
Serum Albumin: A Multifaced Enzyme




Abstract
1. Introduction
2. Human Serum Albumin
2.1. HSA Structure
2.2. HSA Oligomerization
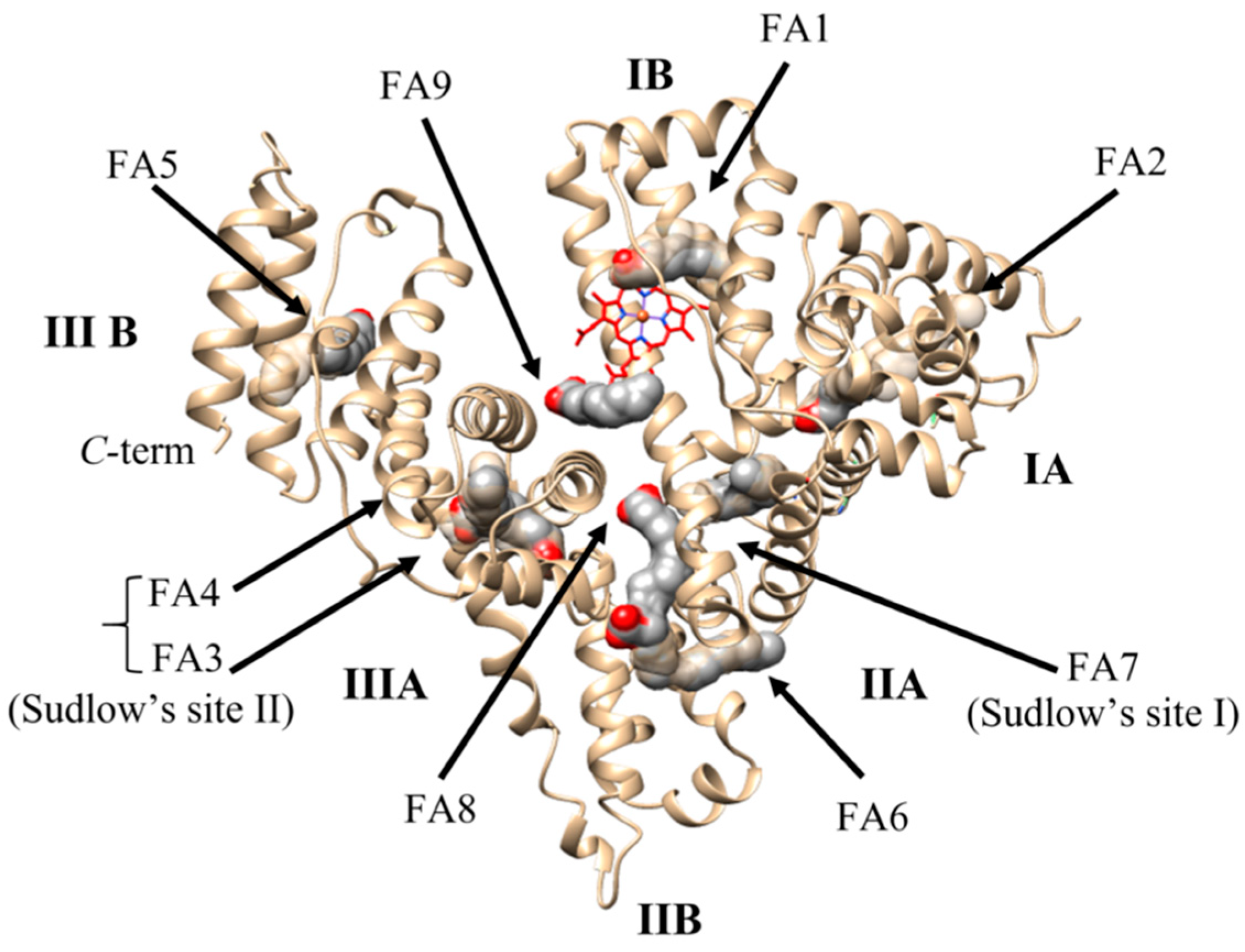
2.3. Ligand Binding Properties of HSA
3. HSA Enzymatic Properties
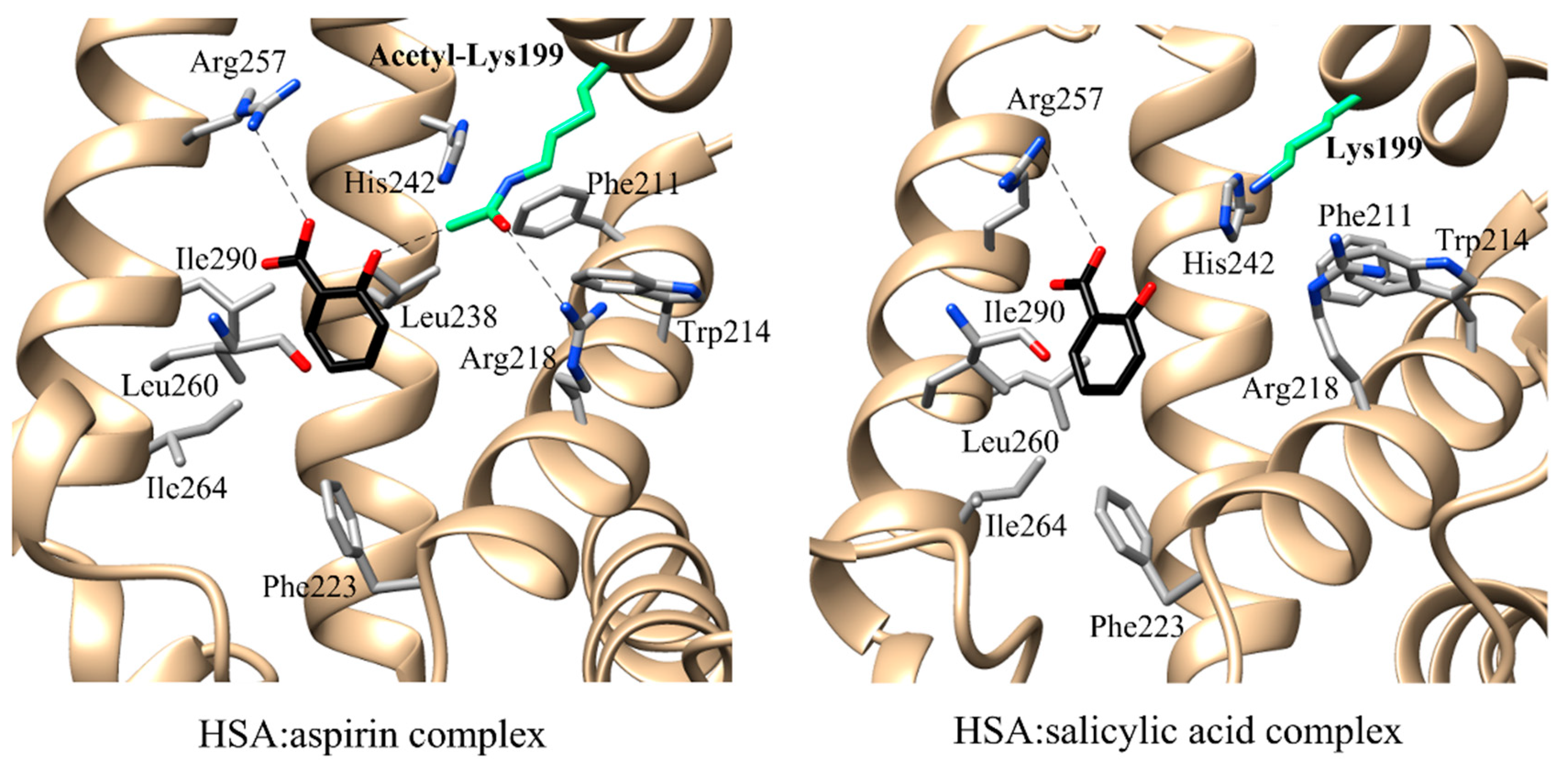
3.1. Esterase (or Pseudo-Esterase) Activity of HSA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymatic Activities | Substrates | Cofactors and/or Coenzyme | Catalytic Sites | Catalytic Residues | Recognition Residues | Kinetic Parameters | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Ks or Km (μM) | k+2 (s−1) | |||||||
| Esterase activity | aspirin | n.n. | FA7 | Lys199 His242 Arg257 | Trp214 Arg218 Leu219 Arg222 Phe223 Leu238 Arg257 Leu260 Ile264 Ser287 Ile290 Ala291 | [97] | ||
| Pseudo-esterase activity | chloropyriphosoxon | n.n. | FA3–FA4 | Tyr411 | Arg410 | 2.6 × 102 b | 6.6 × 10‒4 b | [99] |
| ketoprofen glucuronide | n.n | FA3–FA4 | Tyr411 | Ser489 Leu491 | [76] | |||
| NphOAc | n.n. | FA3–FA4 | Tyr411 | Asn391 Leu407 Arg410 Lys414 Leu430 Leu453 | (4.8 ± 0.5) × 10−4 c | (3.9 ± 0.4) × 10−1 c | [100] | |
| NphODe | n.n. | FA3–FA4 | Tyr411 | Asn391 Leu407 Arg410 Lys414 Leu430 Leu453 | (2.3 ± 0.2) × 10−5 c | (8.1 ± 0.8) × 10−4 c | [86] | |
| NphOHe | n.n. | FA3–FA4 | Tyr411 | Asn391 Leu407 Arg410 Lys414 Leu430 Leu453 | (2.9 ± 0.3) × 10−5 c | (2.8 ± 0.3) × 10−3 c | [86] | |
| NphOMy | n.n. | FA3-FA4 | Tyr411 | Asn391 Leu407 Arg410 Lys414 Leu430 Leu453 | (2.6 ± 0.3) × 10−5 c | (1.6 ± 0.2) × 10−4 c | [101] | |
| p-nitrophenyl-N-methylcarbamate | n.n. | FA3–FA4 | Tyr411 | 1.4 × 102 d | [82] | |||
| paraoxon | n.n. | FA3–FA4 | Tyr411 | Arg410 | 3.7 × 102 b | 2.5 × 10−5 b | [99] | |
| RNA-hydrolyzing activity | RNA | n.n. | FA7 | Lys195 Lys199 | [102] | |||
| n.n. | FA3–FA4 | Lys541 Lys545 | [103] | |||||
| Enolase activity | dihydro-testosterone | n.n. | NTS | His3 | [104] | |||
| Glucuronidase activity | carprofen glucuronides | n.n. | FA7 | Tyr411 | [105] | |||
| oxaprozin glucuronides | n.n. | FA3–FA4 | Tyr411 | Asn391 Leu407 Arg410 Lys414 Leu430 Leu453 | [106,107] | |||
| Peroxidase activity | α-tocopherol | palmitoyl-CoA | FA3–FA4 | Cys392 Cys438 | [108] | |||
| LDL | palmitoyl-CoA | FA3–FA4 | Cys392 Cys438 | [108] | ||||
| Aldolase activity | aromatic aldehydes with acetone | n.n. | FA7 | Lys199 | [109] | |||
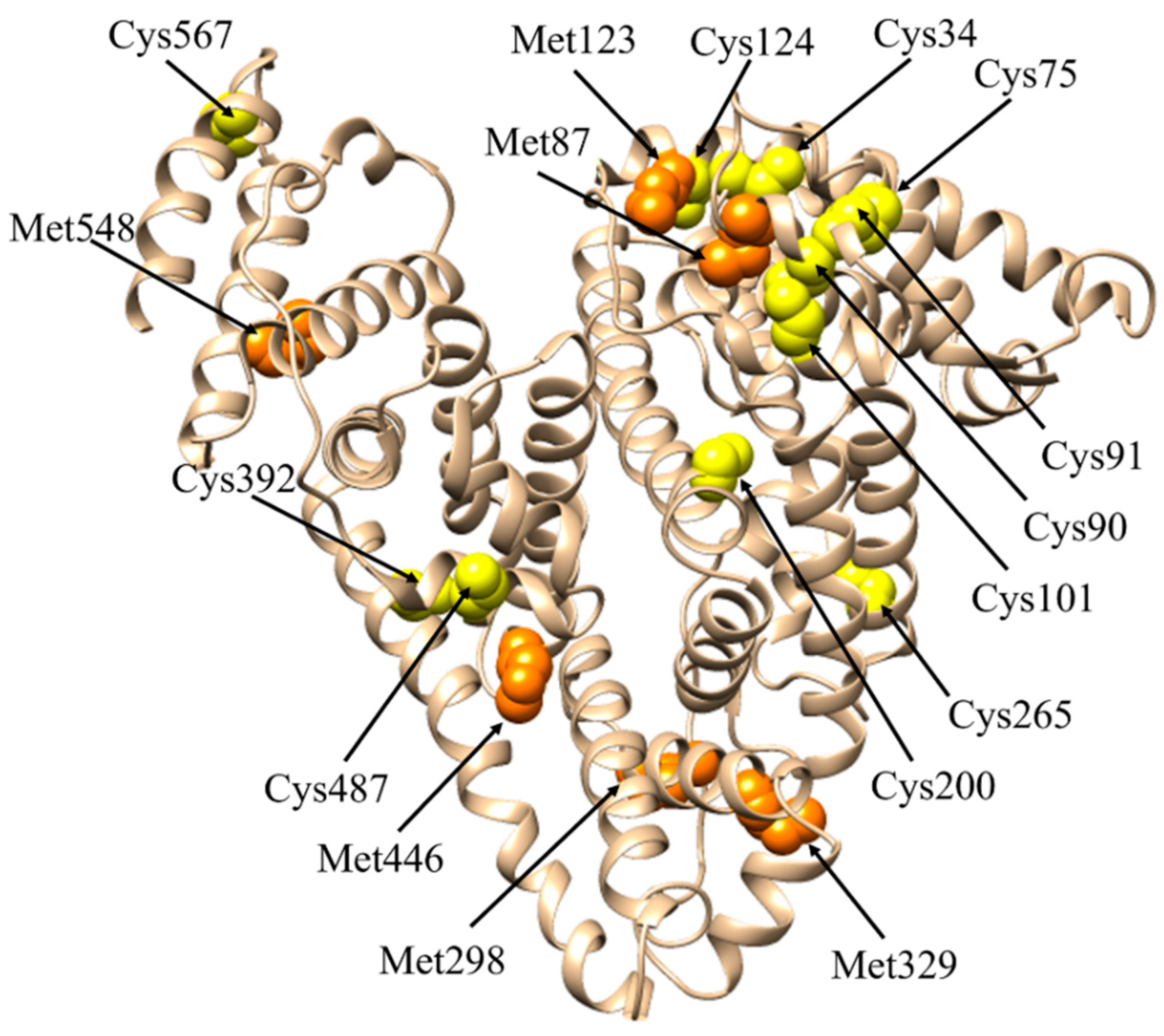
| Direct anti-oxidant activity | ROS (i.e., H2O2, O2−, HOCl) | Cu(I/II) Fe(II/III) | NTS MBS | Cys34 | [22] | |||
| RNS (i.e., ONOO−) | Cu(I/II) Fe(II/III) | Cys34 | [22] | |||||
| Indirect anti-oxidant activity | bilirubin | n.n. | FA1 | Lys190 Lys240 | [110] | |||
| heme | n.n. | FA1 | Tyr161 His142 Lys190 | [111] [112] | ||||
3.2. Enolase Activity of HSA
3.3. Glucuronidase Activity of HSA
3.4. Lipid Peroxidase Activity of HSA
3.5. Aldolase Activity of HSA
3.6. Glutathione-Linked Thiol Peroxidase Activity of HSA
3.7. Anti-Oxidant Activity of HSA
3.7.1. Modulation of the HSA Anti-Oxidant Activity
The Cys34 Residue
HSA Glycation
FAs Binding
Aging
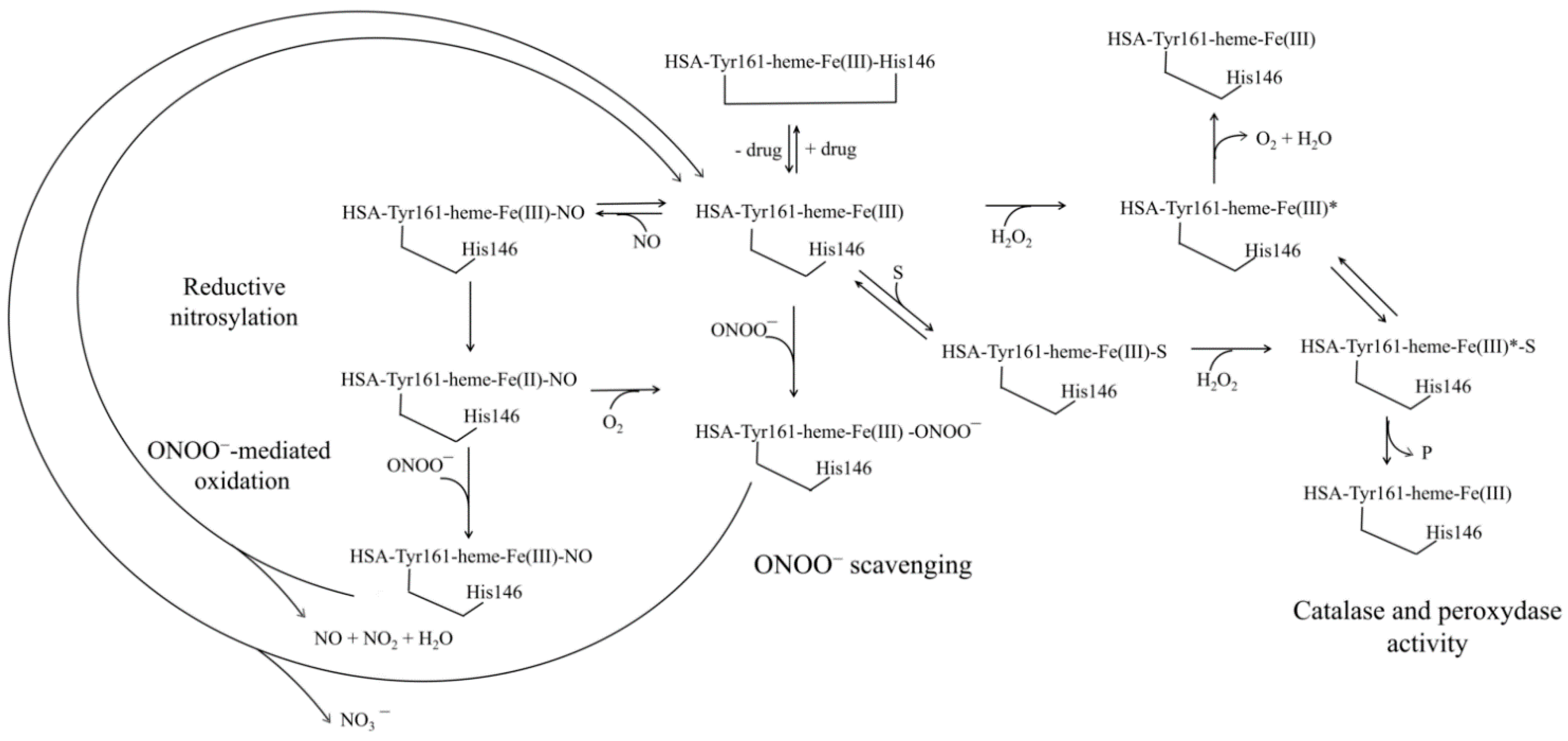
4. Human Serum Heme-Albumin
5. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Peters, T., Jr. All about Albumin: Biochemistry, Genetics and Medical Applications; Academic Press: London, UK; San Diego, CA, USA, 1996. [Google Scholar]
- Evans, T.W. Review article: Albumin as a drug--biological effects of albumin unrelated to oncotic pressure. Aliment. Pharmacol. Ther. 2002, 16 (Suppl. 5), 6–11. [Google Scholar] [CrossRef] [PubMed]
- Mendez, C.M.; McClain, C.J.; Marsano, L.S. Albumin therapy in clinical practice. Nutr. Clin. Pract. 2005, 20, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Asp. Med. 2012, 33, 209–290. [Google Scholar] [CrossRef]
- di Masi, A.; Trezza, V.; Leboffe, L.; Ascenzi, P. Human plasma lipocalins and serum albumin: Plasma alternative carriers? J. Control Release 2016, 228, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Pstras, L.; Waniewski, J.; Lindholm, B. Transcapillary transport of water, small solutes and proteins during hemodialysis. Sci. Rep. 2020, 10, 18736. [Google Scholar] [CrossRef]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol. 1975, 11, 824–832. [Google Scholar]
- Carter, D.C.; Ho, J.X. Structure of serum albumin. Adv. Protein Chem. 1994, 45, 153–203. [Google Scholar] [CrossRef] [PubMed]
- di Masi, A.; Leboffe, L.; Polticelli, F.; Tonon, F.; Zennaro, C.; Caterino, M.; Stano, P.; Fischer, S.; Hagele, M.; Muller, M.; et al. Human Serum Albumin Is an Essential Component of the Host Defense Mechanism Against Clostridium difficile Intoxication. J. Infect. Dis. 2018, 218, 1424–1435. [Google Scholar] [CrossRef]
- Alinovskaya, L.I.; Sedykh, S.E.; Ivanisenko, N.V.; Soboleva, S.E.; Nevinsky, G.A. How human serum albumin recognizes DNA and RNA. Biol. Chem. 2018, 399, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Soboleva, S.E.; Guschina, T.A.; Nevinsky, G.A. Human serum and milk albumins are metal-dependent DNases. IUBMB Life 2018, 70, 501–510. [Google Scholar] [CrossRef]
- Vita, G.M.; De Simone, G.; Leboffe, L.; Montagnani, F.; Mariotti, D.; Di Bella, S.; Luzzati, R.; Gori, A.; Ascenzi, P.; di Masi, A. Human Serum Albumin Binds Streptolysin O (SLO) Toxin Produced by Group A Streptococcus and Inhibits Its Cytotoxic and Hemolytic Effects. Front. Immunol. 2020, 11, 507092. [Google Scholar] [CrossRef]
- De Simone, G.; Pasquadibisceglie, A.; di Masi, A.; Buzzelli, V.; Trezza, V.; Macari, G.; Polticelli, F.; Ascenzi, P. Binding of direct oral anticoagulants to the FA1 site of human serum albumin. J. Mol. Recognit. 2021, 34, e2877. [Google Scholar] [CrossRef]
- Monzani, E.; Bonafe, B.; Fallarini, A.; Redaelli, C.; Casella, L.; Minchiotti, L.; Galliano, M. Enzymatic properties of human hemalbumin. Biochim. Biophys. Acta 2001, 1547, 302–312. [Google Scholar] [CrossRef]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Gianni, S. Functional role of transient conformations: Rediscovering “chronosteric effects” thirty years later. IUBMB Life 2013, 65, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; di Masi, A.; Fanali, G.; Fasano, M. Heme-albumin: An honorary enzyme. Cell Death Dis. 2015, 6, e1895. [Google Scholar] [CrossRef]
- Vlasova, I.I. Peroxidase Activity of Human Hemoproteins: Keeping the Fire under Control. Molecules 2018, 23, 2561. [Google Scholar] [CrossRef]
