
Tumor Immune Microenvironment and Immunosuppressive Therapy in Hepatocellular Carcinoma: A Review


Abstract
1. Introduction
1.1. Hepatocellular Carcinoma (HCC)
1.2. Tumor Microenvironment (TME)
2. The Constituent Cells and the Immune Microenvironment in the Liver
2.1. Kupffer Cells (KCs)
2.2. Liver Endothelial Cells (LSECs)
2.3. Hepatic Stellate Cells (HSCs)
2.4. Pit Cells (Natural Killer Cells)
2.5. Hepatic Lymphocytes
2.6. Dendritic Cells (DCs)
3. The Immune Microenvironment in Liver Inflammation and Fibrosis
3.1. Immune Regulation and Microenvironment in Liver Inflammation
3.2. Hepatic Immune Microenvironment with Progression to Fibrosis
4. Immune Network in HCC Microenvironment
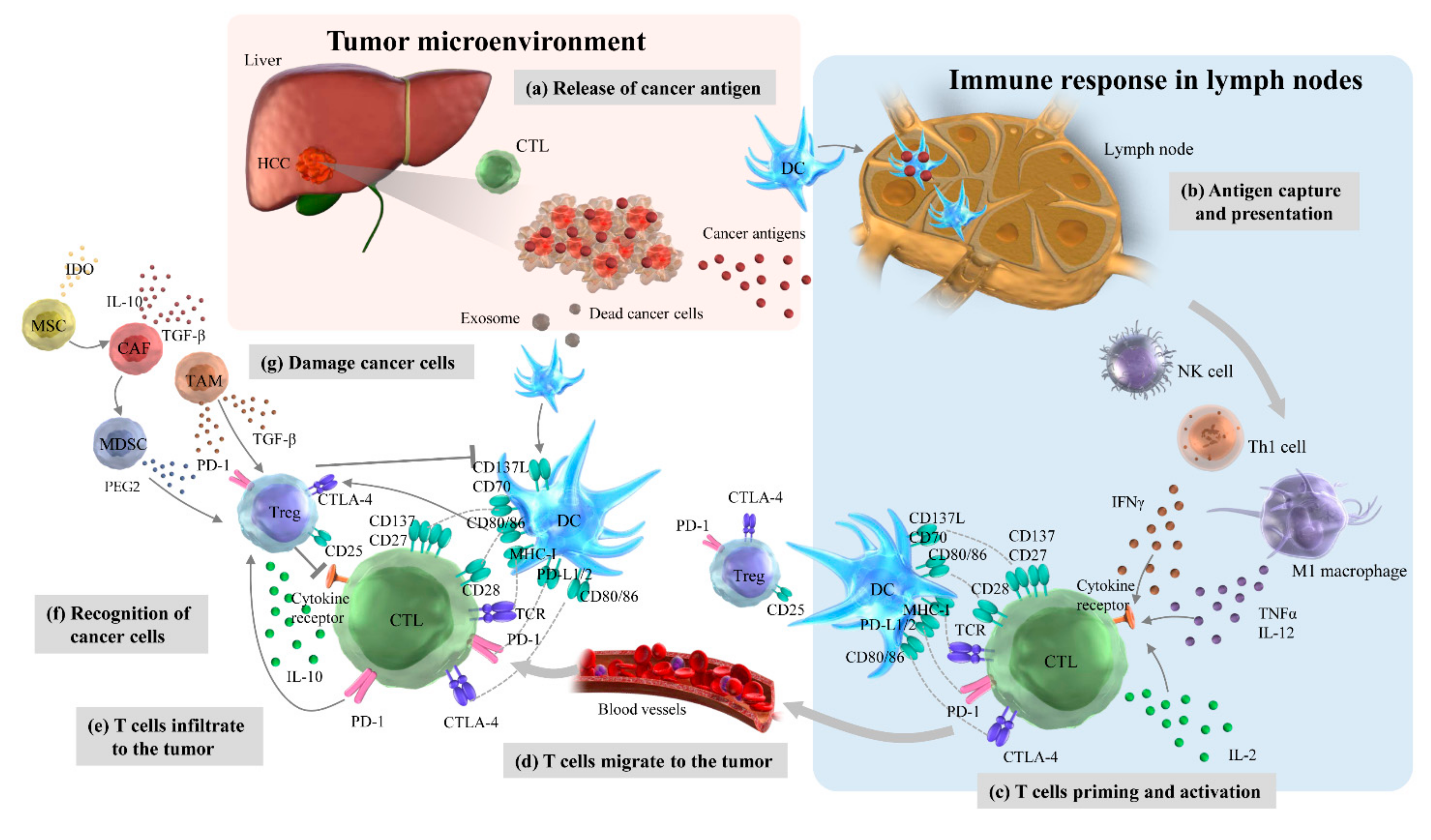
4.1. Cancer-Immunity Cycles
4.2. Myeloid-Derived Suppressor Cells (MDSCs)
4.3. Tumor-Associated Macrophages (TAMs)
4.4. Tumor-Associated Neutrophils (TANs)
4.5. Cancer-Associated Fibroblasts (CAFs)
4.6. Regulatory T Cells (Tregs)
4.7. Tumor-Infiltrating Lymphocytes (TILs)
4.8. CD8+ Cytotoxic T Lymphocytes (CTLs)
5. Current HCC Therapeutic Strategies Targeting Multikinase Activity and the Immune System
6. Immune-Based Therapy for HCC
6.1. Immune Checkpoint Inhibitors (ICIs)
6.2. Combination Therapy Including ICIs
6.3. Adoptive Cell Transfer-Based Therapies (ACTs) in HCC
6.4. Non-Cell-Based Vaccine and Oncolytic Viruse-Based Immunotherapy in HCC
7. Immune-Related Adverse Events (irAE) in HCC
8. The Immunological Classification and Biomarkers for HCC Immunotherapy
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Zhang, W.; He, H.; Zang, M.; Wu, Q.; Zhao, H.; Lu, L.L.; Ma, P.; Zheng, H.; Wang, N.; Zhang, Y.; et al. Genetic features of aflatoxin-associated hepatocellular carcinoma. Gastroenterology 2017, 153, 249–262.e2. [Google Scholar] [CrossRef] [PubMed]
- Altekruse, S.F.; Henley, S.J.; Cucinelli, J.E.; McGlynn, K.A. Changing hepatocellular carcinoma incidence and liver cancer mortality rates in the United States. Am. J. Gastroenterol. 2014, 109, 542–553. [Google Scholar] [CrossRef]
- Xu, L.; Kim, Y.; Spolverato, G.; Gani, F.; Pawlik, T.M. Racial disparities in treatment and survival of patients with hepatocellular carcinoma in the United States. Hepatobiliary Surg. Nutr. 2016, 5, 43–52. [Google Scholar] [PubMed]
- Zhang, G.; Li, R.; Deng, Y.; Zhao, L. Conditional survival of patients with hepatocellular carcinoma: Results from the surveillance, epidemiology, and end results registry. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 515–523. [Google Scholar] [CrossRef]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased alpha-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M. Immuno-Oncology therapy for hepatocellular carcinoma: Current status and ongoing trials. Liver Cancer 2019, 8, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Jeong, W.I.; Tian, Z. Liver: An organ with predominant innate immunity. Hepatology 2008, 47, 729–736. [Google Scholar] [CrossRef]
- Keenan, B.P.; Fong, L.; Kelley, R.K. Immunotherapy in hepatocellular carcinoma: The complex interface between inflammation, fibrosis, and the immune response. J. Immunother. Cancer 2019, 7, 267. [Google Scholar] [CrossRef]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer cells in the liver. Compr. Physiol. 2013, 3, 785–797. [Google Scholar]
- Heymann, F.; Tacke, F. Immunology in the liver—From homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef]
- Brodt, P. Role of the microenvironment in liver metastasis: From pre- to prometastatic niches. Clin. Cancer Res. 2016, 22, 5971–5982. [Google Scholar] [CrossRef]
- Elvevold, K.; Smedsrod, B.; Martinez, I. The liver sinusoidal endothelial cell: A cell type of controversial and confusing identity. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G391–G400. [Google Scholar] [CrossRef]
- Natarajan, V.; Harris, E.N.; Kidambi, S. SECs (Sinusoidal Endothelial Cells), liver microenvironment, and fibrosis. Biomed. Res. Int. 2017, 2017, 4097205. [Google Scholar] [CrossRef]
- Shetty, S.; Lalor, P.F.; Adams, D.H. Liver sinusoidal endothelial cells—Gatekeepers of hepatic immunity. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, A.L.; Qurashi, M.; Shetty, S. The role of sinusoidal endothelial cells in the axis of inflammation and cancer within the liver. Front. Physiol. 2020, 11, 990. [Google Scholar] [CrossRef] [PubMed]
- Wohlleber, D.; Knolle, P.A. The role of liver sinusoidal cells in local hepatic immune surveillance. Clin. Transl. Immunol. 2016, 5, e117. [Google Scholar]
- Patten, D.A.; Wilson, G.K.; Bailey, D.; Shaw, R.K.; Jalkanen, S.; Salmi, M.; Rot, A.; Weston, C.J.; Adams, D.H.; Shetty, S. Human liver sinusoidal endothelial cells promote intracellular crawling of lymphocytes during recruitment: A new step in migration. Hepatology 2017, 65, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Senoo, H.; Mezaki, Y.; Fujiwara, M. The stellate cell system (vitamin A-storing cell system). Anat. Sci. Int. 2017, 92, 387–455. [Google Scholar] [PubMed]
- Gong, Y.; Yang, Y. Activation of Nrf2/AREs-mediated antioxidant signalling, and suppression of profibrotic TGF-beta1/Smad3 pathway: A promising therapeutic strategy for hepatic fibrosis—A review. Life Sci. 2020, 256, 117909. [Google Scholar] [CrossRef]
- Saxena, N.K.; Anania, F.A. Adipocytokines and hepatic fibrosis. Trends Endocrinol. Metab. 2015, 26, 153–161. [Google Scholar] [CrossRef]
- Sun, C.; Sun, H.; Zhang, C.; Tian, Z. NK cell receptor imbalance and NK cell dysfunction in HBV infection and hepatocellular carcinoma. Cell. Mol. Immunol. 2015, 12, 292–302. [Google Scholar]
