
Calmodulin Interactions with Voltage-Gated Sodium Channels



Abstract

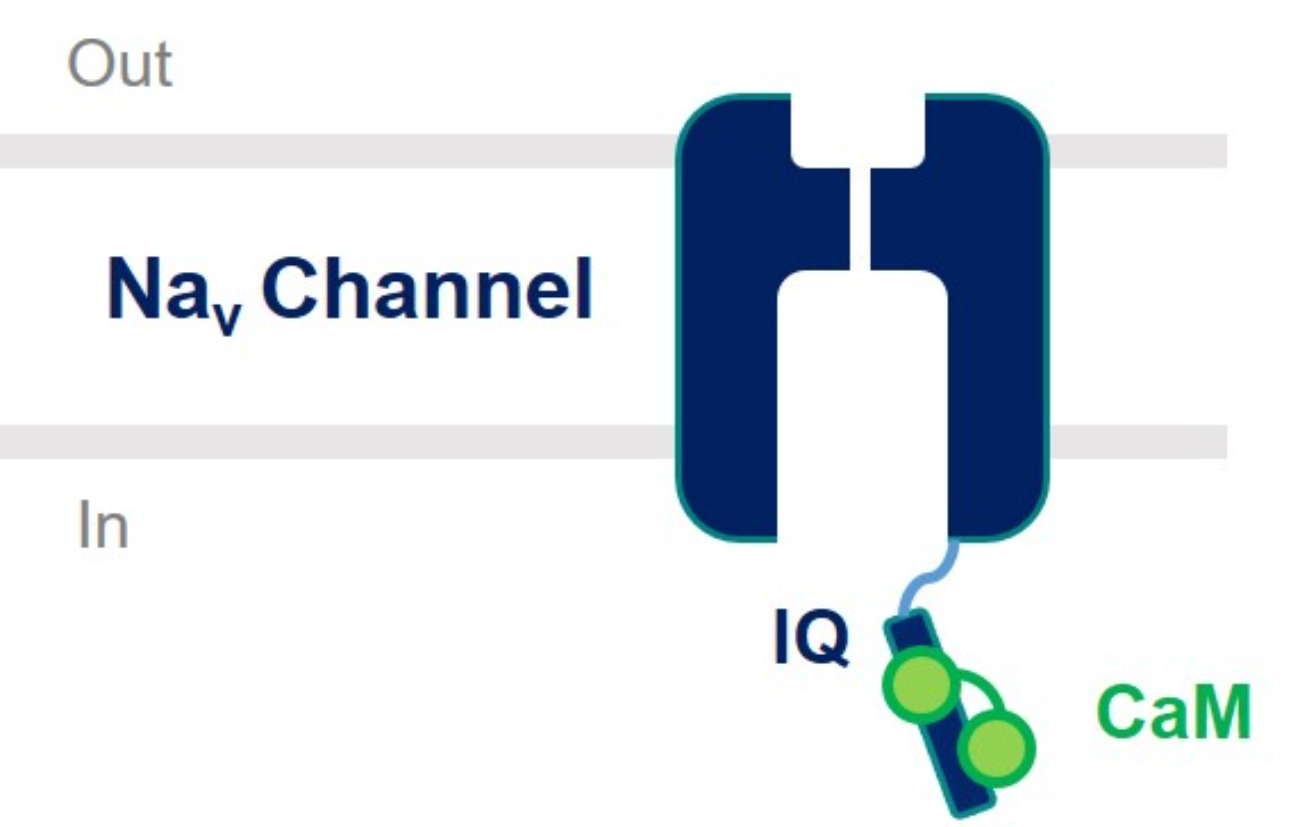
1. Introduction
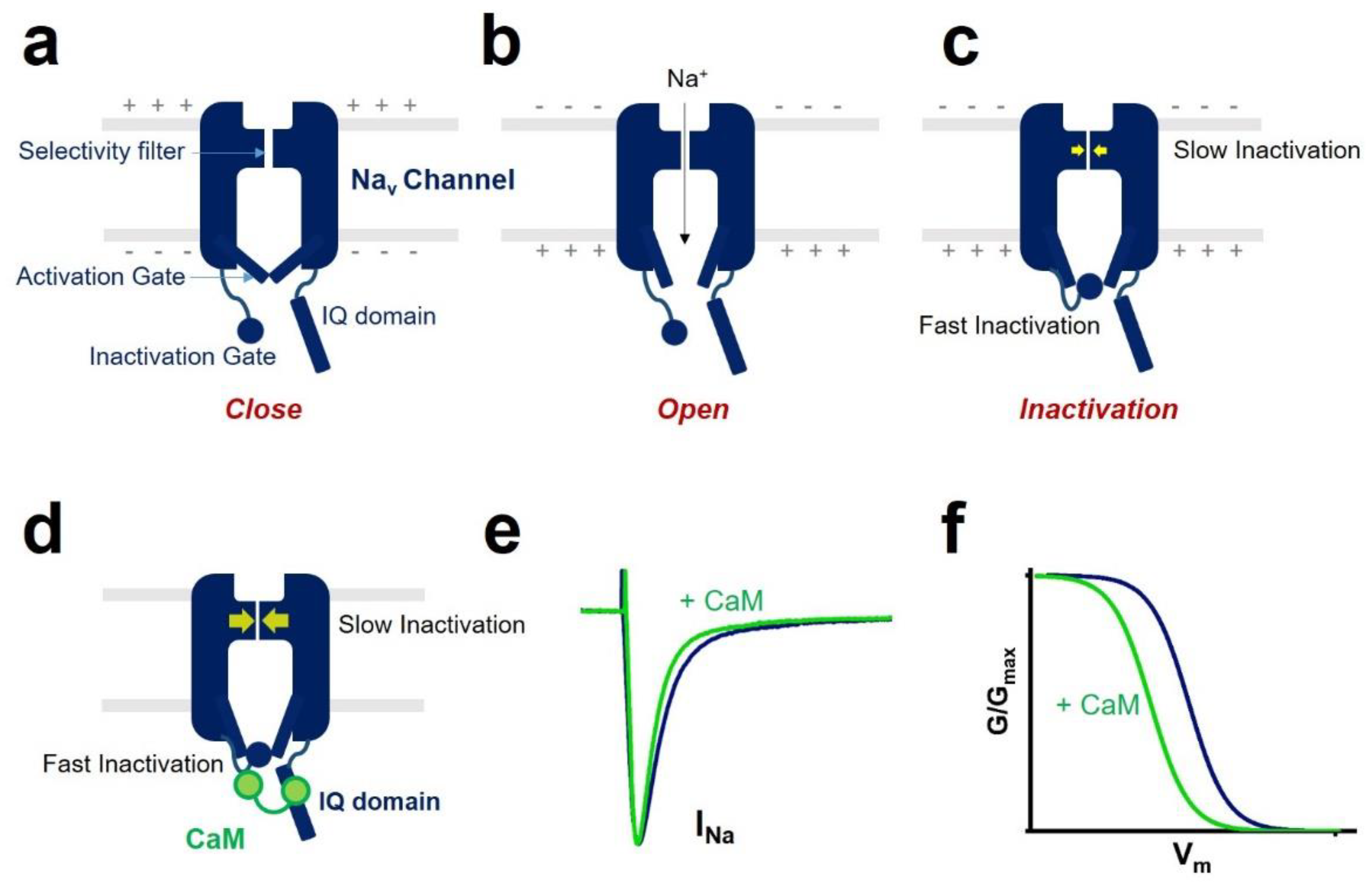
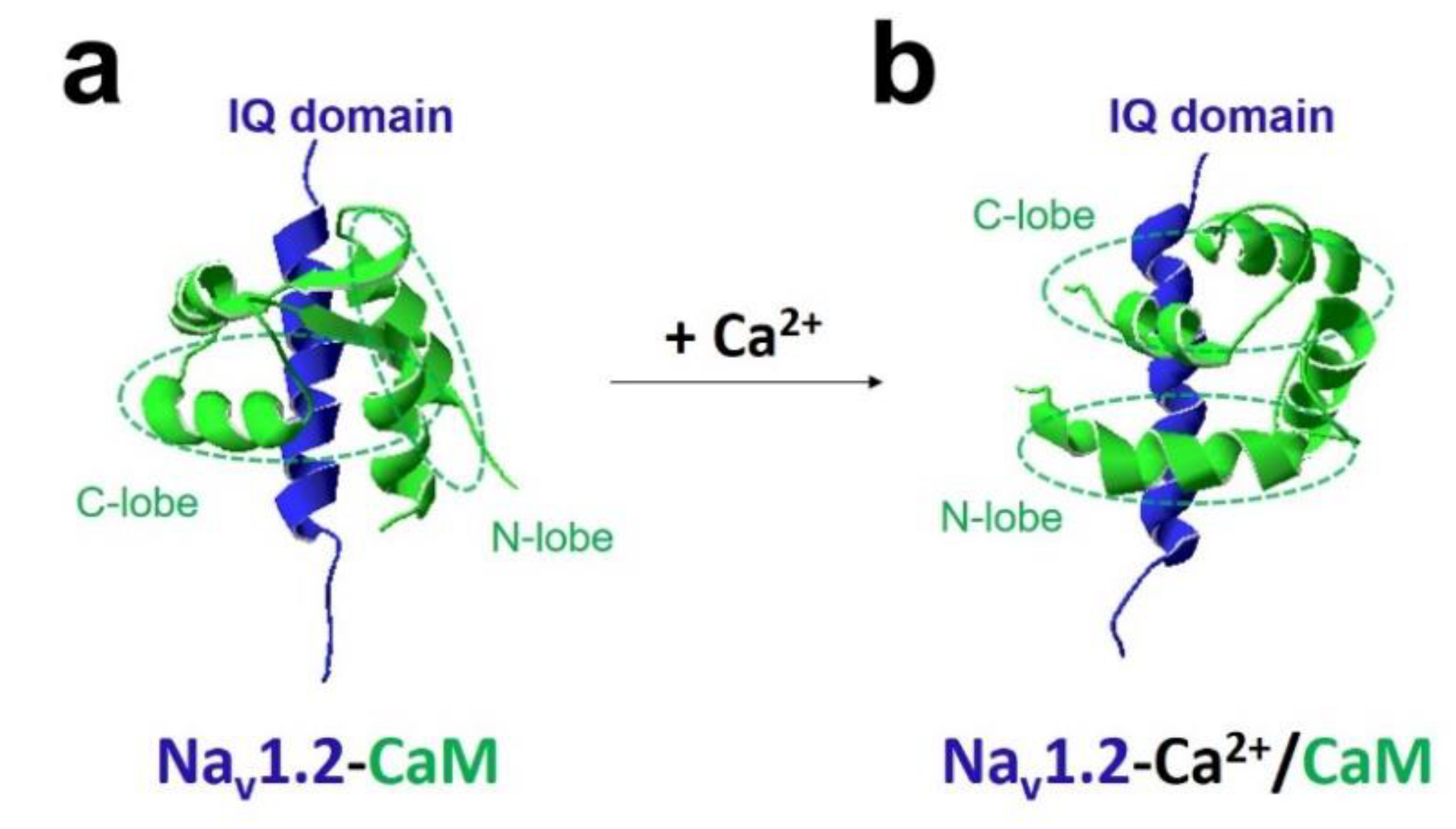
2. CaM Regulation of Voltage-Gated Sodium Channel
3. Modulators Involved in CaM Regulation of Sodium Channel
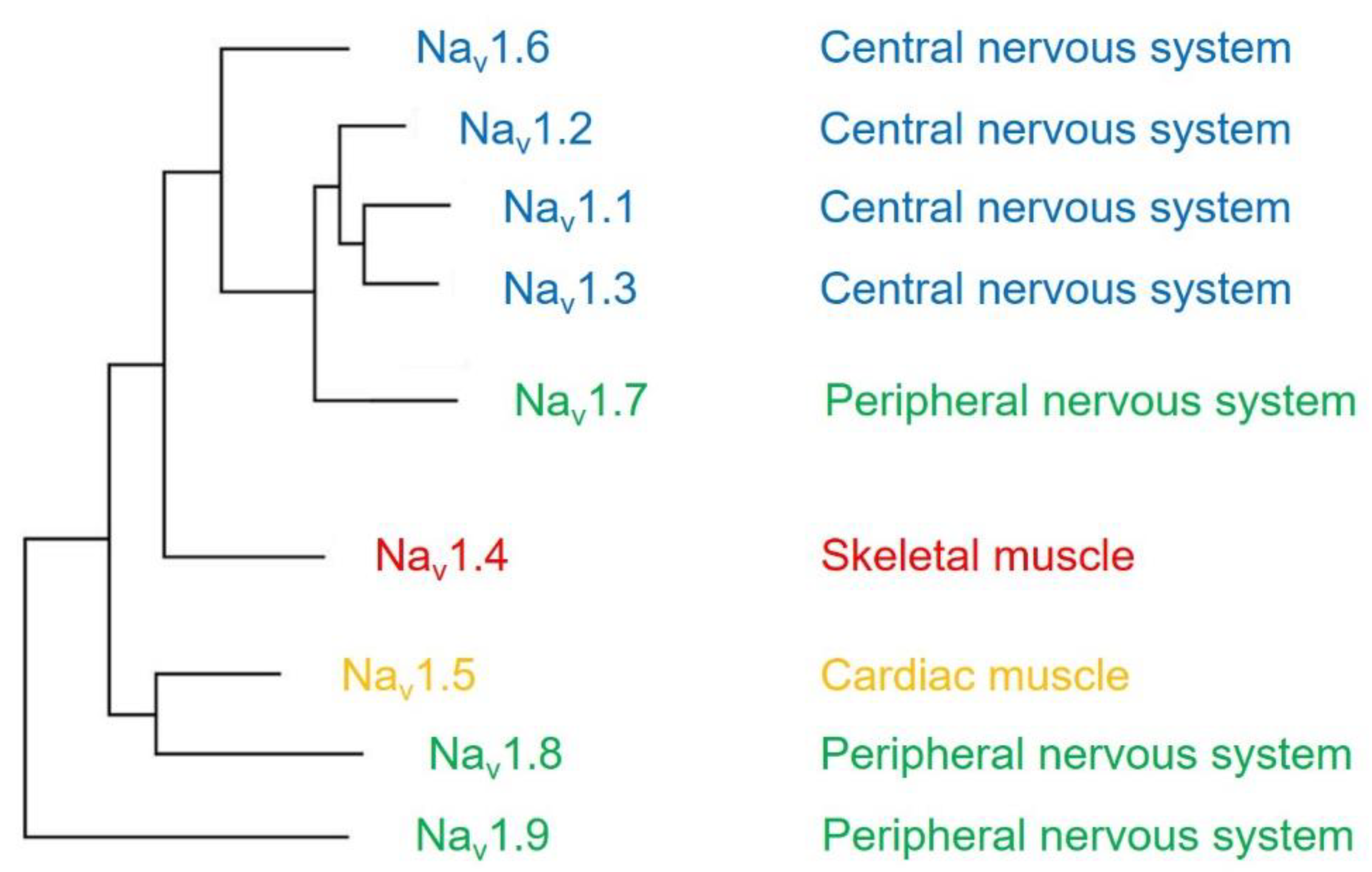
4. Interaction between CaM and Sodium Channel Isoforms
4.1. Nav1.1
4.2. Nav1.2
4.3. Nav1.3
4.4. Nav1.4
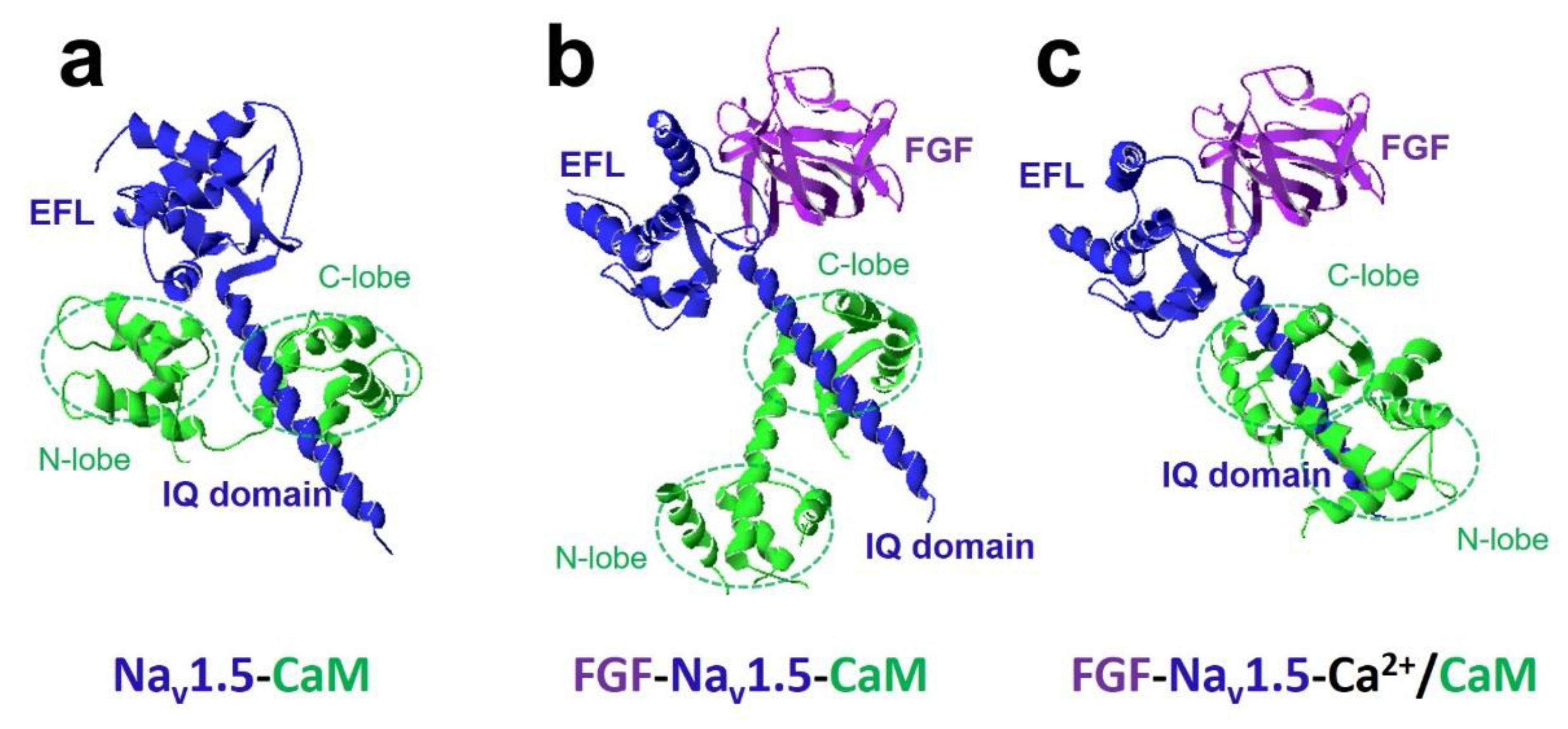
4.5. Nav1.5
4.6. Nav1.6
4.7. Nav1.7
4.8. Nav1.8
4.9. Nav1.9
5. CaM-Binding IQ Domain Mutations in Voltage-Gated Sodium Channel
5.1. Nav1.1 Mutations
5.2. Nav1.2 Mutations
5.3. Nav1.5 Mutations
5.4. Nav1.8 Mutations
6. Conclusions and Perspectives
Funding
Conflicts of Interest
Abbreviations
| Nav | Voltage-gated sodium channel |
| CaM | Calmodulin |
| Apo-CaM | Ca2+-free CaM |
| Ca2+/CaM | Ca2+-binding CaM |
| IQ domain | A domain with highly conserved residues isoleucine (I) and glutamine (Q) |
| CBP | Calmodulin-binding peptide |
| CIP | Calmodulin inhibitory peptide |
| EFL | EF hand-like motifs |
| FHF | Fibroblast growth factor homologous factor |
| FGF | Fibroblast growth factor |
| CaMKII | Calcium/calmodulin-dependent protein kinase II |
| AP | Action potential |
| DRG | Dorsal root ganglia |
| SCG | Superior cervical ganglion |
| NMR | Nuclear magnetic resonance |
| GEFS+ | Generalized epilepsy with febrile seizures plus |
| FHM | Familial hemiplegic migraine |
| SMEI | Severe myoclonic epilepsy of infancy |
| SIDS | Sudden infant death syndrome |
| LQT3 | Type 3 of long-QT syndrome |
References
- Xu, L.; Ding, X.; Wang, T.; Mou, S.; Sun, H.; Hou, T. Voltage-gated sodium channels: Structures, functions, and molecular modeling. Drug Discov. Today 2019, 24, 1389–1397. [Google Scholar] [CrossRef]
- Wood, J.N.; Iseppon, F. Sodium channels. Brain Neurosci. Adv. 2018, 2, 2398212818810684. [Google Scholar] [CrossRef] [PubMed]
- Encinas, A.C.; Watkins, J.C.; Longoria, I.A.; Johnson, J.P.; Hammer, M.F. Variable patterns of mutation density among NaV1.1, NaV1.2 and NaV1.6 point to channel-specific functional differences associated with childhood epilepsy. PLoS ONE 2020, 15, e0238121. [Google Scholar] [CrossRef]
- Wang, Z.; Kuang, P.; Lin, Y.; Liu, W.; Lao, W.; Ji, Y.; Zhu, H. Re-expression of voltage-gated sodium channel subtype Nav1.3 in the substantia nigra after dopamine depletion. Neurosci. Lett. 2018, 687, 146–152. [Google Scholar] [CrossRef]
- Wang, J.T.; Zheng, Y.M.; Chen, Y.T.; Gu, M.; Gao, Z.B.; Nan, F.J. Discovery of aryl sulfonamide-selective Nav1.7 inhibitors with a highly hydrophobic ethanoanthracene core. Acta Pharmacol. Sin. 2020, 41, 293–302. [Google Scholar] [CrossRef]
- Xiao, Y.; Barbosa, C.; Pei, Z.; Xie, W.; Strong, J.A.; Zhang, J.M.; Cummins, T.R. Increased Resurgent Sodium Currents in Nav1.8 Contribute to Nociceptive Sensory Neuron Hyperexcitability Associated with Peripheral Neuropathies. J. Neurosci. 2019, 39, 1539–1550. [Google Scholar] [CrossRef]
- Bai, Q.; Shao, J.; Cao, J.; Ren, X.; Cai, W.; Su, S.; George, S.; Tan, Z.; Zang, W.; Dong, T. Protein kinase C-alpha upregulates sodium channel Nav1.9 in nociceptive dorsal root ganglion neurons in an inflammatory arthritis pain model of rat. J. Cell Biochem. 2020, 121, 768–778. [Google Scholar] [CrossRef]