- Gupta, D.; Lis, C.G. Pretreatment serum albumin as a predictor of cancer survival: A systematic review of the epidemiological literature. Nutr. J. 2010, 9, 69. [Google Scholar] [CrossRef]
- Koga, M.; Kasayama, S. Clinical impact of glycated albumin as another glycemic control marker. Endocr. J. 2010, 57, 751–762. [Google Scholar] [CrossRef]
- Sbarouni, E.; Georgiadou, P.; Voudris, V. Ischemia modified albumin changes–review and clinical implications. Clin. Chem. Lab. Med. 2011, 49, 177–184. [Google Scholar] [CrossRef]
- Belinskaia, D.A.; Voronina, P.A.; Shmurak, V.I.; Vovk, M.A.; Batalova, A.A.; Jenkins, R.O.; Goncharov, N.V. The Universal Soldier: Enzymatic and Non-Enzymatic Antioxidant Functions of Serum Albumin. Antioxidants 2020, 9, 966. [Google Scholar] [CrossRef]
- Tullis, J.L. Albumin. 1. Background and use. JAMA 1977, 237, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.R.; Alexander, B.; Mustion, A.L.; Spector, R.; Wright, C.B. Therapeutic use of albumin: 2. JAMA 1982, 247, 831–833. [Google Scholar] [CrossRef] [PubMed]
- Erstad, B.L.; Gales, B.J.; Rappaport, W.D. The use of albumin in clinical practice. Arch. Intern. Med. 1991, 151, 901–911. [Google Scholar] [CrossRef]
- Hastings, G.E.; Wolf, P.G. The therapeutic use of albumin. Arch. Fam. Med. 1992, 1, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Alderson, P.; Bunn, F.; Lefebvre, C.; Li, W.P.; Li, L.; Roberts, I.; Schierhout, G. Human albumin solution for resuscitation and volume expansion in critically ill patients. Cochrane Database Syst. Rev. 2004, CD001208. [Google Scholar] [CrossRef] [PubMed]
- Liberati, A.; Moja, L.; Moschetti, I.; Gensini, G.F.; Gusinu, R. Human albumin solution for resuscitation and volume expansion in critically ill patients. Intern. Emerg. Med. 2006, 1, 243–245. [Google Scholar] [CrossRef]
- Spinella, R.; Sawhney, R.; Jalan, R. Albumin in chronic liver disease: Structure, functions and therapeutic implications. Hepatol. Int. 2016, 10, 124–132. [Google Scholar] [CrossRef]
- Di Bella, S.; di Masi, A.; Turla, S.; Ascenzi, P.; Gouliouris, T.; Petrosillo, N. The Protective Role of Albumin in Clostridium difficile Infection: A Step Toward Solving the Puzzle. Infect. Control. Hosp. Epidemiol. 2015, 36, 1478–1479. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, S.; Ascenzi, P.; Siarakas, S.; Petrosillo, N.; di Masi, A. Clostridium difficile Toxins A and B: Insights into Pathogenic Properties and Extraintestinal Effects. Toxins 2016, 8, 134. [Google Scholar] [CrossRef] [PubMed]
- Austermeier, S.; Pekmezovic, M.; Porschitz, P.; Lee, S.; Kichik, N.; Moyes, D.L.; Ho, J.; Kotowicz, N.K.; Naglik, J.R.; Hube, B.; et al. Albumin Neutralizes Hydrophobic Toxins and Modulates Candida albicans Pathogenicity. mBio 2021, 12, e0053121. [Google Scholar] [CrossRef]
- Rothschild, M.A.; Oratz, M.; Schreiber, S.S. Ethanol effects on albumin sythesis. Adv. Exp. Med. Biol. 1980, 126, 385–396. [Google Scholar] [CrossRef]
- Rothschild, M.A.; Oratz, M. Albumin metabolism: A brief review. Mt. Sinai J. Med. 1992, 59, 155–156. [Google Scholar]
- Levitt, D.G.; Levitt, M.D. Human serum albumin homeostasis: A new look at the roles of synthesis, catabolism, renal and gastrointestinal excretion, and the clinical value of serum albumin measurements. Int J. Gen. Med. 2016, 9, 229–255. [Google Scholar] [CrossRef]
- Akbarzadehlaleh, P.; Mirzaei, M.; Mashahdi-Keshtiban, M.; Shamsasenjan, K.; Heydari, H. PEGylated Human Serum Albumin: Review of PEGylation, Purification and Characterization Methods. Adv. Pharm. Bull. 2016, 6, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, G.J.; Martin, G.S.; Evans, T.W. Albumin: Biochemical properties and therapeutic potential. Hepatology 2005, 41, 1211–1219. [Google Scholar] [CrossRef]
- Ascenzi, P.; Fasano, M. Allostery in a monomeric protein: The case of human serum albumin. Biophys. Chem. 2010, 148, 16–22. [Google Scholar] [CrossRef]
- Curry, S.; Mandelkow, H.; Brick, P.; Franks, N. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat. Struct. Biol. 1998, 5, 827–835. [Google Scholar] [CrossRef]
- Sugio, S.; Kashima, A.; Mochizuki, S.; Noda, M.; Kobayashi, K. Crystal structure of human serum albumin at 2.5 A resolution. Protein Eng. 1999, 12, 439–446. [Google Scholar] [CrossRef]
- Leboffe, L.; di Masi, A.; Polticelli, F.; Trezza, V.; Ascenzi, P. Structural Basis of Drug Recognition by Human Serum Albumin. Curr. Med. Chem. 2020, 27, 4907–4931. [Google Scholar] [CrossRef]
- Curry, S. Beyond expansion: Structural studies on the transport roles of human serum albumin. Vox Sang. 2002, 83 (Suppl. 1), 315–319. [Google Scholar] [CrossRef] [PubMed]
- Curry, S. Lessons from the crystallographic analysis of small molecule binding to human serum albumin. Drug Metab. Pharm. 2009, 24, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Varshney, A.; Sen, P.; Ahmad, E.; Rehan, M.; Subbarao, N.; Khan, R.H. Ligand binding strategies of human serum albumin: How can the cargo be utilized? Chirality 2010, 22, 77–87. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Pandey, N.K.; Roy, A.; Dasgupta, S. Effect of (-)-epigallocatechin gallate on the fibrillation of human serum albumin. Int. J. Biol. Macromol. 2014, 70, 312–319. [Google Scholar] [CrossRef]
- Chubarov, A.; Spitsyna, A.; Krumkacheva, O.; Mitin, D.; Suvorov, D.; Tormyshev, V.; Fedin, M.; Bowman, M.K.; Bagryanskaya, E. Reversible Dimerization of Human Serum Albumin. Molecules 2020, 26, 108. [Google Scholar] [CrossRef]
- Taguchi, K.; Chuang, V.T.; Maruyama, T.; Otagiri, M. Pharmaceutical aspects of the recombinant human serum albumin dimer: Structural characteristics, biological properties, and medical applications. J. Pharm. Sci. 2012, 101, 3033–3046. [Google Scholar] [CrossRef] [PubMed]
- Brahma, A.; Mandal, C.; Bhattacharyya, D. Characterization of a dimeric unfolding intermediate of bovine serum albumin under mildly acidic condition. Biochim. Biophys. Acta 2005, 1751, 159–169. [Google Scholar] [CrossRef]
- Anand, U.; Mukherjee, S. Binding, unfolding and refolding dynamics of serum albumins. Biochim. Biophys. Acta 2013, 1830, 5394–5404. [Google Scholar] [CrossRef]