- Peng, H.; Wisse, E.; Tian, Z. Liver natural killer cells: Subsets and roles in liver immunity. Cell. Mol. Immunol. 2016, 13, 328–336. [Google Scholar] [PubMed]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef]
- Liu, P.; Chen, L.; Zhang, H. Natural killer cells in liver disease and hepatocellular carcinoma and the NK cell-based immunotherapy. J. Immunol. Res. 2018, 2018, 1206737. [Google Scholar] [CrossRef] [PubMed]
- Racanelli, V.; Rehermann, B. The liver as an immunological organ. Hepatology 2006, 43 (Suppl. 1), S54–S62. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Wang, G.; Yang, Q.; Chen, C.; Deng, J.; Gu, X.; Zhu, H. Natural killer T cells in various mouse models of hepatitis. Biomed. Res. Int. 2021, 2021, 1782765. [Google Scholar] [CrossRef] [PubMed]
- Rossjohn, J.; Pellicci, D.G.; Patel, O.; Gapin, L.; Godfrey, D.I. Recognition of CD1d-restricted antigens by natural killer T cells. Nat. Rev. Immunol. 2012, 12, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, K.; Marrero, I.; Kumar, V. NKT cell subsets as key participants in liver physiology and pathology. Cell. Mol. Immunol. 2016, 13, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tian, Z. Gammadelta T cells in liver diseases. Front. Med. 2018, 12, 262–268. [Google Scholar] [CrossRef]
- Lau, A.H.; Thomson, A.W. Dendritic cells and immune regulation in the liver. Gut 2003, 52, 307–314. [Google Scholar] [CrossRef]
- Lurje, I.; Hammerich, L.; Tacke, F. Dendritic cell and T cell crosstalk in liver fibrogenesis and hepatocarcinogenesis: Implications for prevention and therapy of liver cancer. Int. J. Mol. Sci. 2020, 21, 7378. [Google Scholar] [CrossRef]
- Bonnel, A.R.; Bunchorntavakul, C.; Reddy, K.R. Immune dysfunction and infections in patients with cirrhosis. Clin. Gastroenterol. Hepatol. 2011, 9, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Zazueta, G.; Leon-Garduno, L.A.P.; Aguirre-Valadez, J.; Torre-Delgadillo, A. Bacterial infections in cirrhosis: Current treatment. Ann. Hepatol. 2020, 19, 238–244. [Google Scholar] [CrossRef]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef]
- Bamboat, Z.M.; Stableford, J.A.; Plitas, G.; Burt, B.M.; Nguyen, H.M.; Welles, A.P.; Gonen, M.; Young, J.W.; DeMatteo, R.P. Human liver dendritic cells promote T cell hyporesponsiveness. J. Immunol. 2009, 182, 1901–1911. [Google Scholar] [CrossRef]
- Sander, L.E.; Sackett, S.D.; Dierssen, U.; Beraza, N.; Linke, R.P.; Muller, M.; Blander, J.M.; Tacke, F.; Trautwein, C. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J. Exp. Med. 2010, 207, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Zeromski, J.; Kierepa, A.; Brzezicha, B.; Kowala-Piaskowska, A.; Mozer-Lisewska, I. Pattern recognition receptors: Significance of expression in the liver. Arch. Immunol. Exp. 2020, 68, 29. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Chang, X.; Qian, S.; Liu, C.; Lord, C.C.; Ahmed, N.; Lee, C.E.; Lee, S.; Gautron, L.; Mitchell, M.C.; et al. Hepatocyte toll-like receptor 4 deficiency protects against alcohol-induced fatty liver disease. Mol. Metab. 2018, 14, 121–129. [Google Scholar] [CrossRef]
- Radaeva, S.; Sun, R.; Jaruga, B.; Nguyen, V.T.; Tian, Z.; Gao, B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology 2006, 130, 435–452. [Google Scholar] [CrossRef]
- Gabele, E.; Muhlbauer, M.; Dorn, C.; Weiss, T.S.; Froh, M.; Schnabl, B.; Wiest, R.; Scholmerich, J.; Obermeier, F.; Hellerbrand, C. Role of TLR9 in hepatic stellate cells and experimental liver fibrosis. Biochem. Biophys. Res. Commun. 2008, 376, 271–276. [Google Scholar] [CrossRef]
- Wang, J.; Dong, R.; Zheng, S. Roles of the inflammasome in the gutliver axis (Review). Mol. Med. Rep. 2019, 19, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Yuan, Z.; Wang, G.; Wang, X.; Li, K. The selective NLRP3 inflammasome inhibitor MCC950 alleviates cholestatic liver injury and fibrosis in mice. Int. Immunopharmacol. 2019, 70, 147–155. [Google Scholar] [CrossRef]
- Tian, Z.; Chen, Y.; Gao, B. Natural killer cells in liver disease. Hepatology 2013, 57, 1654–1662. [Google Scholar] [CrossRef]
- Kramer, B.; Korner, C.; Kebschull, M.; Glassner, A.; Eisenhardt, M.; Nischalke, H.D.; Alexander, M.; Sauerbruch, T.; Spengler, U.; Nattermann, J. Natural killer p46High expression defines a natural killer cell subset that is potentially involved in control of hepatitis C virus replication and modulation of liver fibrosis. Hepatology 2012, 56, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.I.; Park, O.; Gao, B. Abrogation of the antifibrotic effects of natural killer cells/interferon-gamma contributes to alcohol acceleration of liver fibrosis. Gastroenterology 2008, 134, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, S. T cells in fibrosis and fibrotic diseases. Front. Immunol. 2020, 11, 1142. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Ikejima, K.; Yamagata, H.; Aoyama, T.; Kon, K.; Arai, K.; Takeda, K.; Watanabe, S. CD1d-Restricted natural killer T cells contribute to hepatic inflammation and fibrogenesis in mice. J. Hepatol. 2011, 54, 1195–1204. [Google Scholar] [CrossRef]
- Park, O.; Jeong, W.I.; Wang, L.; Wang, H.; Lian, Z.X.; Gershwin, M.E.; Gao, B. Diverse roles of invariant natural killer T cells in liver injury and fibrosis induced by carbon tetrachloride. Hepatology 2009, 49, 1683–1694. [Google Scholar] [CrossRef]
- Chew, V.; Lai, L.; Pan, L.; Lim, C.J.; Li, J.; Ong, R.; Chua, C.; Leong, J.Y.; Lim, K.H.; Toh, H.C.; et al. Delineation of an immunosuppressive gradient in hepatocellular carcinoma using high-dimensional proteomic and transcriptomic analyses. Proc. Natl. Acad. Sci. USA 2017, 114, E5900–E5909. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Bottcher, J.P.; Reis e Sousa, C. The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis, E.S.C. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef]