- Mannikko, R.; Wong, L.; Tester, D.J.; Thor, M.G.; Sud, R.; Kullmann, D.M.; Sweeney, M.G.; Leu, C.; Sisodiya, S.M.; FitzPatrick, D.R.; et al. Dysfunction of NaV1.4, a skeletal muscle voltage-gated sodium channel, in sudden infant death syndrome: A case-control study. Lancet 2018, 391, 1483–1492. [Google Scholar] [CrossRef]
- Turan, N.N.; Moshal, K.S.; Roder, K.; Baggett, B.C.; Kabakov, A.Y.; Dhakal, S.; Teramoto, R.; Chiang, D.Y.; Zhong, M.; Xie, A.; et al. The endosomal trafficking regulator LITAF controls the cardiac Nav1.5 channel via the ubiquitin ligase NEDD4-2. J. Biol. Chem. 2020, 295, 18148–18159. [Google Scholar] [CrossRef]
- Nakajima, T.; Kaneko, Y.; Dharmawan, T.; Kurabayashi, M. Role of the voltage sensor module in Nav domain IV on fast inactivation in sodium channelopathies: The implication of closed-state inactivation. Channels (Austin) 2019, 13, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Gabelli, S.B.; Yoder, J.B.; Tomaselli, G.F.; Amzel, L.M. Calmodulin and Ca(2+) control of voltage gated Na(+) channels. Channels (Austin) 2016, 10, 45–54. [Google Scholar] [CrossRef]
- O’Day, D.H.; Taylor, R.J.; Myre, M.A. Calmodulin and Calmodulin Binding Proteins in Dictyostelium: A Primer. Int J. Mol. Sci. 2020, 21, 1210. [Google Scholar] [CrossRef]
- Zoidl, G.R.; Spray, D.C. The Roles of Calmodulin and CaMKII in Cx36 Plasticity. Int J. Mol. Sci. 2021, 22, 4473. [Google Scholar] [CrossRef]
- Villalobo, A.; Ishida, H.; Vogel, H.J.; Berchtold, M.W. Calmodulin as a protein linker and a regulator of adaptor/scaffold proteins. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 507–521. [Google Scholar] [CrossRef]
- Kang, P.W.; Chakouri, N.; Diaz, J.; Tomaselli, G.F.; Yue, D.T.; Ben-Johny, M. Elementary mechanisms of calmodulin regulation of NaV1.5 producing divergent arrhythmogenic phenotypes. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Gabelli, S.B.; Boto, A.; Kuhns, V.H.; Bianchet, M.A.; Farinelli, F.; Aripirala, S.; Yoder, J.; Jakoncic, J.; Tomaselli, G.F.; Amzel, L.M. Regulation of the NaV1.5 cytoplasmic domain by calmodulin. Nat. Commun. 2014, 5, 5126. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, M.F.; Tung, C.C.; Van Petegem, F.; Ahern, C.A. Crystallographic basis for calcium regulation of sodium channels. Proc. Natl. Acad. Sci. USA 2012, 109, 3558–3563. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Hudmon, A.; Waxman, S.G.; Dib-Hajj, S.D. Calmodulin regulates current density and frequency-dependent inhibition of sodium channel Nav1.8 in DRG neurons. J. Neurophysiol. 2006, 96, 97–108. [Google Scholar] [CrossRef]
- Biswas, S.; Deschenes, I.; Disilvestre, D.; Tian, Y.; Halperin, V.L.; Tomaselli, G.F. Calmodulin regulation of Nav1.4 current: Role of binding to the carboxyl terminus. J. Gen. Physiol. 2008, 131, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jin, X.; Wu, T.; Zhao, X.; Wang, W.; Lei, J.; Pan, X.; Yan, N. Structure of human Nav1.5 reveals the fast inactivation-related segments as a mutational hotspot for the long QT syndrome. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Catterall, W.A.; Lenaeus, M.J.; Gamal El-Din, T.M. Structure and Pharmacology of Voltage-Gated Sodium and Calcium Channels. Annu Rev. Pharmacol Toxicol. 2020, 60, 133–154. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Vyas, R.; Chalamalasetti, S.V.; Sahu, I.D.; Clatot, J.; Wan, X.; Lorigan, G.A.; Deschenes, I.; Chakrapani, S. The voltage-gated sodium channel pore exhibits conformational flexibility during slow inactivation. J. Gen. Physiol. 2018, 150, 1333–1347. [Google Scholar] [CrossRef]
