
Keratin 8/18 Regulate the Akt Signaling Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
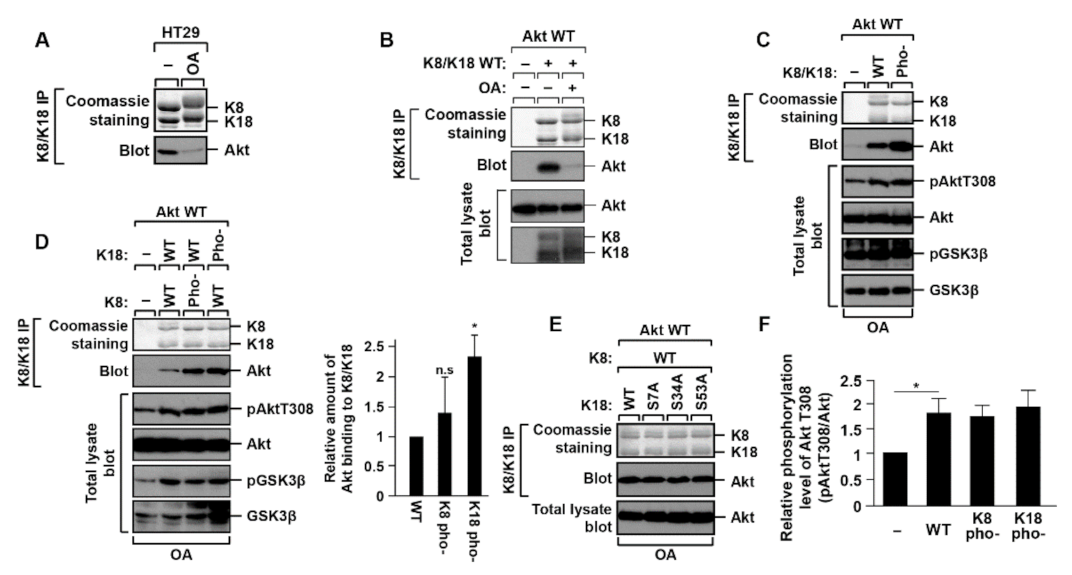
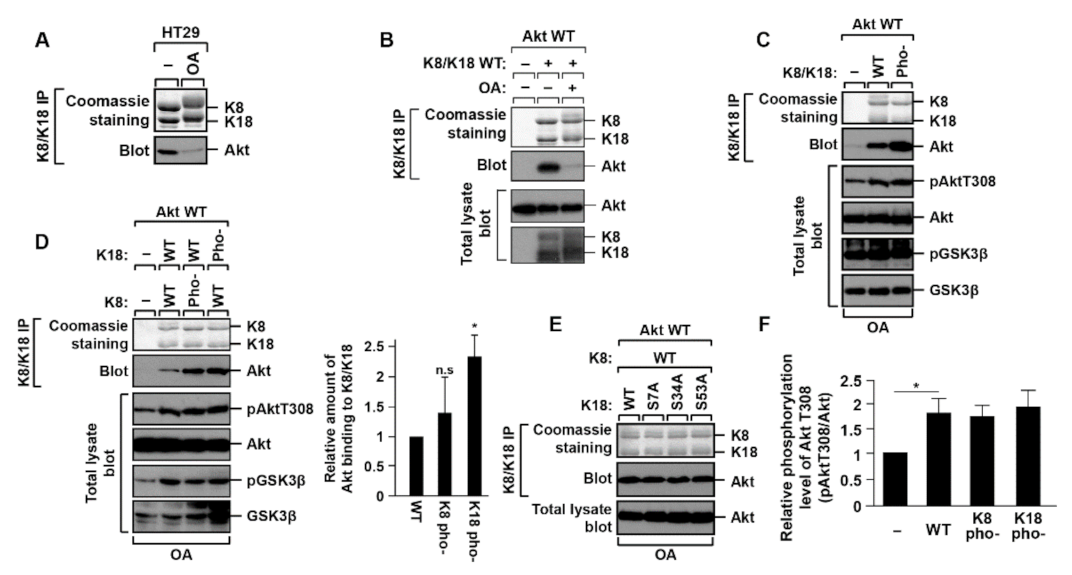
2.1. K18 Phosphorylation Inhibits K8/K18 Interaction with Akt Kinase
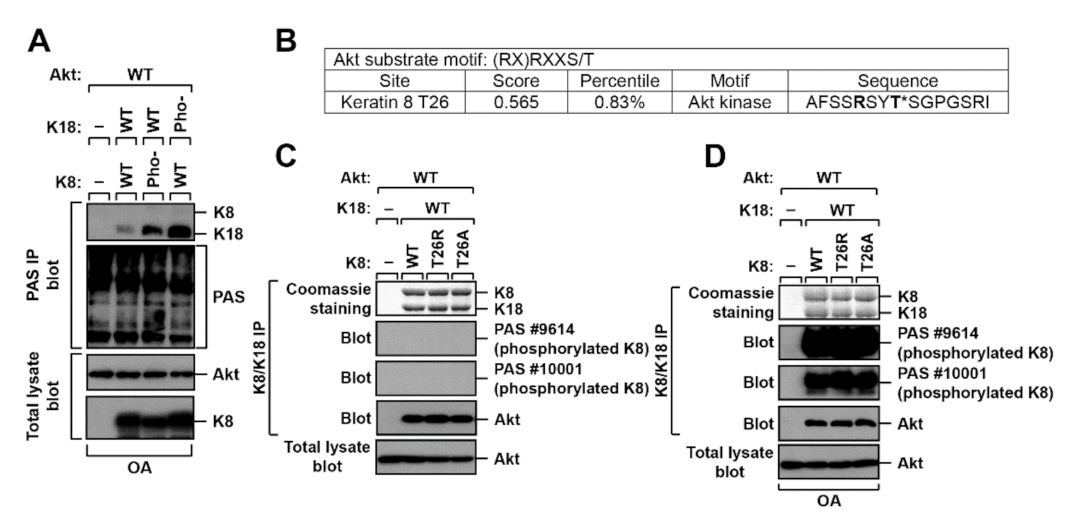
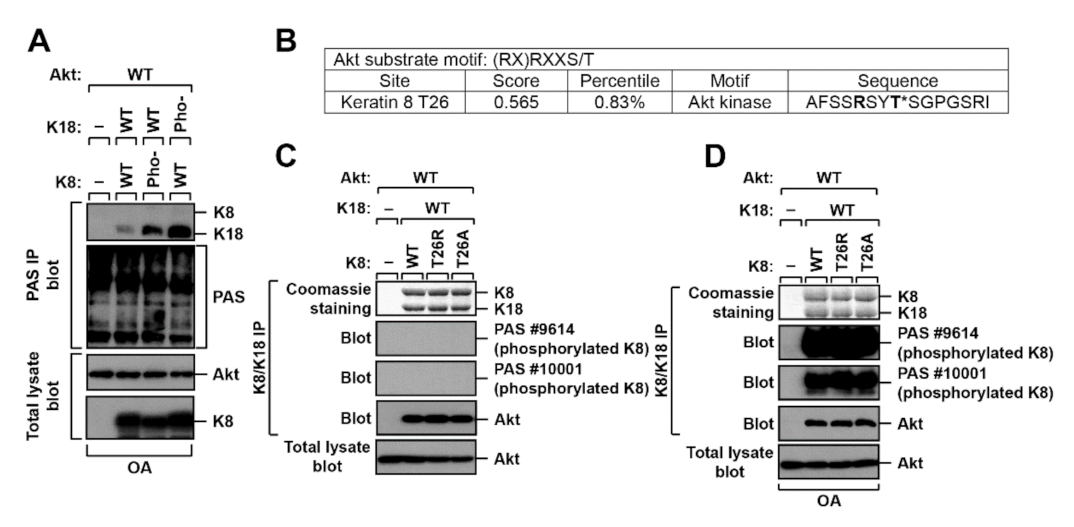
2.2. K8 and K18 as Potential Substrates of Akt
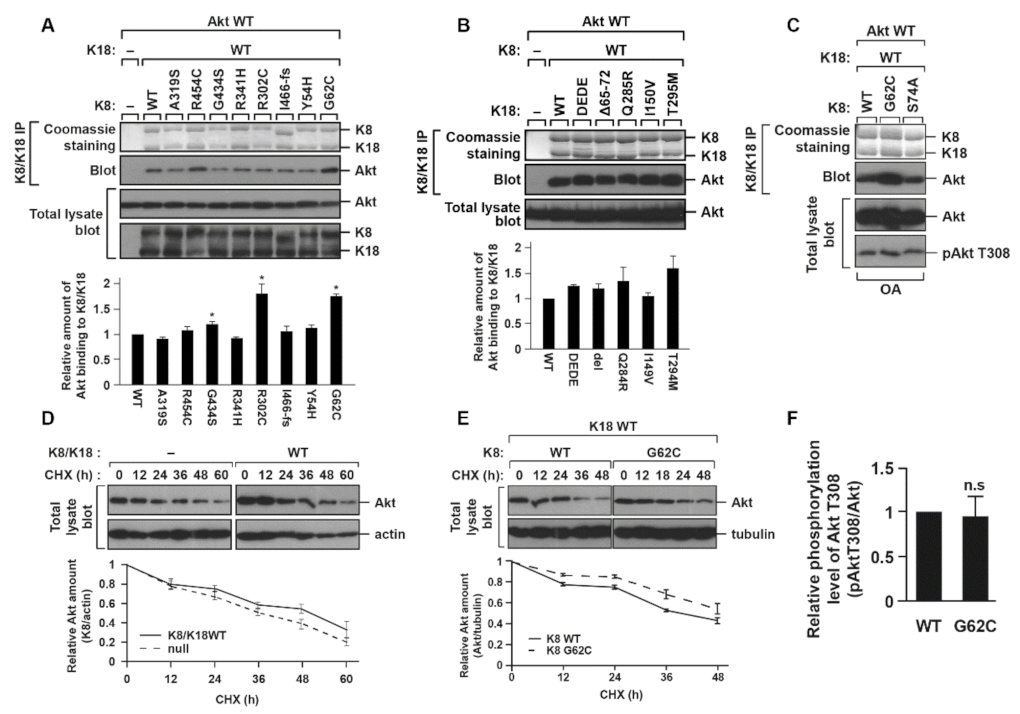
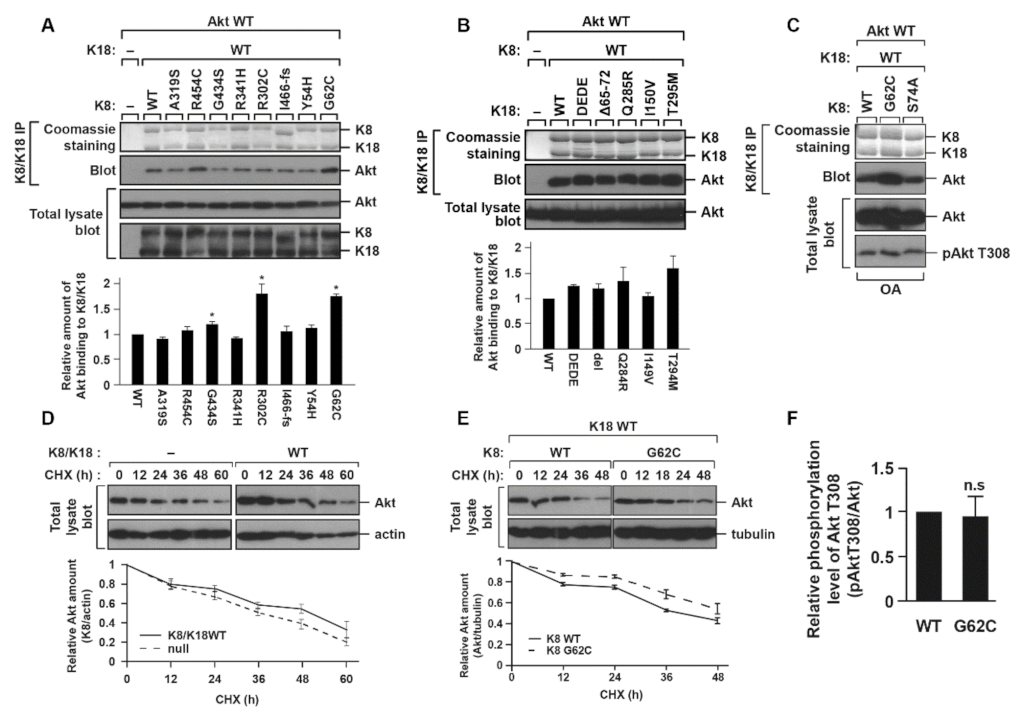
2.3. K8 G62C, R302C, and G434S Mutations, Found in Patients with Liver Diseases, Cause an Increase in the K8/K18–Akt Interaction
2.4. K8/K18–Akt Interaction Tends to Stabilize Akt
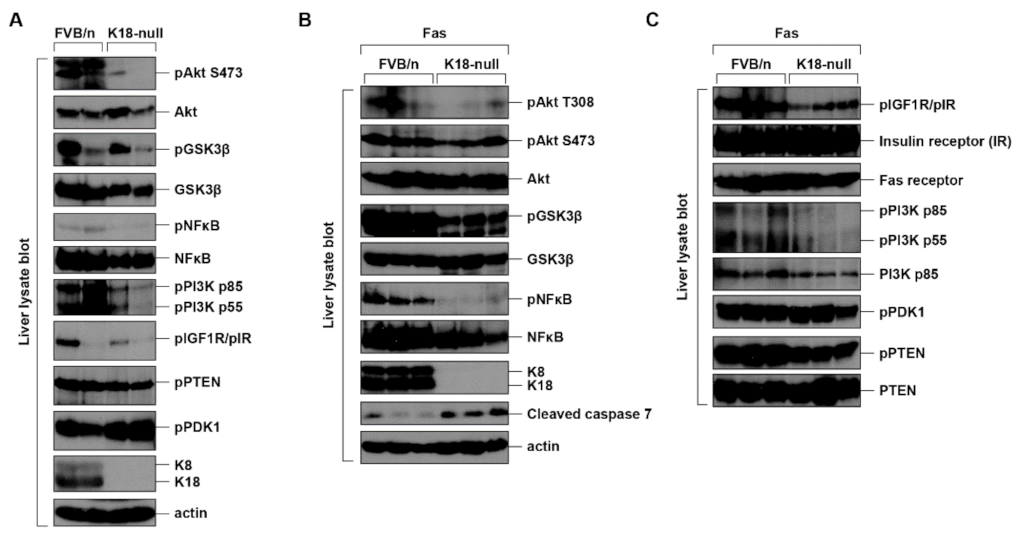
2.5. K8/K18 Expression Enhances the Phosphorylation/Activation of Akt and Its Substrates in the In Vivo Mouse System
3. Discussion
3.1. Akt-Mediated Resistance to Fas-Induced Apoptosis
3.2. A Role of K8/K18 in Tumor Progression
4. Material and Methods
4.1. List of Reagents and Antibodies
4.2. Cell Transfection and Preparation of Cell/Tissue Lysates
4.3. Co-Immunoprecipitation and Immunoblot Analysis
4.4. Protein Stability Analysis
4.5. Animals and TISSUE Collections
4.6. Site-Directed Mutagenesis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Loschke, F.; Seltmann, K.; Bouameur, J.E.; Magin, T.M. Regulation of keratin network organization. Curr. Opin. Cell Biol. 2015, 32, 56–64. [Google Scholar] [CrossRef]
- Omary, M.B. Intermediate filament proteins of digestive organs: Physiology and pathophysiology. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G628–G634. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.T.; Coulombe, P.A.; Kwan, R.; Omary, M.B. Types I and II keratin intermediate filaments. Cold Spring Harb. Perspect. Biol. 2018, 10, a018275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baribault, H.; Price, J.; Miyai, K.; Oshima, R.G. Mid-gestational