- Inarrairaegui, M.; Melero, I.; Sangro, B. Immunotherapy of hepatocellular carcinoma: Facts and hopes. Clin. Cancer Res. 2018, 24, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Kapanadze, T.; Gamrekelashvili, J.; Ma, C.; Chan, C.; Zhao, F.; Hewitt, S.; Zender, L.; Kapoor, V.; Felsher, D.W.; Manns, M.P.; et al. Regulation of accumulation and function of myeloid derived suppressor cells in different murine models of hepatocellular carcinoma. J. Hepatol. 2013, 59, 1007–1013. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, M.; Sun, H.; Feng, Y.; Xu, L.; Chan, A.W.H.; Tong, J.H.; Wong, J.; Chong, C.C.N.; Lai, P.B.S.; et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut 2018, 67, 931–944. [Google Scholar] [CrossRef] [PubMed]
- Chiu, D.K.; Xu, I.M.; Lai, R.K.; Tse, A.P.; Wei, L.L.; Koh, H.Y.; Li, L.L.; Lee, D.; Lo, R.C.; Wong, C.M.; et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology 2016, 64, 797–813. [Google Scholar] [CrossRef] [PubMed]
- Chiu, D.K.; Tse, A.P.; Xu, I.M.; Di Cui, J.; Lai, R.K.; Li, L.L.; Koh, H.Y.; Tsang, F.H.; Wei, L.L.; Wong, C.M.; et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat. Commun. 2017, 8, 517. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yang, L.; Yue, D.; Cao, L.; Li, L.; Wang, D.; Ping, Y.; Shen, Z.; Zheng, Y.; Wang, L.; et al. Macrophage-Derived CCL22 promotes an immunosuppressive tumor microenvironment via IL-8 in malignant pleural effusion. Cancer Lett. 2019, 452, 244–253. [Google Scholar] [CrossRef]
- Mamrot, J.; Balachandran, S.; Steele, E.J.; Lindley, R.A. Molecular model linking Th2 polarized M2 tumour-associated macrophages with deaminase-mediated cancer progression mutation signatures. Scand. J. Immunol. 2019, 89, e12760. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Q.; Lou, Y.; Fu, Q.; Chen, Q.; Wei, T.; Yang, J.; Tang, J.; Wang, J.; Chen, Y.; et al. Hypoxia-Inducible factor-1alpha/interleukin-1beta signaling enhances hepatoma epithelial-mesenchymal transition through macrophages in a hypoxic-inflammatory microenvironment. Hepatology 2018, 67, 1872–1889. [Google Scholar] [CrossRef] [PubMed]
- Kuang, D.M.; Peng, C.; Zhao, Q.; Wu, Y.; Chen, M.S.; Zheng, L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma promote expansion of memory T helper 17 cells. Hepatology 2010, 51, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Liu, X.; Ma, H.; Zhang, H.; Song, X.; Gao, L.; Liang, X.; Ma, C. Tim-3 fosters HCC development by enhancing TGF-beta-mediated alternative activation of macrophages. Gut 2015, 64, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.L.; Dai, Z.; Zhou, Z.J.; Wang, X.Y.; Yang, G.H.; Wang, Z.; Huang, X.W.; Fan, J.; Zhou, J. Overexpression of CXCL5 mediates neutrophil infiltration and indicates poor prognosis for hepatocellular carcinoma. Hepatology 2012, 56, 2242–2254. [Google Scholar] [CrossRef]
- Michaeli, J.; Shaul, M.E.; Mishalian, I.; Hovav, A.H.; Levy, L.; Zolotriov, L.; Granot, Z.; Fridlender, Z.G. Tumor-Associated neutrophils induce apoptosis of non-activated CD8 T-cells in a TNFalpha and NO-dependent mechanism, promoting a tumor-supportive environment. Oncoimmunology 2017, 6, e1356965. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, Y.; Hua, X.; Wang, G.; Liu, W.; Jia, C.; Tai, Y.; Zhang, Q.; Chen, G. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012, 318, 154–161. [Google Scholar] [CrossRef]
- Mano, Y.; Yoshio, S.; Shoji, H.; Tomonari, S.; Aoki, Y.; Aoyanagi, N.; Okamoto, T.; Matsuura, Y.; Osawa, Y.; Kimura, K.; et al. Bone morphogenetic protein 4 provides cancer-supportive phenotypes to liver fibroblasts in patients with hepatocellular carcinoma. J. Gastroenterol. 2019, 54, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Ormandy, L.A.; Hillemann, T.; Wedemeyer, H.; Manns, M.P.; Greten, T.F.; Korangy, F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005, 65, 2457–2464. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, T.; Yuan, Y.; Zhuang, H.; Wang, Z.; Liu, X.; Feng, M. Sorafenib reduces hepatic infiltrated regulatory T cells in hepatocellular carcinoma patients by suppressing TGF-beta signal. J. Surg. Oncol. 2013, 107, 422–427. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Katz, S.C.; Bamboat, Z.M.; Maker, A.V.; Shia, J.; Pillarisetty, V.G.; Yopp, A.C.; Hedvat, C.V.; Gonen, M.; Jarnagin, W.R.; Fong, Y.; et al. Regulatory T cell infiltration predicts outcome following resection of colorectal cancer liver metastases. Ann. Surg. Oncol. 2013, 20, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yan, J.; Xu, J.; Liu, C.Q.; Zhen, Z.J.; Chen, H.W.; Ji, Y.; Wu, Z.P.; Hu, J.Y.; Zheng, L.; et al. CXCL17 expression predicts poor prognosis and correlates with adverse immune infiltration in hepatocellular carcinoma. PLoS ONE 2014, 9, e110064. [Google Scholar] [CrossRef] [PubMed]
- Motz, G.T.; Santoro, S.P.; Wang, L.P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Cubero, E.; Larrubia, J.R. Specific CD8(+) T cell response immunotherapy for hepatocellular carcinoma and viral hepatitis. World J. Gastroenterol. 2016, 22, 6469–6483. [Google Scholar] [CrossRef]
- Han, Y.; Chen, Z.; Yang, Y.; Jiang, Z.; Gu, Y.; Liu, Y.; Lin, C.; Pan, Z.; Yu, Y.; Jiang, M.; et al. Human CD14+ CTLA-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology 2014, 59, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Bruger, A.M.; Dorhoi, A.; Esendagli, G.; Barczyk-Kahlert, K.; van der Bruggen, P.; Lipoldova, M.; Perecko, T.; Santibanez, J.; Saraiva, M.; Van Ginderachter, J.A.; et al. How to measure the immunosuppressive activity of MDSC: Assays, problems and potential solutions. Cancer Immunol. Immunother. 2019, 68, 631–644. [Google Scholar] [CrossRef]