- Gade, A.R.; Marx, S.O.; Pitt, G.S. An interaction between the III-IV linker and CTD in NaV1.5 confers regulation of inactivation by CaM and FHF. J. Gen. Physiol. 2020, 152. [Google Scholar] [CrossRef] [PubMed]
- Gaudioso, C.; Carlier, E.; Youssouf, F.; Clare, J.J.; Debanne, D.; Alcaraz, G. Calmodulin and calcium differentially regulate the neuronal Nav1.1 voltage-dependent sodium channel. Biochem. Biophys Res. Commun. 2011, 411, 329–334. [Google Scholar] [CrossRef]
- Rusconi, R.; Scalmani, P.; Cassulini, R.R.; Giunti, G.; Gambardella, A.; Franceschetti, S.; Annesi, G.; Wanke, E.; Mantegazza, M. Modulatory proteins can rescue a trafficking defective epileptogenic Nav1.1 Na+ channel mutant. J. Neurosci. 2007, 27, 11037–11046. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Abrams, J.; Roybal, D.; Chakouri, N.; Katchman, A.N.; Weinberg, R.; Yang, L.; Chen, B.X.; Zakharov, S.I.; Hennessey, J.A.; Avula, U.M.R.; et al. Fibroblast growth factor homologous factors tune arrhythmogenic late NaV1.5 current in calmodulin binding-deficient channels. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Li, J.; Yu, Z.; Xu, J.; Feng, R.; Gao, Q.; Boczek, T.; Liu, J.; Li, Z.; Wang, Q.; Lei, M.; et al. The Effect of Ca(2+), Lobe-Specificity, and CaMKII on CaM Binding to NaV1.1. Int J. Mol. Sci. 2018, 19, 2495. [Google Scholar] [CrossRef]
- Johnson, C.N. Calcium modulation of cardiac sodium channels. J. Physiol. 2020, 598, 2835–2846. [Google Scholar] [CrossRef]
- Guo, F.; Zhou, P.D.; Gao, Q.H.; Gong, J.; Feng, R.; Xu, X.X.; Liu, S.Y.; Hu, H.Y.; Zhao, M.M.; Adam, H.C.; et al. Low-Mg(2+) treatment increases sensitivity of voltage-gated Na(+) channels to Ca(2+)/calmodulin-mediated modulation in cultured hippocampal neurons. Am. J. Physiol. Cell Physiol. 2015, 308, C594–C605. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Liu, J.; Yu, Z.; Zhang, X.; Xu, X.; Wang, Y.; Gao, Q.; Feng, R.; Wan, Y.; Xu, J.; et al. Properties of Calmodulin Binding to NaV1.2 IQ Motif and Its Autism-Associated Mutation R1902C. Neurochem. Res. 2021, 46, 523–534. [Google Scholar] [CrossRef]
- Deschenes, I.; Neyroud, N.; DiSilvestre, D.; Marban, E.; Yue, D.T.; Tomaselli, G.F. Isoform-specific modulation of voltage-gated Na(+) channels by calmodulin. Circ. Res. 2002, 90, E49–E57. [Google Scholar] [CrossRef]
- Herzog, R.I.; Liu, C.; Waxman, S.G.; Cummins, T.R. Calmodulin binds to the C terminus of sodium channels Nav1.4 and Nav1.6 and differentially modulates their functional properties. J. Neurosci. 2003, 23, 8261–8270. [Google Scholar] [CrossRef] [PubMed]
- Ben-Johny, M.; Yang, P.S.; Niu, J.; Yang, W.; Joshi-Mukherjee, R.; Yue, D.T. Conservation of Ca2+/calmodulin regulation across Na and Ca2+ channels. Cell. 2014, 157, 1657–1670. [Google Scholar] [CrossRef] [PubMed]
- Yoder, J.B.; Ben-Johny, M.; Farinelli, F.; Srinivasan, L.; Shoemaker, S.R.; Tomaselli, G.F.; Gabelli, S.B.; Amzel, L.M. Ca(2+)-dependent regulation of sodium channels NaV1.4 and NaV1.5 is controlled by the post-IQ motif. Nat. Commun. 2019, 10, 1514. [Google Scholar] [CrossRef]
