, G.; Taylor, P.M.; Cantrell, R.A. Single cell analysis of kynurenine and System L amino acid transport in T cells. Nat. Commun. 2018, 9, 1981. [Google Scholar] [CrossRef]
- Seok, S.-H.; Ma, Z.-X.; Feltenberger, J.B.; Chen, H.; Chen, H.; Scarlett, C.; Lin, Z.; Satyshur, K.; Cortopassi, M.; Jefcoate, C.R.; et al. Trace derivatives of kynurenine potently activate the aryl hydrocarbon receptor (AHR). J. Biol. Chem. 2018, 293, 1994–2005. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Harden, J.L.; Anderson, C.D.; Egilmez, N.K.; Anderson, C. Tolerogenic Phenotype of IFN-γ–Induced IDO+ Dendritic Cells Is Maintained via an Autocrine IDO–Kynurenine/AhR–IDO Loop. J. Immunol. 2016, 197, 962–970. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013, 34, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [Green Version]
- Pallotta, M.T.; Orabona, C.; Volpi, C.; Vacca, C.; Belladonna, M.L.; Bianchi, R.; Servillo, G.; Brunacci, C.; Calvitti, M.; Bicciato, S.; et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat. Immunol. 2011, 12, 870–878. [Google Scholar] [CrossRef] [Green Version]
- Lob, S.; Königsrainer, A. Role of IDO in Organ Transplantation: Promises and Difficulties. Int. Rev. Immunol. 2009, 28, 185–206. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, H.; McKenzie, G.; Witting, P.K.; Stasch, J.-P.; Hahn, M.; Changsirivathanathamrong, D.; Wu, B.; Ball, H.; Thomas, S.R.; et al. Kynurenine is an endothelium-derived relaxing factor produced during inflammation. Nat. Med. 2010, 16, 279–285. [Google Scholar] [CrossRef] [Green Version]
- Löb, S.; Königsrainer, A.; Rammensee, H.-G.; Opelz, G.; Terness, P. Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: Can we see the wood for the trees? Nat. Rev. Cancer 2009, 9, 445–452. [Google Scholar] [CrossRef]
- Barth, M.C.; Ahluwalia, N.; Anderson, T.; Hardy, G.J.; Sinha, S.; Alvarez-Cardona, J.A.; Pruitt, I.E.; Rhee, E.P.; Colvin, R.A.; Gerszten, R.E. Kynurenic Acid Triggers Firm Arrest of Leukocytes to Vascular Endothelium under Flow Conditions. J. Biol. Chem. 2009, 284, 19189–19195. [Google Scholar] [CrossRef] [Green Version]
- Romani, L.; Fallarino, F.; DE Luca, A.; Montagnoli, C.; D’Angelo, C.; Zelante, T.; Vacca, C.; Bistoni, F.; Fioretti, M.C.; Grohmann, U.; et al. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nat. Cell Biol. 2008, 451, 211–215. [Google Scholar] [CrossRef]
- Mo, X.; Pi, L.; Yang, J.; Xiang, Z.; Tang, A. Serum indoleamine 2,3-dioxygenase and kynurenine aminotransferase enzyme activity in patients with ischemic stroke. J. Clin. Neurosci. 2014, 21, 482–486. [Google Scholar] [CrossRef]
- Koo, Y.S.; Kim, H.; Park, J.H.; Kim, M.J.; Shin, Y.-I.; Choi, B.T.; Lee, S.-Y.; Shin, H.K. Indoleamine 2,3-Dioxygenase-Dependent Neurotoxic Kynurenine Metabolism Contributes to Poststroke Depression Induced in Mice by Ischemic Stroke along with Spatial Restraint Stress. Oxidative Med. Cell. Longev. 2018, 2018, 2413841. [Google Scholar] [CrossRef]