- Sharma, N.; Sivalingam, V.; Maurya, S.; Prasad, A.; Khandelwal, P.; Yadav, S.C.; Patel, B.K. New insights into in vitro amyloidogenic properties of human serum albumin suggest considerations for therapeutic precautions. FEBS Lett. 2015, 589, 4033–4038. [Google Scholar] [CrossRef] [PubMed]
- Dobson, J.; Kumar, A.; Willis, L.F.; Tuma, R.; Higazi, D.R.; Turner, R.; Lowe, D.C.; Ashcroft, A.E.; Radford, S.E.; Kapur, N.; et al. Inducing protein aggregation by extensional flow. Proc. Natl. Acad. Sci. USA 2017, 114, 4673–4678. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Arakawa, T. Application of native polyacrylamide gel electrophoresis for protein analysis: Bovine serum albumin as a model protein. Int. J. Biol. Macromol. 2019, 125, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Arabi, S.H.; Aghelnejad, B.; Volmer, J.; Hinderberger, D. Hydrogels from serum albumin in a molten globule-like state. Protein Sci. A Publ. Protein Soc. 2020, 29, 2459–2467. [Google Scholar] [CrossRef]
- Watanabe, H.; Imafuku, T.; Otagiri, M.; Maruyama, T. Clinical Implications Associated With the Posttranslational Modification-Induced Functional Impairment of Albumin in Oxidative Stress-Related Diseases. J. Pharm. Sci. 2017, 106, 2195–2203. [Google Scholar] [CrossRef]
- Mimic-Oka, J.; Simic, T.; Djukanovic, L.; Reljic, Z.; Davicevic, Z. Alteration in plasma antioxidant capacity in various degrees of chronic renal failure. Clin. Nephrol. 1999, 51, 233–241. [Google Scholar]
- Ogasawara, Y.; Namai, T.; Togawa, T.; Ishii, K. Formation of albumin dimers induced by exposure to peroxides in human plasma: A possible biomarker for oxidative stress. Biochem. Biophys. Res. Commun. 2006, 340, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.J.; Arnaud, J.; Jurkovitz, C.; Hachache, T.; Meftahi, H.; Laporte, F.; Foret, M.; Favier, A.; Cordonnier, D. Trace elements and lipid peroxidation abnormalities in patients with chronic renal failure. Nephron 1991, 57, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. Further characterization of specific drug binding sites on human serum albumin. Mol. Pharmacol. 1976, 12, 1052–1061. [Google Scholar] [PubMed]
- Ascenzi, P.; Bocedi, A.; Visca, P.; Altruda, F.; Tolosano, E.; Beringhelli, T.; Fasano, M. Hemoglobin and heme scavenging. IUBMB Life 2005, 57, 749–759. [Google Scholar] [CrossRef]
- Bhattacharya, A.A.; Grune, T.; Curry, S. Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin. J. Mol. Biol. 2000, 303, 721–732. [Google Scholar] [CrossRef]
- Simard, J.R.; Zunszain, P.A.; Hamilton, J.A.; Curry, S. Location of high and low affinity fatty acid binding sites on human serum albumin revealed by NMR drug-competition analysis. J. Mol. Biol. 2006, 361, 336–351. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.; Amisaki, T. Fatty acid binding to serum albumin: Molecular simulation approaches. Biochim. et Biophys. Acta 2013, 1830, 5427–5434. [Google Scholar] [CrossRef]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef]
- Petitpas, I.; Petersen, C.E.; Ha, C.E.; Bhattacharya, A.A.; Zunszain, P.A.; Ghuman, J.; Bhagavan, N.V.; Curry, S. Structural basis of albumin-thyroxine interactions and familial dysalbuminemic hyperthyroxinemia. Proc. Natl. Acad. Sci. USA 2003, 100, 6440–6445. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bolli, A.; Gullotta, F.; Fanali, G.; Fasano, M. Drug binding to Sudlow’s site I impairs allosterically human serum heme-albumin-catalyzed peroxynitrite detoxification. IUBMB Life 2010, 62, 776–780. [Google Scholar] [CrossRef]
- Kaneko, K.; Chuang, V.T.; Minomo, A.; Yamasaki, K.; Bhagavan, N.V.; Maruyama, T.; Otagiri, M. Histidine146 of human serum albumin plays a prominent role at the interface of subdomains IA and IIA in allosteric ligand binding. IUBMB Life 2011, 63, 277–285. [Google Scholar] [CrossRef]
- Ascenzi, P.; di Masi, A.; Fanali, G.; Fasano, M. Heme-based catalytic properties of human serum albumin. Cell Death Discov. 2015, 1, 15025. [Google Scholar] [CrossRef] [PubMed]
- di Masi, A.; Leboffe, L.; Trezza, V.; Fanali, G.; Coletta, M.; Fasano, M.; Ascenzi, P. Drugs modulate allosterically heme-Fe-recognition by human serum albumin and heme-fe-mediated reactivity. Curr. Pharm. Des. 2015, 21, 1837–1847. [Google Scholar] [CrossRef]
- Yamasaki, K.; Maruyama, T.; Yoshimoto, K.; Tsutsumi, Y.; Narazaki, R.; Fukuhara, A.; Kragh-Hansen, U.; Otagiri, M. Interactive binding to the two principal ligand binding sites of human serum albumin: Effect of the neutral-to-base transition. Biochim. Biophys. Acta 1999, 1432, 313–323. [Google Scholar] [CrossRef]
- Petitpas, I.; Bhattacharya, A.A.; Twine, S.; East, M.; Curry, S. Crystal structure analysis of warfarin binding to human serum albumin: Anatomy of drug site I. J. Biol. Chem. 2001, 276, 22804–22809. [Google Scholar] [CrossRef] [PubMed]
- Fanali, G.; Fasano, M.; Ascenzi, P.; Zingg, J.M.; Azzi, A. α-Tocopherol binding to human serum albumin. Biofactors 2013, 39, 294–303. [Google Scholar] [CrossRef]
- Zhu, L.; Yang, F.; Chen, L.; Meehan, E.J.; Huang, M. A new drug binding subsite on human serum albumin and drug-drug interaction studied by X-ray crystallography. J. Struct. Biol. 2008, 162, 40–49. [Google Scholar] [CrossRef]
- Dirks, B.M.; Boyer, P.D. Non enzymatic cleavage of p-nitrophenylesters. Cereal Chem. 1951, 28, 483–490. [Google Scholar]
- Kurono, Y.; Kushida, I.; Tanaka, H.; Ikeda, K. Esterase-like activity of human serum albumin. VIII. Reaction with amino acid p-nitrophenyl esters. Chem. Pharm. Bull. 1992, 40, 2169–2172. [Google Scholar] [CrossRef][Green Version]
- Dubois-Presle, N.; Lapicque, F.; Maurice, M.H.; Fournel-Gigleux, S.; Magdalou, J.; Abiteboul, M.; Siest, G.; Netter, P. Stereoselective esterase activity of human serum albumin toward ketoprofen glucuronide. Mol. Pharmacol. 1995, 47, 647–653. [Google Scholar] [PubMed]
- Phuangsawai, O.; Hannongbua, S.; Gleeson, M.P. Elucidating the origin of the esterase activity of human serum albumin using QM/MM calculations. J. Phys. Chem. B 2014, 118, 11886–11894. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, A.; Okada, M.; Inagaki, Y.; Inoue, S.; Hamaguchi, T.; Iwakawa, S. Differences in Esterase Activity to Aspirin and p-Nitrophenyl Acetate among Human Serum Albumin Preparations. Biol. Pharm. Bull. 2016, 39, 1364–1369. [Google Scholar] [CrossRef]