- Li, F.; Zhao, Y.; Wei, L.; Li, S.; Liu, J. Tumor-Infiltrating treg, MDSC, and IDO expression associated with outcomes of neoadjuvant chemotherapy of breast cancer. Cancer Biol. 2018, 19, 695–705. [Google Scholar] [CrossRef]
- Deng, Y.; Cheng, J.; Fu, B.; Liu, W.; Chen, G.; Zhang, Q.; Yang, Y. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene 2017, 36, 1090–1101. [Google Scholar] [CrossRef]
- Dardalhon, V.; Anderson, A.C.; Karman, J.; Apetoh, L.; Chandwaskar, R.; Lee, D.H.; Cornejo, M.; Nishi, N.; Yamauchi, A.; Quintana, F.J.; et al. Tim-3/Galectin-9 pathway: Regulation of Th1 immunity through promotion of CD11b+Ly-6G+ myeloid cells. J. Immunol. 2010, 185, 1383–1392. [Google Scholar] [CrossRef]
- Hoechst, B.; Voigtlaender, T.; Ormandy, L.; Gamrekelashvili, J.; Zhao, F.; Wedemeyer, H.; Lehner, F.; Manns, M.P.; Greten, T.F.; Korangy, F. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 2009, 50, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhou, J.; Liu, X.; Feng, Y.; Yang, W.; Wu, F.; Cheung, O.K.; Sun, H.; Zeng, X.; Tang, W.; et al. Targeting monocyte-intrinsic enhancer reprogramming improves immunotherapy efficacy in hepatocellular carcinoma. Gut 2020, 69, 365–379. [Google Scholar] [CrossRef]
- Wu, C.J.; Tsai, Y.T.; Lee, I.J.; Wu, P.Y.; Lu, L.S.; Tsao, W.S.; Huang, Y.J.; Chang, C.C.; Ka, S.M.; Tao, M.H. Combination of radiation and interleukin 12 eradicates large orthotopic hepatocellular carcinoma through immunomodulation of tumor microenvironment. Oncoimmunology 2018, 7, e1477459. [Google Scholar] [CrossRef]
- Komohara, Y.; Jinushi, M.; Takeya, M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 2014, 105, 1–8. [Google Scholar] [CrossRef]
- Guerriero, J.L. Macrophages: The road less traveled, changing anticancer therapy. Trends Mol. Med. 2018, 24, 472–489. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-Associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Ba, Q.; Li, X.; Li, H.; Zhang, S.; Yuan, Y.; Wang, F.; Duan, X.; Li, J.; Zhang, W.; et al. A natural CCR2 Antagonist relieves tumor-associated macrophage-mediated immunosuppression to produce a therapeutic effect for liver cancer. EBioMedicine 2017, 22, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, H.; Li, X.P.; Zou, H.; Liu, L.; Liu, W.; Duan, T. CC chemokine receptor type 2 promotes epithelialtomesenchymal transition by upregulating matrix metalloproteinase2 in human liver cancer. Oncol. Rep. 2018, 40, 2734–2741. [Google Scholar] [PubMed]
- Li, X.; Yao, W.; Yuan, Y.; Chen, P.; Li, B.; Li, J.; Chu, R.; Song, H.; Xie, D.; Jiang, X.; et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 2017, 66, 157–167. [Google Scholar] [CrossRef]
- Dong, P.; Ma, L.; Liu, L.; Zhao, G.; Zhang, S.; Dong, L.; Xue, R.; Chen, S. CD86(+)/CD206(+), diametrically polarized tumor-associated macrophages, predict hepatocellular carcinoma patient prognosis. Int. J. Mol. Sci. 2016, 17, 320. [Google Scholar] [CrossRef]
- Yang, Y.; Ye, Y.C.; Chen, Y.; Zhao, J.L.; Gao, C.C.; Han, H.; Liu, W.C.; Qin, H.Y. Crosstalk between hepatic tumor cells and macrophages via Wnt/beta-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis. 2018, 9, 793. [Google Scholar] [CrossRef]
- Wu, L.; Saxena, S.; Awaji, M.; Singh, R.K. Tumor-Associated neutrophils in cancer: Going pro. Cancers 2019, 11, 564. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Nicolas-Avila, J.A.; Adrover, J.M.; Hidalgo, A. Neutrophils in homeostasis, immunity, and cancer. Immunity 2017, 46, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Shaul, M.E.; Fridlender, Z.G. Neutrophils as active regulators of the immune system in the tumor microenvironment. J. Leukoc. Biol. 2017, 102, 343–349. [Google Scholar] [CrossRef]
- Andzinski, L.; Kasnitz, N.; Stahnke, S.; Wu, C.F.; Gereke, M.; von Kockritz-Blickwede, M.; Schilling, B.; Brandau, S.; Weiss, S.; Jablonska, J. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int. J. Cancer 2016, 138, 1982–1993. [Google Scholar] [CrossRef]
- Zhou, S.L.; Zhou, Z.J.; Hu, Z.Q.; Huang, X.W.; Wang, Z.; Chen, E.B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-Associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology 2016, 150, 1646–1658.e17. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, P.; Sun, R.; Li, J.; Hu, Z.; Xin, H.; Luo, C.; Zhou, J.; Fan, J.; Zhou, S. Tumor-Associated neutrophils and macrophages interaction contributes to intrahepatic cholangiocarcinoma progression by activating STAT3. J. Immunother. Cancer 2021, 9, e001946. [Google Scholar] [CrossRef]
- Koh, M.Y.; Gagea, M.; Sargis, T.; Lemos, R., Jr.; Grandjean, G.; Charbono, A.; Bekiaris, V.; Sedy, J.; Kiriakova, G.; Liu, X.; et al. A new HIF-1alpha/RANTES-driven pathway to hepatocellular carcinoma mediated by germline haploinsufficiency of SART1/HAF in mice. Hepatology 2016, 63, 1576–1591. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.L.; Yin, D.; Hu, Z.Q.; Luo, C.B.; Zhou, Z.J.; Xin, H.Y.; Yang, X.R.; Shi, Y.H.; Wang, Z.; Huang, X.W.; et al. A positive feedback loop between cancer stem-like cells and tumor-associated neutrophils controls hepatocellular carcinoma progression. Hepatology 2019, 70, 1214–1230. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ren, H.; Dai, B.; Li, J.; Shang, L.; Huang, J.; Shi, X. Hepatocellular carcinoma-derived exosomal miRNA-21 contributes to tumor progression by converting hepatocyte stellate cells to cancer-associated fibroblasts. J. Exp. Clin. Cancer Res. 2018, 37, 324. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, R.; Xiong, S.; Wang, X.; Zhao, Z.; Bai, S.; Wang, Y.; Zhao, Y.; Cheng, B. Cancer-Associated fibroblasts promote the stemness of CD24(+) liver cells via paracrine signaling. J. Mol. Med. 2019, 97, 243–255. [Google Scholar] [CrossRef]