- Wang, Q.; Ding, Y.; Song, P.; Zhu, H.; Okon, I.; Ding, Y.-N.; Chen, H.-Z.; Liu, D.-P.; Zou, M.-H.; Ding, N.-Y. Tryptophan-Derived 3-Hydroxyanthranilic Acid Contributes to Angiotensin II-Induced Abdominal Aortic Aneurysm Formation in Mice In Vivo. Circulation 2017, 136, 2271–2283. [Google Scholar] [CrossRef]
- Metghalchi, S.; Vandestienne, M.; Haddad, Y.; Esposito, B.; Dairou, J.; Tedgui, A.; Mallat, Z.; Potteaux, S.; Taleb, S. Indoleamine 2 3-dioxygenase knockout limits angiotensin II-induced aneurysm in low density lipoprotein receptor-deficient mice fed with high fat diet. PLoS ONE 2018, 13, e0193737. [Google Scholar] [CrossRef] [Green Version]
- Melhem, N.J.; Chajadine, M.; Gomez, I.; Howangyin, K.-Y.; Bouvet, M.; Knosp, C.; Sun, Y.; Rouanet, M.; Laurans, L.; Cazorla, O.; et al. Endothelial Cell Indoleamine 2, 3-Dioxygenase 1 Alters Cardiac Function After Myocardial Infarction Through Kynurenine. Circulation 2021, 143, 566–580. [Google Scholar] [CrossRef]
- Nakajima, K.; Yamashita, T.; Kita, T.; Takeda, M.; Sasaki, N.; Kasahara, K.; Shinohara, M.; Rikitake, Y.; Ishida, T.; Yokoyama, M.; et al. Orally Administered Eicosapentaenoic Acid Induces Rapid Regression of Atherosclerosis Via Modulating the Phenotype of Dendritic Cells in LDL Receptor-Deficient Mice. Arter. Thromb. Vasc. Biol. 2011, 31, 1963–1972. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, K.; Ovchinnikova, O.; Berg, M.; Baumgartner, R.; Agardh, H.; Pirault, J.; Gisterå, A.; Assinger, A.; Fernandez, A.L.; Bäck, M.; et al. Inhibition of indoleamine 2,3-dioxygenase promotes vascular inflammation and increases atherosclerosis in Apoe−/− mice. Cardiovasc. Res. 2015, 106, 295–302. [Google Scholar] [CrossRef]
- Cole, J.; Astola, N.; Cribbs, A.; Goddard, M.E.; Park, I.; Green, P.; Davies, A.H.; Williams, R.O.; Feldmann, M.; Monaco, C. Indoleamine 2,3-dioxygenase-1 is protective in atherosclerosis and its metabolites provide new opportunities for drug development. Proc. Natl. Acad. Sci. USA 2015, 112, 13033–13038. [Google Scholar] [CrossRef] [Green Version]
- Metghalchi, S.; Ponnuswamy, P.; Simon, T.; Haddad, Y.; Laurans, L.; Clement, M.; Dalloz, M.; Romain, M.; Esposito, B.; Koropoulis, V.; et al. Indoleamine 2,3-Dioxygenase Fine-Tunes Immune Homeostasis in Atherosclerosis and Colitis through Repression of Interleukin-10 Production. Cell Metab. 2015, 22, 460–471. [Google Scholar] [CrossRef] [Green Version]
- Brandacher, G.; Winkler, C.; Aigner, F.; Schwelberger, H.; Schroecksnadel, K.; Margreiter, R.; Fuchs, D.; Weiss, H.G. Bariatric Surgery Cannot Prevent Tryptophan Depletion Due to Chronic Immune Activation in Morbidly Obese Patients. Obes. Surg. 2006, 16, 541–548. [Google Scholar] [CrossRef]
- Wolowczuk, I.; Hennart, B.; Leloire, A.; Bessede, A.; Soichot, M.; Taront, S.; Caiazzo, R.; Raverdy, V.; Pigeyre, M.; Guillemin, G.J.; et al. Tryptophan metabolism activation by indoleamine 2,3-dioxygenase in adipose tissue of obese women: An attempt to maintain immune homeostasis and vascular tone. Am. J. Physiol. Integr. Comp. Physiol. 2012, 303, R135–R143. [Google Scholar] [CrossRef] [Green Version]
- Mangge, H.; Stelzer, I.; Reininghaus, E.; Weghuber, D.; Postolache, T.; Fuchs, D. Disturbed Tryptophan Metabolism in Cardiovascular Disease. Curr. Med. Chem. 2014, 21, 1931–1937. [Google Scholar] [CrossRef]