- Kono, K.; Fukuchi, Y.; Okawa, H.; Nunoya, K.I.; Imawaka, H.; Watanabe, H.; Maruyama, T. Unique Hydrolysis of an Ester-Type Prodrug of Levodopa in Human Plasma: Relay-Type Role Sharing between Alpha-1 Acid Glycoprotein and Human Serum Albumin. Mol. Pharm. 2019, 16, 4131–4138. [Google Scholar] [CrossRef]
- Lockridge, O.; Xue, W.; Gaydess, A.; Grigoryan, H.; Ding, S.J.; Schopfer, L.M.; Hinrichs, S.H.; Masson, P. Pseudo-esterase activity of human albumin: Slow turnover on tyrosine 411 and stable acetylation of 82 residues including 59 lysines. J. Biol. Chem. 2008, 283, 22582–22590. [Google Scholar] [CrossRef]
- Kowacz, M.; Warszynski, P. Beyond esterase-like activity of serum albumin. Histidine-(nitro)phenol radical formation in conversion cascade of p-nitrophenyl acetate and the role of infrared light. J. Mol. Recognit. 2019, 32, e2780. [Google Scholar] [CrossRef]
- Casida, J.E.; Augustinsson, K.B. Reaction of plasma albumin with I-naphthyl N-methylcarbamate and certain other esters. Biochim. Biophys. Acta 1959, 36, 411–426. [Google Scholar] [CrossRef]
- Tildon, J.T.; Ogilvie, J.W. The esterase activity of bovine mercaptalbumin. The reaction of the protein with p-nitrophenyl acetate. J. Biol. Chem. 1972, 247, 1265–1271. [Google Scholar] [CrossRef]
- Means, G.E.; Bender, M.L. Acetylation of human serum albumin by p-nitrophenyl acetate. Biochemistry 1975, 14, 4989–4994. [Google Scholar] [CrossRef] [PubMed]
- Tove, S.B. The esterolytic activity of serum albumin. Biochim. Biophys. Acta 1962, 57, 230–235. [Google Scholar] [CrossRef]
- Ascenzi, P.; Leboffe, L.; di Masi, A.; Trezza, V.; Fanali, G.; Gioia, M.; Coletta, M.; Fasano, M. Ligand binding to the FA3-FA4 cleft inhibits the esterase-like activity of human serum albumin. PLoS ONE 2015, 10, e0120603. [Google Scholar] [CrossRef]
- Rainsford, K.D.; Ford, N.L.; Brooks, P.M.; Watson, H.M. Plasma aspirin esterases in normal individuals, patients with alcoholic liver disease and rheumatoid arthritis: Characterization and the importance of the enzymic components. Eur. J. Clin. Investig. 1980, 10, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.H.; Maddison, K.; LoCastro, L.; Borch, R.F. Accelerated decomposition of 4-hydroxycyclophosphamide by human serum albumin. Cancer Res. 1987, 47, 1505–1508. [Google Scholar] [PubMed]
- Salvi, A.; Carrupt, P.A.; Mayer, J.M.; Testa, B. Esterase-like activity of human serum albumin toward prodrug esters of nicotinic acid. Drug Metab. Dispos. 1997, 25, 395–398. [Google Scholar] [PubMed]
- De Vriese, C.; Hacquebard, M.; Gregoire, F.; Carpentier, Y.; Delporte, C. Ghrelin interacts with human plasma lipoproteins. Endocrinology 2007, 148, 2355–2362. [Google Scholar] [CrossRef]
- Manoharan, I.; Boopathy, R. Diisopropylfluorophosphate-sensitive aryl acylamidase activity of fatty acid free human serum albumin. Arch. Biochem. Biophys. 2006, 452, 186–188. [Google Scholar] [CrossRef]
- Masson, P.; Froment, M.T.; Darvesh, S.; Schopfer, L.M.; Lockridge, O. Aryl acylamidase activity of human serum albumin with o-nitrotrifluoroacetanilide as the substrate. J. Enzyme Inhib. Med. Chem. 2007, 22, 463–469. [Google Scholar] [CrossRef]
- Sogorb, M.A.; Monroy, A.; Vilanova, E. Chicken serum albumin hydrolyzes dichlorophenyl phosphoramidates by a mechanism based on transient phosphorylation. Chem. Res. Toxicol. 1998, 11, 1441–1446. [Google Scholar] [CrossRef]
- Goncharov, N.V.; Belinskaia, D.A.; Shmurak, V.I.; Terpilowski, M.A.; Jenkins, R.O.; Avdonin, P.V. Serum Albumin Binding and Esterase Activity: Mechanistic Interactions with Organophosphates. Molecules 2017, 22, 1201. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Tanase, S.; Nakajou, K.; Maruyama, T.; Kragh-Hansen, U.; Otagiri, M. Role of arg-410 and tyr-411 in human serum albumin for ligand binding and esterase-like activity. Biochem. J. 2000, 349 Pt. 3, 813–819. [Google Scholar] [CrossRef]
- Kragh-Hansen, U.; Chuang, V.T.; Otagiri, M. Practical aspects of the ligand-binding and enzymatic properties of human serum albumin. Biol. Pharm. Bull. 2002, 25, 695–704. [Google Scholar] [CrossRef]
- Yang, F.; Bian, C.; Zhu, L.; Zhao, G.; Huang, Z.; Huang, M. Effect of human serum albumin on drug metabolism: Structural evidence of esterase activity of human serum albumin. J. Struct. Biol. 2007, 157, 348–355. [Google Scholar] [CrossRef]
- Liyasova, M.S.; Schopfer, L.M.; Lockridge, O. Reaction of human albumin with aspirin in vitro: Mass spectrometric identification of acetylated lysines 199, 402, 519, and 545. Biochem. Pharmacol. 2010, 79, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Sogorb, M.A.; Vilanova, E. Serum albumins and detoxication of anti-cholinesterase agents. Chem.-Biol. Interact. 2010, 187, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Gioia, M.; Fanali, G.; Coletta, M.; Fasano, M. Pseudo-enzymatic hydrolysis of 4-nitrophenyl acetate by human serum albumin: pH-dependence of rates of individual steps. Biochem. Biophys. Res. Commun. 2012, 424, 451–455. [Google Scholar] [CrossRef]
- Ascenzi, P.; Fasano, M. Pseudo-enzymatic hydrolysis of 4-nitrophenyl myristate by human serum albumin. Biochem. Biophys. Res. Commun. 2012, 422, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Gerasimova, Y.V.; Erchenko, I.A.; Shakirov, M.M.; Godovikova, T.S. Interaction of human serum albumin and its clinically relevant modification with oligoribonucleotides. Bioorg. Med. Chem. Lett. 2008, 18, 4511–4514. [Google Scholar] [CrossRef]
- Gerasimova, Y.V.; Knorre, D.D.; Shakirov, M.M.; Godovikova, T.S. Human serum albumin as a catalyst of RNA cleavage: N-homocysteinylation and N-phosphorylation by oligonucleotide affinity reagent alter the reactivity of the protein. Bioorg. Med. Chem. Lett. 2008, 18, 5396–5398. [Google Scholar] [CrossRef]
- Drmanovic, Z.; Voyatzi, S.; Kouretas, D.; Sahpazidou, D.; Papageorgiou, A.; Antonoglou, O. Albumin possesses intrinsic enolase activity towards dihydrotestosterone which can differentiate benign from malignant breast tumors. Anticancer Res. 1999, 19, 4113–4124. [Google Scholar] [PubMed]
- Rahman, M.H.; Maruyama, T.; Okada, T.; Imai, T.; Otagiri, M. Study of interaction of carprofen and its enantiomers with human serum albumin--II. Stereoselective site-to-site displacement of carprofen by ibuprofen. Biochem. Pharmacol. 1993, 46, 1733–1740. [Google Scholar] [CrossRef]