- Rhee, H.; Kim, H.Y.; Choi, J.H.; Woo, H.G.; Yoo, J.E.; Nahm, J.H.; Choi, J.S.; Park, Y.N. Keratin 19 expression in hepatocellular carcinoma is regulated by fibroblast-derived HGF via a MET-ERK1/2-AP1 and SP1 axis. Cancer Res. 2018, 78, 1619–1631. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, L.; Chen, X.; Cheng, J.; Zhang, H.; Zhang, C.; Shan, J.; Shen, J.; Qian, C. LSD1 stimulates cancer-associated fibroblasts to drive Notch3-dependent self-renewal of liver cancer stem-like cells. Cancer Res. 2018, 78, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Cioplea, M.; Nichita, L.; Georgescu, D.; Sticlaru, L.; Cioroianu, A.; Nedelcu, R.; Turcu, G.; Rauta, A.; Mogodici, C.; Zurac, S.; et al. FOXP3 in melanoma with regression: Between tumoral expression and regulatory T cell upregulation. J. Immunol. Res. 2020, 2020, 5416843. [Google Scholar] [CrossRef]
- Chen, K.J.; Lin, S.Z.; Zhou, L.; Xie, H.Y.; Zhou, W.H.; Taki-Eldin, A.; Zheng, S.S. Selective recruitment of regulatory T cell through CCR6-CCL20 in hepatocellular carcinoma fosters tumor progression and predicts poor prognosis. PLoS ONE 2011, 6, e24671. [Google Scholar] [CrossRef]
- Yang, X.H.; Yamagiwa, S.; Ichida, T.; Matsuda, Y.; Sugahara, S.; Watanabe, H.; Sato, Y.; Abo, T.; Horwitz, D.A.; Aoyagi, Y. Increase of CD4+ CD25+ regulatory T-cells in the liver of patients with hepatocellular carcinoma. J. Hepatol. 2006, 45, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Xu, X.; Qian, Y.; Xue, W.; Wang, Y.; Du, J.; Jin, L.; Tan, Y. Prognostic value of tumor-infiltrating lymphocytes in hepatocellular carcinoma: A meta-analysis. Medicine 2018, 97, e13301. [Google Scholar] [CrossRef] [PubMed]
- Prieto, J.; Melero, I.; Sangro, B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 681–700. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Jing, X.; Li, G.; Wang, T.; Yang, B.; Zhu, Z.; Gao, Y.; Zhang, Q.; Yang, Y.; Wang, Y.; et al. Foxp3+ regulatory T cells are associated with the natural history of chronic hepatitis B and poor prognosis of hepatocellular carcinoma. Liver Int. 2012, 32, 644–655. [Google Scholar] [CrossRef]
- Chew, V.; Chen, J.; Lee, D.; Loh, E.; Lee, J.; Lim, K.H.; Weber, A.; Slankamenac, K.; Poon, R.T.; Yang, H.; et al. Chemokine-Driven lymphocyte infiltration: An early intratumoural event determining long-term survival in resectable hepatocellular carcinoma. Gut 2012, 61, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Ramzan, M.; Sturm, N.; Decaens, T.; Bioulac-Sage, P.; Bancel, B.; Merle, P.; Tran Van Nhieu, J.; Slama, R.; Letoublon, C.; Zarski, J.P.; et al. Liver-Infiltrating CD8(+) lymphocytes as prognostic factor for tumour recurrence in hepatitis C virus-related hepatocellular carcinoma. Liver Int. 2016, 36, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.L.; Yang, X.H.; Cheng, W.; Xu, Y.; Li, J.B.; Sun, Y.X.; Bi, Y.M.; Zhang, L.; Wang, Q.C. Expression of Fas/FasL in CD8+ T and CD3+ Foxp3+ treg cells—Relationship with apoptosis of circulating CD8+ T cells in hepatocellular carcinoma patients. Asian Pac. J. Cancer Prev. 2014, 15, 2613–2618. [Google Scholar] [CrossRef]
- Harsha, C.; Banik, K.; Ang, H.L.; Girisa, S.; Vikkurthi, R.; Parama, D.; Rana, V.; Shabnam, B.; Khatoon, E.; Kumar, A.P.; et al. Targeting AKT/mTOR in oral cancer: Mechanisms and advances in clinical trials. Int. J. Mol. Sci. 2020, 21, 3285. [Google Scholar] [CrossRef]
- Koyama, N.; Saito, K.; Nishioka, Y.; Yusa, W.; Yamamoto, N.; Yamada, Y.; Nokihara, H.; Koizumi, F.; Nishio, K.; Tamura, T. Pharmacodynamic change in plasma angiogenic proteins: A dose-escalation phase 1 study of the multi-kinase inhibitor lenvatinib. BMC Cancer 2014, 14, 530. [Google Scholar] [CrossRef] [PubMed]
- Marino, D.; Zichi, C.; Audisio, M.; Sperti, E.; Di Maio, M. Second-Line treatment options in hepatocellular carcinoma. Drugs Context 2019, 8, 212577. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Thanekar, U.; Amarachintha, S.; Mourya, R.; Nalluri, S.; Bondoc, A.; Shivakumar, P. “Complimenting the complement”: Mechanistic insights and opportunities for therapeutics in hepatocellular carcinoma. Front. Oncol. 2020, 10, 627701. [Google Scholar] [CrossRef]
- Brunetti, O.; Gnoni, A.; Licchetta, A.; Longo, V.; Calabrese, A.; Argentiero, A.; Delcuratolo, S.; Solimando, A.G.; Casadei-Gardini, A.; Silvestris, N. Predictive and prognostic factors in HCC patients treated with sorafenib. Medicine 2019, 55, 707. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K.; Asahina, Y.; Matsuda, S.; Muraoka, M.; Nakata, T.; Suzuki, Y.; Tamaki, N.; Yasui, Y.; Suzuki, S.; Hosokawa, T.; et al. Changes in plasma vascular endothelial growth factor at 8 weeks after sorafenib administration as predictors of survival for advanced hepatocellular carcinoma. Cancer 2014, 120, 229–237. [Google Scholar] [CrossRef]
- Oh, C.R.; Kong, S.Y.; Im, H.S.; Kim, H.J.; Kim, M.K.; Yoon, K.A.; Cho, E.H.; Jang, J.H.; Lee, J.; Kang, J.; et al. Genome-Wide copy number alteration and VEGFA amplification of circulating cell-free DNA as a biomarker in advanced hepatocellular carcinoma patients treated with Sorafenib. BMC Cancer 2019, 19, 292. [Google Scholar] [CrossRef]