- Favennec, M.; Hennart, B.; Caiazzo, R.; Leloire, A.; Yengo, L.; Verbanck, M.; Arredouani, A.; Marre, M.; Pigeyre, M.; Bessede, A.; et al. The kynurenine pathway is activated in human obesity and shifted toward kynurenine monooxygenase activation. Obesity 2015, 23, 2066–2074. [Google Scholar] [CrossRef]
- Natividad, J.M.; Agus, A.; Planchais, J.; Lamas, B.; Jarry, A.C.; Martin, R.; Michel, M.-L.; Chong-Nguyen, C.; Roussel, R.; Straube, M.; et al. Impaired Aryl Hydrocarbon Receptor Ligand Production by the Gut Microbiota Is a Key Factor in Metabolic Syndrome. Cell Metab. 2018, 28, 737–749.e4. [Google Scholar] [CrossRef] [Green Version]
- Chimerel, C.; Emery, E.; Summers, D.K.; Keyser, U.; Gribble, F.; Reimann, F. Bacterial Metabolite Indole Modulates Incretin Secretion from Intestinal Enteroendocrine L Cells. Cell Rep. 2014, 9, 1202–1208. [Google Scholar] [CrossRef] [Green Version]
- Beaumont, M.; Neyrinck, A.; Olivares, M.; Rodriguez, J.; Serra, A.D.R.; Roumain, M.; Bindels, L.B.; Cani, P.D.; Evenepoel, P.; Muccioli, G.G.; et al. The gut microbiota metabolite indole alleviates liver inflammation in mice. FASEB J. 2018, 32, 6681–6693. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic Acid as a Ligand for Orphan G Protein-coupled Receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef] [Green Version]
- Agudelo, L.Z.; Ferreira, D.M.S.; Cervenka, I.; Bryzgalova, G.; Dadvar, S.; Jannig, P.R.; Pettersson-Klein, A.; Lakshmikanth, T.; Sustarsic, E.G.; Porsmyr-Palmertz, M.; et al. Kynurenic Acid and Gpr35 Regulate Adipose Tissue Energy Homeostasis and Inflammation. Cell Metab. 2018, 27, 378–392.e5. [Google Scholar] [CrossRef] [Green Version]
- Crane, J.; Palanivel, R.; Mottillo, E.P.; Bujak, A.L.; Wang, H.; Ford, R.J.; Collins, A.; Blümer, R.M.; Fullerton, M.D.; Yabut, J.M.; et al. Inhibiting peripheral serotonin synthesis reduces obesity and metabolic dysfunction by promoting brown adipose tissue thermogenesis. Nat. Med. 2015, 21, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Nordon, I.M.; Hinchliffe, R.J.; Loftus, I.M.; Thompson, M.M. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat. Rev. Cardiol. 2010, 8, 92–102. [Google Scholar] [CrossRef]
- Hellenthal, F.A.M.V.I.; Buurman, W.A.; Wodzig, W.K.W.H.; Schurink, G.W.H. Biomarkers of abdominal aortic aneurysm progression. Part 2: Inflammation. Nat. Rev. Cardiol. 2009, 6, 543–552. [Google Scholar] [CrossRef]
- Sakalihasan, N.; Limet, R.; Defawe, O. Abdominal aortic aneurysm. Lancet 2005, 365, 1577–1589. [Google Scholar] [CrossRef]
- Thomas, S.; Mohr, D.; Stocker, R. Nitric oxide inhibits indoleamine 2,3-dioxygenase activity in interferon-gamma primed mononuclear phagocytes. J. Biol. Chem. 1994, 269, 14457–14464. [Google Scholar] [CrossRef]