- Wells, D.S.; Janssen, F.W.; Ruelius, H.W. Interactions between oxaprozin glucuronide and human serum albumin. Xenobiotica Fate Foreign Compd. Biol. Syst. 1987, 17, 1437–1449. [Google Scholar] [CrossRef]
- Ruelius, H.W.; Kirkman, S.K.; Young, E.M.; Janssen, F.W. Reactions of oxaprozin-1-O-acyl glucuronide in solutions of human plasma and albumin. Adv. Exp. Med. Biol. 1986, 197, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.K.; Kim, I.H. Disulfide between Cys392 and Cys438 of human serum albumin is redox-active, which is responsible for the thioredoxin-supported lipid peroxidase activity. Arch. Biochem. Biophys. 2006, 445, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, F.; Berti, F.; Bidoggia, S. Aldolase activity of serum albumins. Org. Biomol. Chem. 2011, 9, 4417–4420. [Google Scholar] [CrossRef]
- Zunszain, P.A.; Ghuman, J.; McDonagh, A.F.; Curry, S. Crystallographic analysis of human serum albumin complexed with 4Z,15E-bilirubin-IXalpha. J. Mol. Biol. 2008, 381, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Wardell, M.; Wang, Z.; Ho, J.X.; Robert, J.; Ruker, F.; Ruble, J.; Carter, D.C. The atomic structure of human methemalbumin at 1.9 A. Biochem. Biophys. Res. Commun. 2002, 291, 813–819. [Google Scholar] [CrossRef]
- Zunszain, P.A.; Ghuman, J.; Komatsu, T.; Tsuchida, E.; Curry, S. Crystal structural analysis of human serum albumin complexed with hemin and fatty acid. BMC Struct. Biol. 2003, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Somoza, J.R.; Skene, R.J.; Katz, B.A.; Mol, C.; Ho, J.D.; Jennings, A.J.; Luong, C.; Arvai, A.; Buggy, J.J.; Chi, E.; et al. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 2004, 12, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.E. Lysine residue 199 of human serum albumin is modified by acetylsalicyclic acid. FEBS Lett. 1976, 66, 173–175. [Google Scholar] [CrossRef]
- Diaz, N.; Suarez, D.; Sordo, T.L.; Merz, K.M., Jr. A theoretical study of the aminolysis reaction of lysine 199 of human serum albumin with benzylpenicillin: Consequences for immunochemistry of penicillins. J. Am. Chem. Soc. 2001, 123, 7574–7583. [Google Scholar] [CrossRef] [PubMed]
- Kragh-Hansen, U. Molecular and practical aspects of the enzymatic properties of human serum albumin and of albumin-ligand complexes. Biochim. Biophys. Acta 2013, 1830, 5535–5544. [Google Scholar] [CrossRef]
- Gresner, P.; Dolnik, M.; Waczulikova, I.; Bryszewska, M.; Sikurova, L.; Watala, C. Increased blood plasma hydrolysis of acetylsalicylic acid in type 2 diabetic patients: A role of plasma esterases. Biochim. Biophys. Acta 2006, 1760, 207–215. [Google Scholar] [CrossRef]
- Bojko, B.; Sulkowska, A.; Maciazek, M.; Rownicka, J.; Njau, F.; Sulkowski, W.W. Changes of serum albumin affinity for aspirin induced by fatty acid. Int. J. Biol. Macromol. 2008, 42, 314–323. [Google Scholar] [CrossRef]
- He, X.M.; Carter, D.C. Atomic structure and chemistry of human serum albumin. Nature 1992, 358, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Honma, K.; Nakamura, M.; Ishikawa, Y. Acetylsalicylate-human serum albumin interaction as studied by NMR spectroscopy--antigenicity-producing mechanism of acetylsalicylic acid. Mol. Immunol. 1991, 28, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Ma, S.F.; Watanabe, H.; Yamaotsu, N.; Hirono, S.; Kurono, Y.; Kragh-Hansen, U.; Otagiri, M. Esterase-like activity of serum albumin: Characterization of its structural chemistry using p-nitrophenyl esters as substrates. Pharm. Res. 2004, 21, 285–292. [Google Scholar] [CrossRef]
- Chapuis, N.; Bruhlmann, C.; Reist, M.; Carrupt, P.A.; Mayer, J.M.; Testa, B. The esterase-like activity of serum albumin may be due to cholinesterase contamination. Pharm. Res. 2001, 18, 1435–1439. [Google Scholar] [CrossRef]
- Kumar, D.; Bhattacharyya, R.; Banerjee, D. Pseudosterase activity-based specific detection of human serum albumin on gel. Talanta 2021, 224, 121906. [Google Scholar] [CrossRef]
- Keim, S.A.; Kulkarni, M.M.; McNamara, K.; Geraghty, S.R.; Billock, R.M.; Ronau, R.; Hogan, J.S.; Kwiek, J.J. Cow’s Milk Contamination of Human Milk Purchased via the Internet. Pediatrics 2015, 135, e1157–e1162. [Google Scholar] [CrossRef] [PubMed]
- Mung, D.; Li, L. Applying quantitative metabolomics based on chemical isotope labeling LC-MS for detecting potential milk adulterant in human milk. Anal. Chim. Acta 2018, 1001, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, R.; Kuwahara, K.; Wen, Y.C.; Yen, T.H.; Hiruta, Y.; Cheng, C.M.; Citterio, D. Paper-Based Device for Naked Eye Urinary Albumin/Creatinine Ratio Evaluation. ACS Sens. 2020, 5, 1110–1118. [Google Scholar] [CrossRef]
- Takahashi, S.; Uchino, H.; Shimizu, T.; Kanazawa, A.; Tamura, Y.; Sakai, K.; Watada, H.; Hirose, T.; Kawamori, R.; Tanaka, Y. Comparison of glycated albumin (GA) and glycated hemoglobin (HbA1c) in type 2 diabetic patients: Usefulness of GA for evaluation of short-term changes in glycemic control. Endocr. J. 2007, 54, 139–144. [Google Scholar] [CrossRef] [PubMed]
- di Masi, A.; De Marinis, E.; Ascenzi, P.; Marino, M. Nuclear receptors CAR and PXR: Molecular, functional, and biomedical aspects. Mol. Asp. Med. 2009, 30, 297–343. [Google Scholar] [CrossRef]
- Knadler, M.P.; Hall, S.D. Stereoselective hydrolysis of flurbiprofen conjugates. Drug Metab. Dispos. 1991, 19, 280–282. [Google Scholar] [PubMed]
- Bailey, M.J.; Dickinson, R.G. Acyl glucuronide reactivity in perspective: Biological consequences. Chem.-Biol. Interact. 2003, 145, 117–137. [Google Scholar] [CrossRef]
- Mizuma, T.; Benet, L.Z.; Lin, E.T. Preparative chromatography of furosemide 1-O-acyl-glucuronide from urine using micronized amberiite XAD-2 and its application to other 1-O-acyl-glucuronides. Prep. Biochem. Biotechnol. 1998, 28, 37–47. [Google Scholar] [CrossRef]
- Mizuma, T.; Benet, L.Z.; Lin, E.T. Interaction of human serum albumin with furosemide glucuronide: A role of albumin in isomerization, hydrolysis, reversible binding and irreversible binding of a 1-O-acyl glucuronide metabolite. Biopharm. Drug Dispos. 1999, 20, 131–136. [Google Scholar] [CrossRef]
- Bedford, C.T. Glucuronic acid conjugates. J. Chromatogr. B Biomed. Sci. Appl. 1998, 717, 313–326. [Google Scholar] [CrossRef]