- Faloppi, L.; Puzzoni, M.; Casadei-Gardini, A.; Silvestris, N.; Masi, G.; Marisi, G.; Vivaldi, C.; Gadaleta, C.D.; Ziranu, P.; Bianconi, M.; et al. Angiogenesis genotyping and clinical outcomes in patients with advanced hepatocellular carcinoma receiving Sorafenib: The ALICE-2 study. Target. Oncol. 2020, 15, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Lue, A.; Serrano, M.T.; Bustamante, F.J.; Inarrairaegui, M.; Arenas, J.I.; Testillano, M.; Lorente, S.; Gil, C.; de la Torre, M.; Gomez, A.; et al. Neutrophil-To-Lymphocyte ratio predicts survival in European patients with hepatocellular carcinoma administered sorafenib. Oncotarget 2017, 8, 103077–103086. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Cai, J.; Li, H.; Zeng, K.; He, L.; Fu, H.; Zhang, J.; Chen, L.; Yao, J.; Zhang, Y.; et al. Neutrophil to lymphocyte ratio and platelet to lymphocyte ratio as prognostic predictors for hepatocellular carcinoma patients with various treatments: A meta-analysis and systematic review. Cell. Physiol. Biochem. 2017, 44, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.Y.; Huang, C.C.; Lin, S.D.; Hsu, C.H.; Cheng, A.L. Serum insulin-like growth factor-1 levels predict outcomes of patients with advanced hepatocellular carcinoma receiving antiangiogenic therapy. Clin. Cancer Res. 2012, 18, 3992–3997. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 therapies in cancer: Mechanisms of action, efficacy, and limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Tagliamonte, M.; Mauriello, A.; Cavalluzzo, B.; Ragone, C.; Manolio, C.; Petrizzo, A.; Buonaguro, L. Tackling hepatocellular carcinoma with individual or combinatorial immunotherapy approaches. Cancer Lett. 2020, 473, 25–32. [Google Scholar] [CrossRef]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-Targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Onuma, A.E.; Zhang, H.; Huang, H.; Williams, T.M.; Noonan, A.; Tsung, A. Immune checkpoint inhibitors in hepatocellular cancer: Current understanding on mechanisms of resistance and biomarkers of response to treatment. Gene Expr. 2020, 20, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.H.; Wan, Z.H.; Song, H.; Li, Y.L.; Zhu, B.; Zang, H.; Zhao, Y.; Liu, H.L.; Zhang, A.M.; Xiao, L.; et al. Tim-3 expression on peripheral monocytes and CD3+CD16/CD56+natural killer-like T cells in patients with chronic hepatitis B. Tissue Antigens 2014, 83, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, K.; Tao, K.; Chen, L.; Zheng, Q.; Lu, X.; Liu, J.; Shi, L.; Liu, C.; Wang, G.; et al. Tim-3/Galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology 2012, 56, 1342–1351. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, S.; Zeng, S.; Shen, H. From bench to bed: The tumor immune microenvironment and current immunotherapeutic strategies for hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 396. [Google Scholar] [CrossRef]
- Brunet, J.F.; Denizot, F.; Luciani, M.F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.G.; Golstein, P. A new member of the immunoglobulin superfamily—CTLA-4. Nature 1987, 328, 267–270. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Greten, T.F.; Lai, C.W.; Li, G.; Staveley-O’Carroll, K.F. Targeted and immune-based therapies for hepatocellular carcinoma. Gastroenterology 2019, 156, 510–524. [Google Scholar] [CrossRef]
- Cho, H.; Kang, H.; Lee, H.H.; Kim, C.W. Programmed cell death 1 (PD-1) and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) in viral hepatitis. Int. J. Mol. Sci. 2017, 18, 1517. [Google Scholar] [CrossRef] [PubMed]
- Sangro, B.; Gomez-Martin, C.; de la Mata, M.; Inarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.H.; Harding, J.J.; Merle, P.; et al. LBA38_PR-CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 2019, 30, v874–v875. [Google Scholar] [CrossRef]
- Lee, H.W.; Cho, K.J.; Park, J.Y. Current status and future direction of immunotherapy in hepatocellular carcinoma: What do the data suggest? Immune Netw. 2020, 20, e11. [Google Scholar] [CrossRef]
- Lee, C.H.; Lee, Y.B.; Kim, M.A.; Jang, H.; Oh, H.; Kim, S.W.; Cho, E.J.; Lee, K.H.; Lee, J.H.; Yu, S.J.; et al. Effectiveness of nivolumab versus regorafenib in hepatocellular carcinoma patients who failed sorafenib treatment. Clin. Mol. Hepatol. 2020, 26, 328–339. [Google Scholar] [CrossRef]
- Qin, S.; Finn, R.S.; Kudo, M.; Meyer, T.; Vogel, A.; Ducreux, M.; Macarulla, T.M.; Tomasello, G.; Boisserie, F.; Hou, J.; et al. RATIONALE 301 study: Tislelizumab versus sorafenib as first-line treatment for unresectable hepatocellular carcinoma. Future Oncol. 2019, 15, 1811–1822. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: A randomized, double-blind, phase III trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Pinyol, R.; Sia, D.; Llovet, J.M. Immune exclusion-Wnt/CTNNB1 class predicts resistance to immunotherapies in HCC. Clin. Cancer. Res. 2019, 25, 2021–2023. [Google Scholar] [CrossRef]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective genotyping of hepatocellular carcinoma: Clinical implications of next-generation sequencing for matching patients to targeted and immune therapies. Clin. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef] [PubMed]