- Gold, A.B.; Herrmann, N.; Swardfager, W.; Black, S.E.; Aviv, R.I.; Tennen, G.; Kiss, A.; Lanctôt, K.L. The relationship between indoleamine 2,3-dioxygenase activity and post-stroke cognitive impairment. J. Neuroinflamm. 2011, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, M.; Yamashita, A.; Matsuura, Y.; Okutsu, J.; Fukahori, A.; Hirata, T.; Nishizawa, T.; Ishii, H.; Maekawa, K.; Nakamura, E.; et al. Upregulated Kynurenine Pathway Enzymes in Aortic Atherosclerotic Aneurysm: Macrophage Kynureninase Downregulates Inflammation. J. Atheroscler. Thromb. 2020, 58248. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, M.; Ding, Y.; Wang, Q.; Zhang, W.; Song, P.; Zou, M.-H. Activation of NAD(P)H oxidase by tryptophan-derived 3-hydroxykynurenine accelerates endothelial apoptosis and dysfunction in vivo. Circ. Res. 2014, 114, 480–492. [Google Scholar] [CrossRef] [Green Version]
- Taleb, S. Inflammation in atherosclerosis. Arch. Cardiovasc. Dis. 2016, 109, 708–715. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Daissormont, I.T.M.N.; Christ, A.; Temmerman, L.; Millares, S.S.; Seijkens, T.; Manca, M.; Rousch, M.; Poggi, M.; Boon, L.; Van Der Loos, C.; et al. Plasmacytoid Dendritic Cells Protect Against Atherosclerosis by Tuning T-Cell Proliferation and Activity. Circ. Res. 2011, 109, 1387–1395. [Google Scholar] [CrossRef]
- Yun, T.J.; Lee, J.S.; Machmach, K.; Shim, D.; Choi, J.; Wi, Y.J.; Jang, H.S.; Jung, I.-H.; Kim, K.; Yoon, W.K.; et al. Indoleamine 2,3-Dioxygenase-Expressing Aortic Plasmacytoid Dendritic Cells Protect against Atherosclerosis by Induction of Regulatory T Cells. Cell Metab. 2016, 23, 852–866. [Google Scholar] [CrossRef]
- MacRitchie, N.; Grassia, G.; Sabir, S.R.; Maddaluno, M.; Welsh, P.; Sattar, N.; Ialenti, A.; Kurowska-Stolarska, M.; McInnes, I.B.; Brewer, J.; et al. Plasmacytoid Dendritic Cells Play a Key Role in Promoting Atherosclerosis in Apolipoprotein E–Deficient Mice. Arter. Thromb. Vasc. Biol. 2012, 32, 2569–2579. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Ovchinnikova, O.; Jönsson, A.; Lundberg, A.M.; Berg, M.; Hansson, G.K.; Ketelhuth, D.F. The tryptophan metabolite 3-hydroxyanthranilic acid lowers plasma lipids and decreases atherosclerosis in hypercholesterolaemic mice. Eur. Hear. J. 2012, 33, 2025–2034. [Google Scholar] [CrossRef]
- Jung, I.D.; Lee, M.-G.; Chang, J.H.; Lee, J.S.; Jeong, Y.-I.; Lee, C.-M.; Park, W.S.; Han, J.; Seo, S.-K.; Lee, S.Y.; et al. Blockade of Indoleamine 2,3-Dioxygenase Protects Mice against Lipopolysaccharide-Induced Endotoxin Shock. J. Immunol. 2009, 182, 3146–3154. [Google Scholar] [CrossRef] [Green Version]
- Shinde, R.; Shimoda, M.; Chaudhary, K.; Liu, H.; Mohamed, E.; Bradley, J.; Kandala, S.; Li, X.; Liu, K.; McGaha, T.L. B Cell–Intrinsic IDO1 Regulates Humoral Immunity to T Cell–Independent Antigens. J. Immunol. 2015, 195, 2374–2382. [Google Scholar] [CrossRef]
- Von Bubnoff, D.; Scheler, M.; Wilms, H.; Fimmers, R.; Bieber, T. Identification of IDO-Positive and IDO-Negative Human Dendritic Cells after Activation by Various Proinflammatory Stimuli. J. Immunol. 2011, 186, 6701–6709. [Google Scholar] [CrossRef] [Green Version]