- Martinez-Losa, M.; Tracy, T.E.; Ma, K.; Verret, L.; Clemente-Perez, A.; Khan, A.S.; Cobos, I.; Ho, K.; Gan, L.; Mucke, L.; et al. Nav1.1-Overexpressing Interneuron Transplants Restore Brain Rhythms and Cognition in a Mouse Model of Alzheimer’s Disease. Neuron 2018, 98, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Isbell, H.M.; Kilpatrick, A.M.; Lin, Z.; Mahling, R.; Shea, M.A. Backbone resonance assignments of complexes of apo human calmodulin bound to IQ motif peptides of voltage-dependent sodium channels NaV1.1, NaV1.4 and NaV1.7. Biomol. NMR Assign. 2018, 12, 283–289. [Google Scholar] [CrossRef]
- Nunes, D.; Kuner, T. Axonal sodium channel NaV1.2 drives granule cell dendritic GABA release and rapid odor discrimination. PLoS Biol. 2018, 16, e2003816. [Google Scholar] [CrossRef]
- Hu, X.L.; Wang, X.X.; Zhu, Y.M.; Xuan, L.N.; Peng, L.W.; Liu, Y.Q.; Yang, H.; Yang, C.; Jiao, L.; Hang, P.Z.; et al. MicroRNA-132 regulates total protein of Nav1.1 and Nav1.2 in the hippocampus and cortex of rat with chronic cerebral hypoperfusion. Behav. Brain Res. 2019, 366, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Ghosh, S.; Liu, H.; Tateyama, M.; Kass, R.S.; Pitt, G.S. Calmodulin mediates Ca2+ sensitivity of sodium channels. J. Biol. Chem. 2004, 279, 45004–45012. [Google Scholar] [CrossRef]
- Theoharis, N.T.; Sorensen, B.R.; Theisen-Toupal, J.; Shea, M.A. The neuronal voltage-dependent sodium channel type II IQ motif lowers the calcium affinity of the C-domain of calmodulin. Biochemistry 2008, 47, 112–123. [Google Scholar] [CrossRef]
- Mahling, R.; Hovey, L.; Isbell, H.M.; Marx, D.C.; Miller, M.S.; Kilpatrick, A.M.; Weaver, L.D.; Yoder, J.B.; Kim, E.H.; Andresen, C.N.J.; et al. NaV1.2 EFL domain allosterically enhances Ca(2+) binding to sites I and II of WT and pathogenic calmodulin mutants bound to the channel CTD. Structure 2021. [Google Scholar] [CrossRef]
- Wang, C.; Chung, B.C.; Yan, H.; Wang, H.G.; Lee, S.Y.; Pitt, G.S. Structural analyses of Ca(2)(+)/CaM interaction with NaV channel C-termini reveal mechanisms of calcium-dependent regulation. Nat. Commun. 2014, 5, 4896. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Wang, C.; Marx, S.O.; Pitt, G.S. Calmodulin limits pathogenic Na+ channel persistent current. J. Gen. Physiol. 2017, 149, 277–293. [Google Scholar] [CrossRef]
- Ghovanloo, M.R.; Peters, C.H.; Ruben, P.C. Effects of acidosis on neuronal voltage-gated sodium channels: Nav1.1 and Nav1.3. Channels (Austin) 2018, 12, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Lee-Kwon, W.; Goo, J.H.; Zhang, Z.; Silldorff, E.P.; Pallone, T.L. Vasa recta voltage-gated Na+ channel Nav1.3 is regulated by calmodulin. Am. J. Physiol. Renal Physiol. 2007, 292, F404–F414. [Google Scholar] [CrossRef]
- Horie, R.; Kubota, T.; Koh, J.; Tanaka, R.; Nakamura, Y.; Sasaki, R.; Ito, H.; Takahashi, M.P. EF hand-like motif mutations of Nav1.4 C-terminus cause myotonic syndrome by impairing fast inactivation. Muscle Nerve 2020, 61, 808–814. [Google Scholar] [CrossRef]
- Dong, C.; Wang, Y.; Ma, A.; Wang, T. Life Cycle of the Cardiac Voltage-Gated Sodium Channel NaV1.5. Front. Physiol. 2020, 11, 609733. [Google Scholar] [CrossRef]