- Georges, H.; Presle, N.; Buronfosse, T.; Fournel-Gigleux, S.; Netter, P.; Magdalou, J.; Lapicque, F. In vitro stereoselective degradation of carprofen glucuronide by human serum albumin. Characterization of sites and reactive amino acids. Chirality 2000, 12, 53–62. [Google Scholar] [CrossRef]
- Bueno, C.J.; Jimenez, M.C.; Miranda, M.A. In situ transient spectroscopy for the study of glucuronidase activity within serum albumin. J. Phys. Chem. B 2009, 113, 6861–6865. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, I.H. Thioredoxin-linked lipid hydroperoxide peroxidase activity of human serum albumin in the presence of palmitoyl coenzyme A. Free Radic. Biol. Med. 2001, 30, 327–333. [Google Scholar] [CrossRef]
- Kakizoe, T.; Komatsu, H.; Honma, Y.; Niijima, T.; Kawachi, T.; Sugimura, T.; Nagase, S. High susceptibility of analbuminaemic rats to induced bladder cancer. Br. J. Cancer 1982, 45, 474–476. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yamamoto, Y.; Wakabayashi, K.; Niki, E.; Nagao, M. Comparison of plasma levels of lipid hydroperoxides and antioxidants in hyperlipidemic Nagase analbuminemic rats, Sprague-Dawley rats, and humans. Biochem. Biophys. Res. Commun. 1992, 189, 518–523. [Google Scholar] [CrossRef]
- Alvarez, J.G.; Storey, B.T. Taurine, hypotaurine, epinephrine and albumin inhibit lipid peroxidation in rabbit spermatozoa and protect against loss of motility. Biol. Reprod. 1983, 29, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Soejima, A.; Matsuzawa, N.; Miyake, N.; Karube, M.; Fukuoka, K.; Nakabayashi, K.; Kitamoto, K.; Nagasawa, T. Hypoalbuminemia accelerates erythrocyte membrane lipid peroxidation in chronic hemodialysis patients. Clin. Nephrol. 1999, 51, 92–97. [Google Scholar]
- Luisi, I.; Pavan, S.; Fontanive, G.; Tossi, A.; Benedetti, F.; Savoini, A.; Maurizio, E.; Sgarra, R.; Sblattero, D.; Berti, F. An albumin-derived peptide scaffold capable of binding and catalysis. PLoS ONE 2013, 8, e56469. [Google Scholar] [CrossRef]
- Cha, M.K.; Kim, I.H. Glutathione-linked thiol peroxidase activity of human serum albumin: A possible antioxidant role of serum albumin in blood plasma. Biochem. Biophys. Res. Commun. 1996, 222, 619–625. [Google Scholar] [CrossRef]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, E.; Blache, D. The importance of proteins in defense against oxidation. Antioxid. Redox Signal. 2001, 3, 293–311. [Google Scholar] [CrossRef]
- Iwao, Y.; Ishima, Y.; Yamada, J.; Noguchi, T.; Kragh-Hansen, U.; Mera, K.; Honda, D.; Suenaga, A.; Maruyama, T.; Otagiri, M. Quantitative evaluation of the role of cysteine and methionine residues in the antioxidant activity of human serum albumin using recombinant mutants. IUBMB Life 2012, 64, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Berlett, B.S.; Stadtman, E.R. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 1997, 272, 20313–20316. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Mosoni, L.; Berlett, B.S.; Stadtman, E.R. Methionine residues as endogenous antioxidants in proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 15036–15040. [Google Scholar] [CrossRef] [PubMed]
- Moskovitz, J.; Flescher, E.; Berlett, B.S.; Azare, J.; Poston, J.M.; Stadtman, E.R. Overexpression of peptide-methionine sulfoxide reductase in Saccharomyces cerevisiae and human T cells provides them with high resistance to oxidative stress. Proc. Natl. Acad. Sci. USA 1998, 95, 14071–14075. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, F.; Shibata, T.; Kamiya, K.; Yoshitake, J.; Kikuchi, R.; Matsushita, T.; Ishii, I.; Gimenez-Bastida, J.A.; Schneider, C.; Uchida, K. Structural and functional insights into S-thiolation of human serum albumins. Sci. Rep. 2018, 8, 932. [Google Scholar] [CrossRef]
- Nakashima, F.; Shibata, T.; Uchida, K. A unique mechanism for thiolation of serum albumins by disulphide molecules. J. Biochem. 2020, 167, 165–171. [Google Scholar] [CrossRef]
- Lloyd, D.R.; Phillips, D.H. Oxidative DNA damage mediated by copper(II), iron(II) and nickel(II) fenton reactions: Evidence for site-specific mechanisms in the formation of double-strand breaks, 8-hydroxydeoxyguanosine and putative intrastrand cross-links. Mutat. Res. 1999, 424, 23–36. [Google Scholar] [CrossRef]
- Halliwell, B. Albumin--an important extracellular antioxidant? Biochem. Pharmacol. 1988, 37, 569–571. [Google Scholar] [CrossRef]
- Bar-Or, D.; Rael, L.T.; Lau, E.P.; Rao, N.K.; Thomas, G.W.; Winkler, J.V.; Yukl, R.L.; Kingston, R.G.; Curtis, C.G. An analog of the human albumin N-terminus (Asp-Ala-His-Lys) prevents formation of copper-induced reactive oxygen species. Biochem. Biophys. Res. Commun. 2001, 284, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Gum, E.T.; Swanson, R.A.; Alano, C.; Liu, J.; Hong, S.; Weinstein, P.R.; Panter, S.S. Human serum albumin and its N-terminal tetrapeptide (DAHK) block oxidant-induced neuronal death. Stroke 2004, 35, 590–595. [Google Scholar] [CrossRef]
- Sendzik, M.; Pushie, M.J.; Stefaniak, E.; Haas, K.L. Structure and Affinity of Cu(I) Bound to Human Serum Albumin. Inorg. Chem. 2017, 56, 15057–15065. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bocedi, A.; Visca, P.; Minetti, M.; Clementi, E. Does CO2 modulate peroxynitrite specificity? IUBMB Life 2006, 58, 611–613. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; di Masi, A.; De Sanctis, G.; Coletta, M.; Fasano, M. Ibuprofen modulates allosterically NO dissociation from ferrous nitrosylated human serum heme-albumin by binding to three sites. Biochem. Biophys. Res. Commun. 2009, 387, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; di Masi, A.; Coletta, M.; Ciaccio, C.; Fanali, G.; Nicoletti, F.P.; Smulevich, G.; Fasano, M. Ibuprofen impairs allosterically peroxynitrite isomerization by ferric human serum heme-albumin. J. Biol. Chem. 2009, 284, 31006–31017. [Google Scholar] [CrossRef]
- Roche, M.; Rondeau, P.; Singh, N.R.; Tarnus, E.; Bourdon, E. The antioxidant properties of serum albumin. FEBS Lett. 2008, 582, 1783–1787. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.W.; Wu, J.; Li, R.K.; Mickle, D.; Carey, D. Albumin-bound bilirubins protect human ventricular myocytes against oxyradical damage. Biochem. Cell Biol. 1991, 69, 683–688. [Google Scholar] [CrossRef]
- Neuzil, J.; Stocker, R. Bilirubin attenuates radical-mediated damage to serum albumin. FEBS Lett. 1993, 331, 281–284. [Google Scholar] [CrossRef]