- Van de Velde, L.-A.; Gingras, S.; Pelletier, S.; Murray, P.J. Issues with the Specificity of Immunological Reagents for Murine IDO1. Cell Metab. 2016, 23, 389–390. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.S.; Gensollen, T.; Gandhi, A.; Oh, S.F.; das Neves, J.F.P.; Collin, F.; Lavin, R.; Serra, C.; Glickman, J.; de Silva, P.S.; et al. Dietary and Microbial Oxazoles Induce Intestinal Inflammation by Modulating Aryl Hydrocarbon Receptor Responses. Cell 2018, 173, 1123–1134.e11. [Google Scholar] [CrossRef] [Green Version]
- Baumgartner, R.; Berg, M.; Matic, L.; Polyzos, K.P.; Forteza, M.J.; Hjorth, S.A.; Schwartz, T.W.; Paulsson-Berne, G.; Hansson, G.K.; Hedin, U.; et al. Evidence that a deviation in the kynurenine pathway aggravates atherosclerotic disease in humans. J. Intern. Med. 2021, 289, 53–68. [Google Scholar] [CrossRef]
- Baumgartner, R.; Casagrande, F.; Mikkelsen, R.; Berg, M.; Polyzos, K.; Forteza, M.; Arora, A.; Schwartz, T.; Hjorth, S.; Ketelhuth, D. Disruption of GPR35 Signaling in Bone Marrow-Derived Cells Does Not Influence Vascular Inflammation and Atherosclerosis in Hyperlipidemic Mice. Metabolites. 2021, 11, 411. [Google Scholar] [CrossRef]
- Rami, M.; Guillamat-Prats, R.; Rinne, P.; Salvermoser, M.; Ring, L.; Bianchini, M.; Blanchet, X.; Megens, R.T.; Döring, Y.; Walzog, B.; et al. Chronic Intake of the Selective Serotonin Reuptake Inhibitor Fluoxetine Enhances Atherosclerosis. Arter. Thromb. Vasc. Biol. 2018, 38, 1007–1019. [Google Scholar] [CrossRef] [Green Version]
- Vikenes, K.; Farstad, M.; Nordrehaug, J.E. Serotonin Is Associated with Coronary Artery Disease and Cardiac Events. Circulation 1999, 100, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, T.; Dohi, Y.; Yamashita, S.; Hirowatari, Y.; Fujii, S.; Ohte, N. Serotonin in peripheral blood reflects oxidative stress and plays a crucial role in atherosclerosis: Novel insights toward holistic anti-atherothrombotic strategy. Atherosclerosis 2016, 246, 157–160. [Google Scholar] [CrossRef]
- Fu, Z.; Jiao, Y.; Wang, J.; Zhang, Y.; Shen, M.; Reiter, R.J.; Xi, Q.; Chen, Y. Cardioprotective Role of Melatonin in Acute Myocardial Infarction. Front. Physiol. 2020, 11, 366. [Google Scholar] [CrossRef]
- Hu, Z.-P.; Fang, X.-L.; Fang, N.; Wang, X.-B.; Qian, H.-Y.; Cao, Z.; Cheng, Y.; Wang, B.-N.; Wang, Y. Melatonin ameliorates vascular endothelial dysfunction, inflammation, and atherosclerosis by suppressing the TLR4/NF-κB system in high-fat-fed rabbits. J. Pineal Res. 2013, 55, 388–398. [Google Scholar] [CrossRef]
- Ding, S.; Lin, N.; Sheng, X.; Zhao, Y.; Su, Y.; Xu, L.; Tong, R.; Yan, Y.; Fu, Y.; He, J.; et al. Melatonin stabilizes rupture-prone vulnerable plaques via regulating macrophage polarization in a nuclear circadian receptor RORα-dependent manner. J. Pineal Res. 2019, 67, e12581. [Google Scholar] [CrossRef]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Simoons, M.L.; Chaitman, B.R.; White, H.D.; Katus, H.A.; Apple, F.S.; Lindahl, B.; Morrow, D.A.; et al. Third Universal Definition of Myocardial Infarction. J. Am. Coll. Cardiol. 2012, 60, 1581–1598. [Google Scholar] [CrossRef] [Green Version]