- Hong, L.; Zhang, M.; Ly, O.T.; Chen, H.; Sridhar, A.; Lambers, E.; Chalazan, B.; Youn, S.W.; Maienschein-Cline, M.; Feferman, L.; et al. Human induced pluripotent stem cell-derived atrial cardiomyocytes carrying an SCN5A mutation identify nitric oxide signaling as a mediator of atrial fibrillation. Stem Cell Rep. 2021, 16, 1542–1554. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.L.; Kupershmidt, S.; Zhang, R.; Stepanovic, S.; Roden, D.M.; Wilde, A.A.; Anderson, M.E.; Balser, J.R. A calcium sensor in the sodium channel modulates cardiac excitability. Nature 2002, 415, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.N.; Wingo, T.L.; Weiss, K.L.; Williams, C.K.; Balser, J.R.; Chazin, W.J. Calcium-dependent regulation of the voltage-gated sodium channel hH1: Intrinsic and extrinsic sensors use a common molecular switch. Proc. Natl. Acad. Sci. USA 2006, 103, 3592–3597. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.N.; Potet, F.; Thompson, M.K.; Kroncke, B.M.; Glazer, A.M.; Voehler, M.W.; Knollmann, B.C.; George, A.L.; Chazin, W.J. A Mechanism of Calmodulin Modulation of the Human Cardiac Sodium Channel. Structure 2018, 26, 683–694. [Google Scholar] [CrossRef]
- Potet, F.; Chagot, B.; Anghelescu, M.; Viswanathan, P.C.; Stepanovic, S.Z.; Kupershmidt, S.; Chazin, W.J.; Balser, J.R. Functional Interactions between Distinct Sodium Channel Cytoplasmic Domains through the Action of Calmodulin. J. Biol. Chem. 2009, 284, 8846–8854. [Google Scholar] [CrossRef] [PubMed]
- Aiba, T.; Hesketh, G.G.; Liu, T.; Carlisle, R.; Villa-Abrille, M.C.; O’Rourke, B.; Akar, F.G.; Tomaselli, G.F. Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes. Cardiovasc. Res. 2010, 85, 454–463. [Google Scholar] [CrossRef]
- Wang, Z.; Vermij, S.H.; Sottas, V.; Shestak, A.; Ross-Kaschitza, D.; Zaklyazminskaya, E.V.; Hudmon, A.; Pitt, G.S.; Rougier, J.S.; Abriel, H. Calmodulin binds to the N-terminal domain of the cardiac sodium channel Nav1.5. Channels (Austin) 2020, 14, 268–286. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, J.; Benson, C.; Lankford, K.L.; Zhao, P.; Carrara, J.; Tan, A.M.; Kocsis, J.D.; Waxman, S.G.; Dib-Hajj, S.D. Sodium channel Nav1.6 in sensory neurons contributes to vincristine-induced allodynia. Brain 2020, 143, 2421–2436. [Google Scholar] [CrossRef] [PubMed]
- Reddy Chichili, V.P.; Xiao, Y.; Seetharaman, J.; Cummins, T.R.; Sivaraman, J. Structural basis for the modulation of the neuronal voltage-gated sodium channel NaV1.6 by calmodulin. Sci. Rep. 2013, 3, 2435. [Google Scholar] [CrossRef]
- Chen, L.; Wimalasena, N.K.; Shim, J.; Han, C.; Lee, S.I.; Gonzalez-Cano, R.; Estacion, M.; Faber, C.G.; Lauria, G.; Dib-Hajj, S.D.; et al. Two independent mouse lines carrying the Nav1.7 I228M gain-of-function variant display dorsal root ganglion neuron hyperexcitability but a minimal pain phenotype. Pain 2021, 162, 1758–1770. [Google Scholar] [CrossRef]
- Macri, V.; Brody, J.A.; Arking, D.E.; Hucker, W.J.; Yin, X.; Lin, H.; Mills, R.W.; Sinner, M.F.; Lubitz, S.A.; Liu, C.T.; et al. Common Coding Variants in SCN10A Are Associated With the Nav1.8 Late Current and Cardiac Conduction. Circ. Genom. Precis. Med. 2018, 11, e001663. [Google Scholar] [CrossRef] [PubMed]
- Odening, K.E. The Role of Nav1.8 in Cardiac Electrophysiology-a Matter of the Heart or the Nerve? Cardiovasc. Drugs Ther. 2019, 33, 645–647. [Google Scholar] [CrossRef]
- Hong, L.; Zhang, M.; Sridhar, A.; Darbar, D. Pathogenic mutations perturb calmodulin regulation of Nav1.8 channel. Biochem. Biophys. Res. Commun. 2020. [Google Scholar] [CrossRef]
- Bonnet, C.; Hao, J.; Osorio, N.; Donnet, A.; Penalba, V.; Ruel, J.; Delmas, P. Maladaptive activation of Nav1.9 channels by nitric oxide causes triptan-induced medication overuse headache. Nat. Commun. 2019, 10, 4253. [Google Scholar] [CrossRef] [PubMed]