- Neuzil, J.; Stocker, R. Free and albumin-bound bilirubin are efficient co-antioxidants for α-tocopherol, inhibiting plasma and low density lipoprotein lipid peroxidation. J. Biol. Chem. 1994, 269, 16712–16719. [Google Scholar] [CrossRef]
- Papatheodorou, L.; Weiss, N. Vascular oxidant stress and inflammation in hyperhomocysteinemia. Antioxid. Redox Signal. 2007, 9, 1941–1958. [Google Scholar] [CrossRef]
- Fujii, R.; Ueyama, J.; Aoi, A.; Ichino, N.; Osakabe, K.; Sugimoto, K.; Suzuki, K.; Hamajima, N.; Wakai, K.; Kondo, T. Oxidized human serum albumin as a possible correlation factor for atherosclerosis in a rural Japanese population: The results of the Yakumo Study. Environ. Health Prev. Med. 2018, 23, 1. [Google Scholar] [CrossRef]
- van Reyk, D.M.; Brown, A.J.; Hult’en, L.M.; Dean, R.T.; Jessup, W. Oxysterols in biological systems: Sources, metabolism and pathophysiological relevance. Redox Rep. 2006, 11, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Ishima, Y.; Kinoshita, R.; Chuang, V.T.G.; Tasaka, N.; Matsuo, N.; Watanabe, H.; Shimizu, T.; Ishida, T.; Otagiri, M.; et al. A novel S-sulfhydrated human serum albumin preparation suppresses melanin synthesis. Redox Biol. 2018, 14, 354–360. [Google Scholar] [CrossRef]
- Taverna, M.; Marie, A.L.; Mira, J.P.; Guidet, B. Specific antioxidant properties of human serum albumin. Ann. Intensive Care 2013, 3, 4. [Google Scholar] [CrossRef]
- Turell, L.; Botti, H.; Carballal, S.; Ferrer-Sueta, G.; Souza, J.M.; Duran, R.; Freeman, B.A.; Radi, R.; Alvarez, B. Reactivity of sulfenic acid in human serum albumin. Biochemistry 2008, 47, 358–367. [Google Scholar] [CrossRef]
- Bocedi, A.; Cattani, G.; Stella, L.; Massoud, R.; Ricci, G. Thiol disulfide exchange reactions in human serum albumin: The apparent paradox of the redox transitions of Cys34. FEBS J. 2018, 285, 3225–3237. [Google Scholar] [CrossRef]
- Sharma, G.S.; Kumar, T.; Singh, L.R. N-homocysteinylation induces different structural and functional consequences on acidic and basic proteins. PLoS ONE 2014, 9, e116386. [Google Scholar] [CrossRef]
- Perla-Kajan, J.; Twardowski, T.; Jakubowski, H. Mechanisms of homocysteine toxicity in humans. Amino Acids 2007, 32, 561–572. [Google Scholar] [CrossRef]
- Sikora, M.; Marczak, L.; Twardowski, T.; Stobiecki, M.; Jakubowski, H. Direct monitoring of albumin lysine-525 N-homocysteinylation in human serum by liquid chromatography/mass spectrometry. Anal. Biochem. 2010, 405, 132–134. [Google Scholar] [CrossRef]
- Zinellu, A.; Sotgia, S.; Scanu, B.; Arru, D.; Cossu, A.; Posadino, A.M.; Giordo, R.; Mangoni, A.A.; Pintus, G.; Carru, C. N- and S-homocysteinylation reduce the binding of human serum albumin to catechins. Eur. J. Nutr. 2017, 56, 785–791. [Google Scholar] [CrossRef]
- Jakubowski, H. Metabolism of homocysteine thiolactone in human cell cultures. Possible mechanism for pathological consequences of elevated homocysteine levels. J. Biol. Chem. 1997, 272, 1935–1942. [Google Scholar] [CrossRef]
- Hasan, T.; Arora, R.; Bansal, A.K.; Bhattacharya, R.; Sharma, G.S.; Singh, L.R. Disturbed homocysteine metabolism is associated with cancer. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Nagumo, K.; Tanaka, M.; Chuang, V.T.; Setoyama, H.; Watanabe, H.; Yamada, N.; Kubota, K.; Tanaka, M.; Matsushita, K.; Yoshida, A.; et al. Cys34-cysteinylated human serum albumin is a sensitive plasma marker in oxidative stress-related chronic diseases. PLoS ONE 2014, 9, e85216. [Google Scholar] [CrossRef] [PubMed]
- Tabata, F.; Wada, Y.; Kawakami, S.; Miyaji, K. Serum Albumin Redox States: More Than Oxidative Stress Biomarker. Antioxidants 2021, 10, 503. [Google Scholar] [CrossRef]
- Oettl, K.; Marsche, G. Redox state of human serum albumin in terms of cysteine-34 in health and disease. Methods Enzymol. 2010, 474, 181–195. [Google Scholar] [CrossRef]
- Brioschi, M.; Gianazza, E.; Mallia, A.; Zoanni, B.; Altomare, A.; Martinez Fernandez, A.; Agostoni, P.; Aldini, G.; Banfi, C. S-Thiolation Targets Albumin in Heart Failure. Antioxidants 2020, 9, 763. [Google Scholar] [CrossRef]
- Grounds, M.D.; Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Radley-Crabb, H.G.; Arthur, P.G. Biomarkers for Duchenne muscular dystrophy: Myonecrosis, inflammation and oxidative stress. Dis. Models Mech. 2020, 13, dmm043638. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Horrillo, R.; Ortiz, A.M.; Perez, A.; Mestre, A.; Ruiz, A.; Boada, M.; Grancha, S. Increased Albumin Oxidation in Cerebrospinal Fluid and Plasma from Alzheimer’s Disease Patients. J. Alzheimers Dis. JAD 2018, 63, 1395–1404. [Google Scholar] [CrossRef]
- Ueno, S.I.; Hatano, T.; Okuzumi, A.; Saiki, S.; Oji, Y.; Mori, A.; Koinuma, T.; Fujimaki, M.; Takeshige-Amano, H.; Kondo, A.; et al. Nonmercaptalbumin as an oxidative stress marker in Parkinson’s and PARK2 disease. Ann. Clin. Transl. Neurol. 2020, 7, 307–317. [Google Scholar] [CrossRef]
- Nasif, W.A.; Mukhtar, M.H.; El-Emshaty, H.M.; Alwazna, A.H. Redox State of Human Serum Albumin and Inflammatory Biomarkers in Hemodialysis Patients with Secondary Hyperparathyroidism During Oral Calcitriol Supplementation for Vitamin D. Open Med. Chem. J. 2018, 12, 98–110. [Google Scholar] [CrossRef]
- Rael, L.T.; Leonard, J.; Salottolo, K.; Bar-Or, R.; Bartt, R.E.; Wagner, J.C.; Bar-Or, D. Plasma Oxidized Albumin in Acute Ischemic Stroke Is Associated With Better Outcomes. Front. Neurol. 2019, 10, 709. [Google Scholar] [CrossRef] [PubMed]
- Rahmani-Kukia, N.; Abbasi, A.; Pakravan, N.; Hassan, Z.M. Measurement of oxidized albumin: An opportunity for diagnoses or treatment of COVID-19. Bioorg. Chem. 2020, 105, 104429. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Morales, A.J.; Cardona-Ospina, J.A.; Gutierrez-Ocampo, E.; Villamizar-Pena, R.; Holguin-Rivera, Y.; Escalera-Antezana, J.P.; Alvarado-Arnez, L.E.; Bonilla-Aldana, D.K.; Franco-Paredes, C.; Henao-Martinez, A.F.; et al. Clinical, laboratory and imaging features of COVID-19: A systematic review and meta-analysis. Travel Med. Infect. Dis. 2020, 34, 101623. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Cangemi, R.; Romiti, G.F.; Ceccarelli, G.; Oliva, A.; Alessandri, F.; Pirro, M.; Pignatelli, P.; Lichtner, M.; Carraro, A.; et al. Is Albumin Predictor of Mortality in COVID-19? Antioxid. Redox Signal. 2021, 35, 139–142. [Google Scholar] [CrossRef]