- Curley, D.; Plaza, B.L.; Shah, A.M.; Botnar, R. Molecular imaging of cardiac remodelling after myocardial infarction. Basic Res. Cardiol. 2018, 113, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Ezekowitz, J.A.; Kaul, P.; Bakal, J.A.; Armstrong, P.; Welsh, R.C.; McAlister, F.A. Declining In-Hospital Mortality and Increasing Heart Failure Incidence in Elderly Patients With First Myocardial Infarction. J. Am. Coll. Cardiol. 2009, 53, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Naghavi, M.; Allen, C.; Barber, R.M.; Bhutta, Z.A.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; Coates, M.M.; et al. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Kwok, M.K.; Fong, S.S.M.; Schooling, C.M. Indoleamine 2,3-dioxygenase and ischemic heart disease: A Mendelian Randomization study. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; Dos Remedios, C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Perbellini, F.; Watson, S.A.; Bardi, I.; Terracciano, C.M. Heterocellularity and Cellular Cross-Talk in the Cardiovascular System. Front. Cardiovasc. Med. 2018, 5, 143. [Google Scholar] [CrossRef]
- Talman, V.; Kivelä, R. Cardiomyocyte—Endothelial Cell Interactions in Cardiac Remodeling and Regeneration. Front. Cardiovasc. Med. 2018, 5, 101. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.U.; Dimmeler, S. Cellular cross-talks in the diseased and aging heart. J. Mol. Cell. Cardiol. 2020, 138, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Hausenloy, D.J.; Chilian, W.; Crea, F.; Davidson, S.M.; Ferdinandy, P.; Garcia-Dorado, D.; Van Royen, N.; Schulz, R.; Heusch, G. The coronary circulation in acute myocardial ischaemia/reperfusion injury: A target for cardioprotection. Cardiovasc. Res. 2019, 115, 1143–1155. [Google Scholar] [CrossRef]
- Nian, M.; Lee, P.; Khaper, N.; Liu, P. Inflammatory Cytokines and Postmyocardial Infarction Remodeling. Circ. Res. 2004, 94, 1543–1553. [Google Scholar] [CrossRef]
- Mohib, K.; Wang, S.; Guan, Q.; Mellor, A.L.; Sun, H.; Du, C.; Jevnikar, A.M. Indoleamine 2,3-dioxygenase expression promotes renal ischemia-reperfusion injury. Am. J. Physiol. Physiol. 2008, 295, F226–F234. [Google Scholar] [CrossRef] [Green Version]
- Eleftheriadis, T.; Pissas, G.; Golfinopoulos, S.; Liakopoulos, V.; Stefanidis, I. Role of indoleamine 2,3-dioxygenase in ischemia-reperfusion injury of renal tubular epithelial cells. Mol. Med. Rep. 2021, 23, 1–13. [Google Scholar] [CrossRef]
- Duerschmied, D.; Suidan, G.L.; Demers, M.; Herr, N.; Carbo, C.; Brill, A.; Cifuni, S.M.; Mauler, M.; Cicko, S.; Bader, M.; et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood 2013, 121, 1008–1015. [Google Scholar] [CrossRef] [Green Version]
- Sauer, W.H.; Berlin, J.A.; Kimmel, S.E. Selective serotonin reuptake inhibitors and myocardial infarction. Circulation 2001, 104, 1894–1898. [Google Scholar] [CrossRef] [Green Version]
- Sauer, W.H.; Berlin, J.A.; Kimmel, S.E. Effect of Antidepressants and Their Relative Affinity for the Serotonin Transporter on the Risk of Myocardial Infarction. Circulation 2003, 108, 32–36. [Google Scholar] [CrossRef] [Green Version]