lethality in mice lacking keratin 8. Genes Dev. 1993, 7, 1191–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baribault, H.; Penner, J.; Iozzo, R.V.; Wilson-Heiner, M. Colorectal hyperplasia and inflammation in keratin 8-deficient FVB/N mice. Genes Dev. 1994, 8, 2964–2973. [Google Scholar] [CrossRef] [Green Version]
- Strnad, P.; Stumptner, C.; Zatloukal, K.; Denk, H. Intermediate filament cytoskeleton of the liver in health and disease. Histochem. Cell Biol. 2008, 129, 735. [Google Scholar] [CrossRef] [Green Version]
- Magin, T.M.; Schröder, R.; Leitgeb, S.; Wanninger, F.; Zatloukal, K.; Grund, C.; Melton, D.W. Lessons from keratin 18 knockout mice: Formation of novel keratin filaments, secondary loss of keratin 7 and accumulation of liver-specific keratin 8-positive aggregates. J. Cell Biol. 1998, 140, 1441–1451. [Google Scholar] [CrossRef]
- Staal, S.P. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: Amplification of AKT1 in a primary human gastric adenocarcinoma. Proc. Natl. Acad. Sci. USA 1987, 84, 5034–5037. [Google Scholar] [CrossRef] [Green Version]
- Bellacosa, A.; Staal, S.; Tsichlis, P. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science 1991, 254, 274–277. [Google Scholar] [CrossRef]
- Coffer, P.J.; Woodgett, J.R. Molecular cloning and characterisation of a novel putative protein-serine kinase related to the cAMP-dependent and protein kinase C families. Eur. J. Biochem. 1991, 201, 475–481. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Chen, W.S.; Xu, P.-Z.; Gottlob, K.; Chen, M.-L.; Sokol, K.; Shiyanova, T.; Roninson, I.; Weng, W.; Suzuki, R.; Tobe, K. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001, 15, 2203–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, T. PI3K/Akt: Getting it right matters. Oncogene 2008, 27, 6473–6488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.S.; Rosenblatt, K.; Huang, K.L.; Lahat, G.; Brobey, R.; Bolshakov, S.; Nguyen, T.; Ding, Z.; Belousov, R.; Bill, K.; et al. Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene 2011, 30, 457–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cenni, V.; Bertacchini, J.; Beretti, F.; Lattanzi, G.; Bavelloni, A.; Riccio, M.; Ruzzene, M.; Marin, O.; Arrigoni, G.; Parnaik, V.; et al. Lamin A Ser404 is a nuclear target of Akt phosphorylation in C2C12 cells. J. Proteome Res. 2008, 7, 4727–4735. [Google Scholar] [CrossRef]
- Paramio, J.M.; Segrelles, C.; Ruiz, S.; Jorcano, J.L. Inhibition of protein kinase B (PKB) and PKCζ mediates keratin K10-induced cell cycle arrest. Mol. Cell. Biol. 2001, 21, 7449–7459. [Google Scholar] [CrossRef] [Green Version]
- Santos, M.; Paramio, J.M.; Bravo, A.; Ramirez, A.; Jorcano, J.L. The expression of keratin k10 in the basal layer of the epidermis inhibits cell proliferation and prevents skin tumorigenesis. J. Biol. Chem. 2002, 277, 19122–19130. [Google Scholar] [CrossRef] [Green Version]
- Tseng, Y.H.; Yang, C.C.; Lin, S.C.; Cheng, C.C.; Lin, S.H.; Liu, C.J.; Chang, K.W. Areca nut extract upregulates vimentin by activating PI3K/AKT signaling in oral carcinoma. J. Oral Pathol. Med. 2011, 40, 160–166. [Google Scholar] [CrossRef]
- Kidd, M.E.; Shumaker, D.K.; Ridge, K.M. The role of Vimentin intermediate filaments in the progression of lung cancer. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1–6. [Google Scholar] [CrossRef]
- Kong, L.; Schafer, G.; Bu, H.; Zhang, Y.; Zhang, Y.; Klocker, H. Lamin A/C protein is overexpressed in tissue-invading prostate cancer and promotes prostate cancer cell growth, migration and invasion through the PI3K/AKT/PTEN pathway. Carcinogenesis 2012, 33, 751–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Wong, P.; Coulombe, P.A. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature 2006, 441, 362–365. [Google Scholar] [CrossRef]