- Vetter, S.W.; Indurthi, V.S. Moderate glycation of serum albumin affects folding, stability, and ligand binding. Clin. Chim. Acta Int. J. Clin. Chem. 2011, 412, 2105–2116. [Google Scholar] [CrossRef]
- Sattarahmady, N.; Moosavi-Movahedi, A.A.; Ahmad, F.; Hakimelahi, G.H.; Habibi-Rezaei, M.; Saboury, A.A.; Sheibani, N. Formation of the molten globule-like state during prolonged glycation of human serum albumin. Biochim. Biophys. Acta 2007, 1770, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Bouma, B.; Kroon-Batenburg, L.M.; Wu, Y.P.; Brunjes, B.; Posthuma, G.; Kranenburg, O.; de Groot, P.G.; Voest, E.E.; Gebbink, M.F. Glycation induces formation of amyloid cross-beta structure in albumin. J. Biol. Chem. 2003, 278, 41810–41819. [Google Scholar] [CrossRef]
- Bourdon, E.; Loreau, N.; Blache, D. Glucose and free radicals impair the antioxidant properties of serum albumin. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1999, 13, 233–244. [Google Scholar] [CrossRef]
- Rondeau, P.; Singh, N.R.; Caillens, H.; Tallet, F.; Bourdon, E. Oxidative stresses induced by glycoxidized human or bovine serum albumin on human monocytes. Free Radic. Biol. Med. 2008, 45, 799–812. [Google Scholar] [CrossRef]
- Gryzunov, Y.A.; Arroyo, A.; Vigne, J.L.; Zhao, Q.; Tyurin, V.A.; Hubel, C.A.; Gandley, R.E.; Vladimirov, Y.A.; Taylor, R.N.; Kagan, V.E. Binding of fatty acids facilitates oxidation of cysteine-34 and converts copper-albumin complexes from antioxidants to prooxidants. Arch. Biochem. Biophys. 2003, 413, 53–66. [Google Scholar] [CrossRef]
- Rossi, R.; Giustarini, D.; Milzani, A.; Dalle-Donne, I. Cysteinylation and homocysteinylation of plasma protein thiols during ageing of healthy human beings. J. Cell. Mol. Med. 2009, 13, 3131–3140. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Suzuki, R.; Yasukawa, K.; Oba, K.; Yamauchi, T.; Yatomi, Y.; Kadowaki, T. Oxidized albumin in blood reflects the severity of multiple vascular complications in diabetes mellitus. Metab. Open 2020, 6, 100032. [Google Scholar] [CrossRef]
- Muller-Eberhard, U.; Javid, J.; Liem, H.H.; Hanstein, A.; Hanna, M. Plasma concentrations of hemopexin, haptoglobin and heme in patients with various hemolytic diseases. Blood 1968, 32, 811–815. [Google Scholar] [CrossRef]
- Miller, Y.I.; Shaklai, N. Kinetics of hemin distribution in plasma reveals its role in lipoprotein oxidation. Biochim. Biophys. Acta 1999, 1454, 153–164. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M. Role of free radicals and catalytic metal ions in human disease: An overview. Methods Enzymol. 1990, 186, 1–85. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Fasano, M. Heme-hemopexin: A ‘chronosteric’ heme-protein. IUBMB Life 2007, 59, 700–708. [Google Scholar] [CrossRef]
- di Masi, A.; Gullotta, F.; Bolli, A.; Fanali, G.; Fasano, M.; Ascenzi, P. Ibuprofen binding to secondary sites allosterically modulates the spectroscopic and catalytic properties of human serum heme-albumin. FEBS J. 2011, 278, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Kamal, J.K.; Behere, D.V. Spectroscopic studies on human serum albumin and methemalbumin: Optical, steady-state, and picosecond time-resolved fluorescence studies, and kinetics of substrate oxidation by methemalbumin. J. Biol. Inorg. Chem. 2002, 7, 273–283. [Google Scholar] [CrossRef]
- Antonini, E.; Brunori, M. Hemoglobin and Myoglobin in Their Reactions with Ligands; North Holland Publishing Co.: Amsterdam, The Netherlands; London, UK, 1971. [Google Scholar]
- Alayash, A.I.; Patel, R.P.; Cashon, R.E. Redox reactions of hemoglobin and myoglobin: Biological and toxicological implications. Antioxid. Redox Signal. 2001, 3, 313–327. [Google Scholar] [CrossRef]
- Meneghini, C.; Leboffe, L.; Bionducci, M.; Fanali, G.; Meli, M.; Colombo, G.; Fasano, M.; Ascenzi, P.; Mobilio, S. The five-to-six-coordination transition of ferric human serum heme-albumin is allosterically-modulated by ibuprofen and warfarin: A combined XAS and MD study. PLoS ONE 2014, 9, e104231. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, F.P.; Howes, B.D.; Fittipaldi, M.; Fanali, G.; Fasano, M.; Ascenzi, P.; Smulevich, G. Ibuprofen induces an allosteric conformational transition in the heme complex of human serum albumin with significant effects on heme ligation. J. Am. Chem. Soc. 2008, 130, 11677–11688. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Fasano, M. Abacavir modulates peroxynitrite-mediated oxidation of ferrous nitrosylated human serum heme-albumin. Biochem. Biophys. Res. Commun. 2007, 353, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, S.; Lind, J.; Merenyi, G. Chemistry of peroxynitrites as compared to peroxynitrates. Chem. Rev. 2005, 105, 2457–2470. [Google Scholar] [CrossRef]
- Papina, A.A.; Koppenol, W.H. Two pathways of carbon dioxide catalyzed oxidative coupling of phenol by peroxynitrite. Chem. Res. Toxicol. 2006, 19, 382–391. [Google Scholar] [CrossRef]
- Ascenzi, P.; Visca, P. Scavenging of reactive nitrogen species by mycobacterial truncated hemoglobins. Methods Enzymol. 2008, 436, 317–337. [Google Scholar] [CrossRef]
- Goldstein, S.; Merenyi, G. The chemistry of peroxynitrite: Implications for biological activity. Methods Enzymol. 2008, 436, 49–61. [Google Scholar] [CrossRef]
- Grinberg, L.N.; O’Brien, P.J.; Hrkal, Z. The effects of heme-binding proteins on the peroxidative and catalatic activities of hemin. Free Radic. Biol. Med. 1999, 27, 214–219. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Simone, G.; di Masi, A.; Ascenzi, P. Serum Albumin: A Multifaced Enzyme. Int. J. Mol. Sci. 2021, 22, 10086. https://doi.org/10.3390/ijms221810086
De Simone G, di Masi A, Ascenzi P. Serum Albumin: A Multifaced Enzyme. International Journal of Molecular Sciences. 2021; 22(18):10086. https://doi.org/10.3390/ijms221810086
Chicago/Turabian StyleDe Simone, Giovanna, Alessandra di Masi, and Paolo Ascenzi. 2021. "Serum Albumin: A Multifaced Enzyme" International Journal of Molecular Sciences 22, no. 18: 10086. https://doi.org/10.3390/ijms221810086
APA StyleDe Simone, G., di Masi, A., & Ascenzi, P. (2021). Serum Albumin: A Multifaced Enzyme. International Journal of Molecular Sciences, 22(18), 10086. https://doi.org/10.3390/ijms221810086