- Matthews, E.; Balestrini, S.; Sisodiya, S.M.; Hanna, M.G. Muscle and brain sodium channelopathies: Genetic causes, clinical phenotypes, and management approaches. Lancet Child Adolesc. Health 2020, 4, 536–547. [Google Scholar] [CrossRef]
- Lossin, C. A catalog of SCN1A variants. Brain Dev. 2009, 31, 114–130. [Google Scholar] [CrossRef] [PubMed]
- Harkin, L.A.; McMahon, J.M.; Iona, X.; Dibbens, L.; Pelekanos, J.T.; Zuberi, S.M.; Sadleir, L.G.; Andermann, E.; Gill, D.; Farrell, K.; et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain 2007, 130, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Zucca, C.; Redaelli, F.; Epifanio, R.; Zanotta, N.; Romeo, A.; Lodi, M.; Veggiotti, P.; Airoldi, G.; Panzeri, C.; Romaniello, R.; et al. Cryptogenic epileptic syndromes related to SCN1A: Twelve novel mutations identified. Arch. Neurol. 2008, 65, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Wallace, R.H.; Scheffer, I.E.; Barnett, S.; Richards, M.; Dibbens, L.; Desai, R.R.; Lerman-Sagie, T.; Lev, D.; Mazarib, A.; Brand, N.; et al. Neuronal sodium-channel alpha1-subunit mutations in generalized epilepsy with febrile seizures plus. Am. J. Hum. Genet. 2001, 68, 859–865. [Google Scholar] [CrossRef] [PubMed]
- de Vries, B.; Freilinger, T.; Vanmolkot, K.R.; Koenderink, J.B.; Stam, A.H.; Terwindt, G.M.; Babini, E.; van den Boogerd, E.H.; van den Heuvel, J.J.; Frants, R.R.; et al. Systematic analysis of three FHM genes in 39 sporadic patients with hemiplegic migraine. Neurology 2007, 69, 2170–2176. [Google Scholar] [CrossRef]
- Depienne, C.; Trouillard, O.; Saint-Martin, C.; Gourfinkel-An, I.; Bouteiller, D.; Carpentier, W.; Keren, B.; Abert, B.; Gautier, A.; Baulac, S.; et al. Spectrum of SCN1A gene mutations associated with Dravet syndrome: Analysis of 333 patients. J. Med. Genet. 2009, 46, 183–191. [Google Scholar] [CrossRef]
- Weiss, L.A.; Escayg, A.; Kearney, J.A.; Trudeau, M.; MacDonald, B.T.; Mori, M.; Reichert, J.; Buxbaum, J.D.; Meisler, M.H. Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol. Psychiatry 2003, 8, 186–194. [Google Scholar] [CrossRef]
- Haug, K.; Hallmann, K.; Rebstock, J.; Dullinger, J.; Muth, S.; Haverkamp, F.; Pfeiffer, H.; Rau, B.; Elger, C.E.; Propping, P.; et al. The voltage-gated sodium channel gene SCN2A and idiopathic generalized epilepsy. Epilepsy Res. 2001, 47, 243–246. [Google Scholar] [CrossRef]
- Te Riele, A.S.; Agullo-Pascual, E.; James, C.A.; Leo-Macias, A.; Cerrone, M.; Zhang, M.; Lin, X.; Lin, B.; Sobreira, N.L.; Amat-Alarcon, N.; et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 2017, 113, 102–111. [Google Scholar] [CrossRef]
- Selga, E.; Campuzano, O.; Pinsach-Abuin, M.L.; Perez-Serra, A.; Mademont-Soler, I.; Riuro, H.; Pico, F.; Coll, M.; Iglesias, A.; Pagans, S.; et al. Comprehensive Genetic Characterization of a Spanish Brugada Syndrome Cohort. PLoS ONE 2015, 10, e0132888. [Google Scholar] [CrossRef]
- Kapplinger, J.D.; Tester, D.J.; Salisbury, B.A.; Carr, J.L.; Harris-Kerr, C.; Pollevick, G.D.; Wilde, A.A.; Ackerman, M.J. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm 2009, 6, 1297–1303. [Google Scholar] [CrossRef]
- Kapplinger, J.D.; Tester, D.J.; Alders, M.; Benito, B.; Berthet, M.; Brugada, J.; Brugada, P.; Fressart, V.; Guerchicoff, A.; Harris-Kerr, C.; et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010, 7, 33–46. [Google Scholar] [CrossRef]