- Chivu-Economescu, M.; Dragu, D.L.; Necula, L.G.; Matei, L.; Enciu, A.M.; Bleotu, C.; Diaconu, C.C. Knockdown of KRT17 by siRNA induces antitumoral effects on gastric cancer cells. Gastric Cancer 2017, 20, 948–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, N.-O.; Toivola, D.M.; Strnad, P.; Omary, M.B. Cytoskeletal keratin glycosylation protects epithelial tissue from injury. Nat. Cell Biol. 2010, 12, 876–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walz, H.A.; Shi, X.; Chouinard, M.; Bue, C.A.; Navaroli, D.M.; Hayakawa, A.; Zhou, Q.L.; Nadler, J.; Leonard, D.M.; Corvera, S. Isoform-specific regulation of Akt signaling by the endosomal protein WDFY2. J. Biol. Chem. 2010, 285, 14101–14108. [Google Scholar] [CrossRef] [Green Version]
- Strnad, P.; Lienau, T.C.; Tao, G.Z.; Lazzeroni, L.C.; Stickel, F.; Schuppan, D.; Omary, M.B. Keratin variants associate with progression of fibrosis during chronic hepatitis C infection. Hepatology 2006, 43, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Ku, N.O.; Strnad, P.; Bantel, H.; Omary, M.B. Keratins: Biomarkers and modulators of apoptotic and necrotic cell death in the liver. Hepatology 2016, 64, 966–976. [Google Scholar] [CrossRef] [Green Version]
- Omary, M.B.; Coulombe, P.A.; McLean, W.H. Intermediate filament proteins and their associated diseases. N. Engl. J. Med. 2004, 351, 2087–2100. [Google Scholar] [CrossRef] [PubMed]
- Toivola, D.M.; Boor, P.; Alam, C.; Strnad, P. Keratins in health and disease. Curr. Opin. Cell Biol. 2015, 32, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Ku, N.-O.; Omary, M.B. A disease- and phosphorylation-related nonmechanical function for keratin 8. J. Cell Biol. 2006, 174, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Jang, K.H.; Kim, H.; Lim, Y.; Kim, S.; Yoon, H.N.; Chung, I.K.; Roth, J.; Ku, N.O. Predisposition to apoptosis in keratin 8-null liver is related to inactivation of NF-kappaB and SAPKs but not decreased c-Flip. Biol. Open 2013, 2, 695–702. [Google Scholar] [CrossRef] [Green Version]