- Mauler, M.; Herr, N.; Schoenichen, C.; Witsch, T.; Marchini, T.; Härdtner, C.; Koentges, C.; Kienle, K.; Ollivier, V.; Schell, M.; et al. Platelet Serotonin Aggravates Myocardial Ischemia/Reperfusion Injury via Neutrophil Degranulation. Circulation 2019, 139, 918–931. [Google Scholar] [CrossRef]
- Douros, A.; Dell’Aniello, S.; Dehghan, G.; Boivin, J.-F.; Renoux, C. Degree of serotonin reuptake inhibition of antidepressants and ischemic risk. Neurology 2019, 93, e1010–e1020. [Google Scholar] [CrossRef]
- Snider, J.C.; Riley, L.A.; Mallory, N.T.; Bersi, M.R.; Umbarkar, P.; Gautam, R.; Zhang, Q.; Mahadevan-Jansen, A.; Hatzopoulos, A.K.; Maroteaux, L.; et al. Targeting 5-HT 2B Receptor Signaling Prevents Border Zone Expansion and Improves Microstructural Remodeling After Myocardial Infarction. Circulation 2021, 143, 1317–1330. [Google Scholar] [CrossRef]
- Zhou, H.; Li, D.; Zhu, P.; Hu, S.; Hu, N.; Ma, S.; Zhang, Y.; Han, T.; Ren, J.; Cao, F.; et al. Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARγ/FUNDC1/mitophagy pathways. J. Pineal Res. 2017, 63, e12438. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Xu, J.; Tian, F.; Hu, S.; Chen, Y.; Fu, Z. Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. J. Pineal Res. 2019, 66, e12542. [Google Scholar] [CrossRef]
- Dominguez-Rodriguez, A. The role of melatonin in acute myocardial infarction. Front. Biosci. 2012, 17, 2433–2441. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M. How Common Is Residual Inflammatory Risk? Circ. Res. 2017, 120, 617–619. [Google Scholar] [CrossRef]
- Alfaddagh, A.; Martin, S.S.; Leucker, T.M.; Michos, E.D.; Blaha, M.J.; Lowenstein, C.J.; Jones, S.R.; Toth, P.P. Inflammation and cardiovascular disease: From mechanisms to therapeutics. Am. J. Prev. Cardiol. 2020, 4, 100130. [Google Scholar] [CrossRef]
- Van den Eynde, B.J.; Van Baren, N.; Baurain, J.-F. Is There a Clinical Future for IDO1 Inhibitors After the Failure of Epacadostat in Melanoma? Annu. Rev. Cancer Biol. 2020, 4, 241–256. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Shao, M.; Wu, T. Kynurenine-3-monooxygenase: A new direction for the treatment in different diseases. Food Sci. Nutr. 2020, 8, 711–719. [Google Scholar] [CrossRef]
- Bader, M. Inhibition of serotonin synthesis: A novel therapeutic paradigm. Pharmacol. Ther. 2020, 205, 107423. [Google Scholar] [CrossRef]
- Kostoglou-Athanassiou, I. Therapeutic applications of melatonin. Ther. Adv. Endocrinol. Metab. 2013, 4, 13–24. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melhem, N.J.; Taleb, S. Tryptophan: From Diet to Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 22, 9904. https://doi.org/10.3390/ijms22189904
Melhem NJ, Taleb S. Tryptophan: From Diet to Cardiovascular Diseases. International Journal of Molecular Sciences. 2021; 22(18):9904. https://doi.org/10.3390/ijms22189904
Chicago/Turabian StyleMelhem, Nada Joe, and Soraya Taleb. 2021. "Tryptophan: From Diet to Cardiovascular Diseases" International Journal of Molecular Sciences 22, no. 18: 9904. https://doi.org/10.3390/ijms22189904
APA StyleMelhem, N. J., & Taleb, S. (2021). Tryptophan: From Diet to Cardiovascular Diseases. International Journal of Molecular Sciences, 22(18), 9904. https://doi.org/10.3390/ijms22189904