- Pitt, G.S.; Lee, S.Y. Current view on regulation of voltage-gated sodium channels by calcium and auxiliary proteins. Protein Sci. 2016, 25, 1573–1584. [Google Scholar] [CrossRef]
- Bankston, J.R.; Sampson, K.J.; Kateriya, S.; Glaaser, I.W.; Malito, D.L.; Chung, W.K.; Kass, R.S. A novel LQT-3 mutation disrupts an inactivation gate complex with distinct rate-dependent phenotypic consequences. Channels (Austin) 2007, 1, 273–280. [Google Scholar] [CrossRef][Green Version]
- Winkel, B.G.; Yuan, L.; Olesen, M.S.; Sadjadieh, G.; Wang, Y.; Risgaard, B.; Jabbari, R.; Haunso, S.; Holst, A.G.; Hollegaard, M.V.; et al. The role of the sodium current complex in a nonreferred nationwide cohort of sudden infant death syndrome. Heart Rhythm 2015, 12, 1241–1249. [Google Scholar] [CrossRef]
- Tester, D.J.; Will, M.L.; Haglund, C.M.; Ackerman, M.J. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm 2005, 2, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Abdelsayed, M.; Baruteau, A.E.; Gibbs, K.; Sanatani, S.; Krahn, A.D.; Probst, V.; Ruben, P.C. Differential calcium sensitivity in NaV 1.5 mixed syndrome mutants. J. Physiol. 2017, 595, 6165–6186. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, C.; Priori, S.G.; Schwartz, P.J.; Bloise, R.; Ronchetti, E.; Nastoli, J.; Bottelli, G.; Cerrone, M.; Leonardi, S. Genetic testing in the long QT syndrome: Development and validation of an efficient approach to genotyping in clinical practice. JAMA 2005, 294, 2975–2980. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Splawski, I.; Makielski, J.C.; Tester, D.J.; Will, M.L.; Timothy, K.W.; Keating, M.T.; Jones, G.; Chadha, M.; Burrow, C.R.; et al. Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: Implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm 2004, 1, 600–607. [Google Scholar] [CrossRef]
- Rook, M.B.; Bezzina Alshinawi, C.; Groenewegen, W.A.; van Gelder, I.C.; van Ginneken, A.C.; Jongsma, H.J.; Mannens, M.M.; Wilde, A.A. Human SCN5A gene mutations alter cardiac sodium channel kinetics and are associated with the Brugada syndrome. Cardiovasc. Res. 1999, 44, 507–517. [Google Scholar] [CrossRef]
- Nof, E.; Vysochek, L.; Meisel, E.; Burashnikov, E.; Antzelevitch, C.; Clatot, J.; Beinart, R.; Luria, D.; Glikson, M.; Oz, S. Mutations in NaV1.5 Reveal Calcium-Calmodulin Regulation of Sodium Channel. Front. Physiol. 2019, 10, 700. [Google Scholar] [CrossRef] [PubMed]
- Gardill, B.R.; Rivera-Acevedo, R.E.; Tung, C.C.; Okon, M.; McIntosh, L.P.; Van Petegem, F. The voltage-gated sodium channel EF-hands form an interaction with the III-IV linker that is disturbed by disease-causing mutations. Sci Rep. 2018, 8, 4483. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, M.; Ohno, S.; Makiyama, T.; Horie, M. Novel SCN10A variants associated with Brugada syndrome. Europace 2016, 18, 905–911. [Google Scholar] [CrossRef]
- Hu, D.; Barajas-Martinez, H.; Pfeiffer, R.; Dezi, F.; Pfeiffer, J.; Buch, T.; Betzenhauser, M.J.; Belardinelli, L.; Kahlig, K.M.; Rajamani, S.; et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J. Am. Coll. Cardiol. 2014, 64, 66–79. [Google Scholar] [CrossRef]
- Savio-Galimberti, E.; Weeke, P.; Muhammad, R.; Blair, M.; Ansari, S.; Short, L.; Atack, T.C.; Kor, K.; Vanoye, C.G.; Olesen, M.S.; et al. SCN10A/Nav1.8 modulation of peak and late sodium currents in patients with early onset atrial fibrillation. Cardiovasc. Res. 2014, 104, 355–363. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathological Mutation in CaM-Binding IQ Domain | Change in Nucleotide | Phenotype | Effect On Nav Function |