- Gibson, S.; Tu, S.; Oyer, R.; Anderson, S.M.; Johnson, G.L. Epidermal growth factor protects epithelial cells against Fas-induced apoptosis: Requirement for Akt activation. J. Biol. Chem. 1999, 274, 17612–17618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, A.; Hayashida, M.; Kawano, H.; Sugimoto, K.; Nakano, T.; Shiraki, K. Hepatocyte growth factor promotes cell survival from Fas-mediated cell death in hepatocellular carcinoma cells via Akt activation and Fas-death-inducing signaling complex suppression. Hepatology 2000, 32, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Hatano, E.; Brenner, D.A. Akt protects mouse hepatocytes from TNF-α- and Fas-mediated apoptosis through NK-κB activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1357–G1368. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Bjerke, L.; Clarke, P.A.; Workman, P. Drugging PI3K in cancer: Refining targets and therapeutic strategies. Curr. Opin. Pharmacol. 2015, 23, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase–AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Moll, R.; Divo, M.; Langbein, L. The human keratins: Biology and pathology. Histochem. Cell Biol. 2008, 129, 705–733. [Google Scholar] [CrossRef] [Green Version]
- Werner, S.; Keller, L.; Pantel, K. Epithelial keratins: Biology and implications as diagnostic markers for liquid biopsies. Mol. Asp. Med. 2020, 72, 100817. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.; Sahu, I.; Soni, B.L.; Sathe, G.J.; Thapa, P.; Patel, P.; Sinha, S.; Vadivel, C.K.; Patel, S.; Jamghare, S.N.; et al. Depletion of keratin 8/18 modulates oncogenic potential by governing multiple signaling pathways. FEBS J. 2018, 285, 1251–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, N.O.; Fu, H.; Omary, M.B. Raf-1 activation disrupts its binding to keratins during cell stress. J. Cell Biol. 2004, 166, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Ku, N.O.; Michie, S.; Oshima, R.G.; Omary, M.B. Chronic hepatitis, hepatocyte fragility, and increased soluble phosphoglycokeratins in transgenic mice expressing a keratin 18 conserved arginine mutant. J. Cell Biol. 1995, 131, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Ku, N.O.; Lim, J.K.; Krams, S.M.; Esquivel, C.O.; Keeffe, E.B.; Wright, T.L.; Parry, D.A.D.; Omary, M.B. Keratins as susceptibility genes for end-stage liver disease. Gastroenterology 2005, 129, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Kim, S.; Lim, Y.; Yoon, H.-N.; Ku, N.-O. Keratins regulate Hsp70-mediated nuclear localization of p38 mitogen-activated protein kinase. J. Cell Sci. 2019, 132, jcs229534. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, Y.; Kim, S.; Yoon, H.-N.; Ku, N.-O. Keratin 8/18 Regulate the Akt Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 9227. https://doi.org/10.3390/ijms22179227
Lim Y, Kim S, Yoon H-N, Ku N-O. Keratin 8/18 Regulate the Akt Signaling Pathway. International Journal of Molecular Sciences. 2021; 22(17):9227. https://doi.org/10.3390/ijms22179227
Chicago/Turabian StyleLim, Younglan, Sujin Kim, Han-Na Yoon, and Nam-On Ku. 2021. "Keratin 8/18 Regulate the Akt Signaling Pathway" International Journal of Molecular Sciences 22, no. 17: 9227. https://doi.org/10.3390/ijms22179227
APA StyleLim, Y., Kim, S., Yoon, H.-N., & Ku, N.-O. (2021). Keratin 8/18 Regulate the Akt Signaling Pathway. International Journal of Molecular Sciences, 22(17), 9227. https://doi.org/10.3390/ijms22179227