- Kitao, A.; Matsui, O.; Yoneda, N.; Kozaka, K.; Kobayashi, S.; Sanada, J.; Koda, W.; Minami, T.; Inoue, D.; Yoshida, K.; et al. Hepatocellular carcinoma with beta-catenin mutation: Imaging and pathologic characteristics. Radiology 2015, 275, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.A.; Kerbel, R.S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 2018, 15, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Atezolizumab: First global approval. Drugs 2016, 76, 1227–1232. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: The CheckMate 040 randomized clinical trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Duffy, A.G.; Mabry-Hrones, D.; Wood, B.; Levy, E.; Krishnasamy, V.; Khan, J.; Wei, J.S.; Agdashian, D.; Tyagi, M.; et al. Tremelimumab in combination with microwave ablation in patients with refractory biliary tract cancer. Hepatology 2019, 69, 2048–2060. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Ikeda, M.; Zhu, A.X.; Sung, M.W.; Baron, A.D.; Kudo, M.; Okusaka, T.; Kobayashi, M.; Kumada, H.; Kaneko, S.; et al. Phase Ib study of lenvatinib plus pembrolizumab in patients with unresectable hepatocellular carcinoma. J. Clin. Oncol. 2020, 38, 2960–2970. [Google Scholar] [CrossRef]
- Osa, A.; Uenami, T.; Koyama, S.; Fujimoto, K.; Okuzaki, D.; Takimoto, T.; Hirata, H.; Yano, Y.; Yokota, S.; Kinehara, Y.; et al. Clinical implications of monitoring nivolumab immunokinetics in non-small cell lung cancer patients. JCI Insight 2018, 3, e59125. [Google Scholar] [CrossRef]
- Kudo, M. Scientific rationale for combined immunotherapy with PD-1/PD-L1 antibodies and VEGF inhibitors in advanced hepatocellular carcinoma. Cancers 2020, 12, 1089. [Google Scholar] [CrossRef]
- Joerger, M.; Guller, U.; Bastian, S.; Driessen, C.; von Moos, R. Prolonged tumor response associated with sequential immune checkpoint inhibitor combination treatment and regorafenib in a patient with advanced pretreated hepatocellular carcinoma. J. Gastrointest. Oncol. 2019, 10, 373–378. [Google Scholar] [CrossRef]
- Tan, A.T.; Yang, N.; Lee-Krishnamoorthy, T.; Oei, V.; Chua, A.; Zhao, X.; Tan, H.S.; Chia, A.; Le Bert, N.; Low, D.; et al. Use of expression profiles of HBV-DNA integrated into genomes of hepatocellular carcinoma cells to select T cells for immunotherapy. Gastroenterology 2019, 156, 1862–1876.e9. [Google Scholar] [CrossRef]
- Qasim, W.; Brunetto, M.; Gehring, A.J.; Xue, S.A.; Schurich, A.; Khakpoor, A.; Zhan, H.; Ciccorossi, P.; Gilmour, K.; Cavallone, D.; et al. Immunotherapy of HCC metastases with autologous T cell receptor redirected T cells, targeting HBsAg in a liver transplant patient. J. Hepatol. 2015, 62, 486–491. [Google Scholar] [CrossRef]
- Schmidt, T.L.; Negrin, R.S.; Contag, C.H. A killer choice for cancer immunotherapy. Immunol. Res. 2014, 58, 300–306. [Google Scholar] [CrossRef]
- Chang, B.; Shen, L.; Wang, K.; Jin, J.; Huang, T.; Chen, Q.; Li, W.; Wu, P. High number of PD-1 positive intratumoural lymphocytes predicts survival benefit of cytokine-induced killer cells for hepatocellular carcinoma patients. Liver Int. 2018, 38, 1449–1458. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.H.; Lim, Y.S.; Yeon, J.E.; Song, T.J.; Yu, S.J.; Gwak, G.Y.; Kim, K.M.; Kim, Y.J.; Lee, J.W.; et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology 2015, 148, 1383–1391.e6. [Google Scholar] [CrossRef]
- Szoor, A.; Vaidya, A.; Velasquez, M.P.; Mei, Z.; Galvan, D.L.; Torres, D.; Gee, A.; Heczey, A.; Gottschalk, S. T cell-activating mesenchymal stem cells as a biotherapeutic for HCC. Mol. Ther. Oncolytics 2017, 6, 69–79. [Google Scholar] [CrossRef]
- Jiang, Z.; Jiang, X.; Chen, S.; Lai, Y.; Wei, X.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; Liang, Q.; et al. Anti-GPC3-CAR T cells suppress the growth of tumor cells in patient-derived xenografts of hepatocellular carcinoma. Front. Immunol. 2016, 7, 690. [Google Scholar] [CrossRef]
- Yu, M.; Luo, H.; Fan, M.; Wu, X.; Shi, B.; Di, S.; Liu, Y.; Pan, Z.; Jiang, H.; Li, Z. Development of GPC3-specific chimeric antigen receptor-engineered natural killer cells for the treatment of hepatocellular carcinoma. Mol. Ther. 2018, 26, 366–378. [Google Scholar] [CrossRef]
- Makarova-Rusher, O.V.; Medina-Echeverz, J.; Duffy, A.G.; Greten, T.F. The yin and yang of evasion and immune activation in HCC. J. Hepatol. 2015, 62, 1420–1429. [Google Scholar] [CrossRef]
- Tada, Y.; Yoshikawa, T.; Shimomura, M.; Sawada, Y.; Sakai, M.; Shirakawa, H.; Nobuoka, D.; Nakatsura, T. Analysis of cytotoxic T lymphocytes from a patient with hepatocellular carcinoma who showed a clinical response to vaccination with a glypican3derived peptide. Int. J. Oncol. 2013, 43, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Greten, T.F.; Forner, A.; Korangy, F.; N’Kontchou, G.; Barget, N.; Ayuso, C.; Ormandy, L.A.; Manns, M.P.; Beaugrand, M.; Bruix, J. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer 2010, 10, 209. [Google Scholar] [CrossRef] [PubMed]
- Caballero, O.L.; Chen, Y.T. Cancer/Testis (CT) antigens: Potential targets for immunotherapy. Cancer Sci. 2009, 100, 2014–2021. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Nouso, K.; Noguchi, Y.; Higashi, T.; Ono, T.; Jungbluth, A.; Chen, Y.T.; Old, L.J.; Nakayama, E.; Shiratori, Y. Expression and immunogenicity of NY-ESO-1 in hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2006, 21, 1281–1285. [Google Scholar] [CrossRef]
- Jiang, H.; Rivera-Molina, Y.; Gomez-Manzano, C.; Clise-Dwyer, K.; Bover, L.; Vence, L.M.; Yuan, Y.; Lang, F.F.; Toniatti, C.; Hossain, M.B.; et al. Oncolytic adenovirus and tumor-targeting immune modulatory therapy improve autologous cancer vaccination. Cancer Res. 2017, 77, 3894–3907. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, F.; Cai, H.; Zhong, S.; Liu, X.; Tan, W.S. Potent antitumor effect of TRAIL mediated by a novel adeno-associated viral vector targeting to telomerase activity for human hepatocellular carcinoma. J. Gene Med. 2008, 10, 518–526. [Google Scholar] [CrossRef]
- Khan, S.; Gerber, D.E. Autoimmunity, checkpoint inhibitor therapy and immune-related adverse events: A review. Semin. Cancer Biol. 2020, 64, 93–101. [Google Scholar] [CrossRef]