|---|---|---|---|
| Nav1.1-I1922T | c.5765T>C | Dravet syndrome | unknown |
| Nav1.1-R1928G | c.5782C>G | Familial epilepsy syndrome Familial hemiplegic migraine Cryptogenic epileptic syndrome Dravet syndrome | unknown |
| Nav1.2-R1902C | c.5704C>T | Familial autism | (1) Abolished Ca2+-dependence of CaM binding; (2) Induced hyperpolarization shift of the voltage dependence of activation and inactivation of the channel. |
| Nav1.2-R1918H | c.5753G>A | Idiopathic generalized epilepsy | (1) Induced an increased late sodium current; (2) Overexpression of CaM reduced the late sodium current. |
| Nav1.5-R1898C | c.5692C>T | Brugada syndrome | unknown |
| Nav1.5-R1898H | c.5693G>A | Arrhythmogenic right ventricular dysplasia/cardiomyopathy | (1) Caused a reduction in peak sodium current; (2) Caused a structural deficit and decreased abundance of Nav1.5 and N-Cadherin clusters. |
| Nav1.5-E1901K | c.5701G>A | Brugada syndrome | unknown |
| Nav1.5-E1901Q | c.5701G>C | Long QT syndrome | (1) Induced an increased late sodium current; (2) Overexpression of CaM reduced the late sodium current. |
| Nav1.5-S1904L | c.5711C>T | Long QT syndrome Brugada syndrome | (1) Reduced binding affinity between channel and CaM; (2) Induced an increased late sodium current. |
| Nav1.5-Q1909R | c.5726A>G | Long QT syndrome Sudden infant death syndrome | (1) Induced an increased late sodium current; (2) Overexpression of CaM reduced the late sodium current. |
| Nav1.5-R1913H | c.5738G>A | Long QT syndrome | (1) Induced an increased late sodium current; (2) Overexpression of CaM reduced the late sodium current. |
| Nav1.5-R1919C | c.5755C>T | Long QT syndrome Brugada syndrome | unknown |
| Nav1.5-A1924T | c.5770G>A | Brugada syndrome | (1) Induced a hyperpolarization shift in voltage-state activation; (2) Generated a reduced slow inactivation that was rescued by CaM. |
| Nav1.8-R1863Q | c.5588 G>A | Brugada syndrome | (1) Reduced CaM-induced hyperpolarization shift of the voltage dependence of inactivation of the channel. |
| Nav1.8-R1869C | c.5605 C>T | Brugada syndrome | (1) Disrupted CaM-induced hyperpolarization shift; (2) Attenuated effects of CaM on development and recovery from slow inactivation. |
| Nav1.8-R1869G | c.5605 C>G | Atrial fibrillation | (1) Disrupted CaM-induced hyperpolarization shift; (2) Attenuated effects of CaM on development and recovery from slow inactivation. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Hong, L. Calmodulin Interactions with Voltage-Gated Sodium Channels. Int. J. Mol. Sci. 2021, 22, 9798. https://doi.org/10.3390/ijms22189798
Wu X, Hong L. Calmodulin Interactions with Voltage-Gated Sodium Channels. International Journal of Molecular Sciences. 2021; 22(18):9798. https://doi.org/10.3390/ijms22189798
Chicago/Turabian StyleWu, Xin, and Liang Hong. 2021. "Calmodulin Interactions with Voltage-Gated Sodium Channels" International Journal of Molecular Sciences 22, no. 18: 9798. https://doi.org/10.3390/ijms22189798
APA StyleWu, X., & Hong, L. (2021). Calmodulin Interactions with Voltage-Gated Sodium Channels. International Journal of Molecular Sciences, 22(18), 9798. https://doi.org/10.3390/ijms22189798