- Wang, D.Y.; Salem, J.E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal toxic effects associated with immune checkpoint inhibitors: A systematic review and meta-analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef]
- Haanen, J.; Carbonnel, F.; Robert, C.; Kerr, K.M.; Peters, S.; Larkin, J.; Jordan, K.; Committee, E.G. Management of toxicities from immunotherapy: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28 (Suppl. 4), iv119–iv142. [Google Scholar] [CrossRef]
- Bertrand, A.; Kostine, M.; Barnetche, T.; Truchetet, M.E.; Schaeverbeke, T. Immune related adverse events associated with anti-CTLA-4 antibodies: Systematic review and meta-analysis. BMC Med. 2015, 13, 211. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, Q.; Zhou, Y.L.; Guo, X.; Ge, J.; Fu, J. Immune-Related adverse events from combination immunotherapy in cancer patients: A comprehensive meta-analysis of randomized controlled trials. Int. Immunopharmacol. 2018, 63, 292–298. [Google Scholar] [CrossRef]
- Sangro, B.; Chan, S.L.; Meyer, T.; Reig, M.; El-Khoueiry, A.; Galle, P.R. Diagnosis and management of toxicities of immune checkpoint inhibitors in hepatocellular carcinoma. J. Hepatol. 2020, 72, 320–341. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, S.; Yang, F.; Qi, X.; Wang, X.; Guan, X.; Shen, C.; Duma, N.; Vera Aguilera, J.; Chintakuntlawar, A.; et al. Treatment-Related adverse events of PD-1 and PD-L1 inhibitors in clinical trials: A systematic review and meta-analysis. JAMA Oncol. 2019, 5, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I.A. PD-1 inhibitors monotherapy in hepatocellular carcinoma: Meta-analysis and systematic review. Hepatobiliary Pancreat. Dis. Int. 2019, 18, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.M.; Liu, Y.; Wang, J.B.; Liu, L.X. Adverse effects of immune-checkpoint inhibitors in hepatocellular carcinoma. Onco Targets Ther. 2020, 13, 11725–11740. [Google Scholar] [CrossRef] [PubMed]
- Kadota, H.; Gono, T.; Shirai, Y.; Okazaki, Y.; Takeno, M.; Kuwana, M. Immune checkpoint inhibitor-induced myositis: A case report and literature review. Curr. Rheumatol. Rep. 2019, 21, 10. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Oba, A.; Shimada, S.; Akiyama, Y.; Nishikawaji, T.; Mogushi, K.; Ito, H.; Matsumura, S.; Aihara, A.; Mitsunori, Y.; Ban, D.; et al. ARID2 modulates DNA damage response in human hepatocellular carcinoma cells. J. Hepatol. 2017, 66, 942–951. [Google Scholar] [CrossRef]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [PubMed]
- Kurebayashi, Y.; Ojima, H.; Tsujikawa, H.; Kubota, N.; Maehara, J.; Abe, Y.; Kitago, M.; Shinoda, M.; Kitagawa, Y.; Sakamoto, M. Landscape of immune microenvironment in hepatocellular carcinoma and its additional impact on histological and molecular classification. Hepatology 2018, 68, 1025–1041. [Google Scholar] [CrossRef]
- Shimada, S.; Mogushi, K.; Akiyama, Y.; Furuyama, T.; Watanabe, S.; Ogura, T.; Ogawa, K.; Ono, H.; Mitsunori, Y.; Ban, D.; et al. Comprehensive molecular and immunological characterization of hepatocellular carcinoma. EBioMedicine 2019, 40, 457–470. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Immune Cell | Molecule(s) | Major Effects | Reference(s) |
|---|---|---|---|
| MDSC | GM-CSF, IL-β, IL-6, VEGF, MCP-1 | MDSC accumulation and migration adjustment | [65] |
| EZH2 enzyme | Activation of NF-κB Accumulation of polymorphonuclear MDSCs | [66] | |
| CCL26 | Invasion to the hypoxic region of HCC tissues | [67] | |
| ENTPD2 | MDSC accumulation by converting extracellular ATP to 5′ AMP | [68] | |
| TAM | CCL17, CCL18, CCL22 | Attraction of Treg cells to HCC tissues Activation of CTLs | [69,70] |
| IL-1β | Promoting EMT and HCC immune evasion | [71] | |
| TNF-α, IL-β, IL-6, IL-23 | Expansion of IL-17-producing CD4+ Th17 cells | [72] | |
| TGF-β | Promotion of TIM-3 expression in TAMs | [73] | |
| TAN | CXCL5 | TAN infiltration, indicating a poor prognosis | [74] |
| CCL2, CCL17 | Associated with tumor size, microvascular invasion, differentiation, and clinical stage | [75] | |
| HIF-1α | TAN infiltration associated with HCC progression | [75] | |
| CAF | IDO, PGE2 | TNF-α and INF-γ production by NK cells Associated with HCC development | [76] |
| BNP-4 | Activation of hepatic fibroblasts Enhancing CAF invasiveness | [77] | |
| Treg | AP-1, NFAT1 | Promotion of immunosuppression in HCC | [78] |
| TGF-β | Treg infiltration into the liver | [79] | |
| TIL | FOXP3 | Master transcription factor of Treg Associated with poor prognosis | [80,81] |
| CTL | VEGF, CXCL17, IL-10, IDO | Causing poor production of INF-γ by CTLs | [67,82,83,84] |
| IL-2, IDO | CTL suppression | [85] |
| Regimen | Target Population | Design | Registration Number | Result | Reference |
|---|---|---|---|---|---|
| Anti-CTLA-4 antibody | |||||
| Tremelimumab | Second-line for advanced HCC | Single group assignment | NCT01008358 | Effective | [152] |
| Tremelimumab | Neoadjuvant before ablation | Single group assignment | NCT01853618 | Effective | [153] |
| Anti-PD-1 antibody | |||||
| Nivolumab | First- and second-line for advanced HCC | Single group assignment | NCT01658878 | Positive | [154] |
| Nivolumab | First-line for advanced HCC | Versus sorafenib | NCT02576509 | Negative | [155] |
| Nivolumab | Adjuvant after curative treatment | Versus placebo | NCT03383458 | Ongoing | |
| Nivolumab | Improvement after TACE | Versus placebo | NCT04268888 | Ongoing | |
| Tislelizumab | First-line for unresectable HCC | Versus sorafenib | NCT03412773 | Ongoing | [158] |
| Anti-PD-L1 antibody | |||||
| Pembrolizumab | Second-line for advanced HCC | Single group assignment | NCT02702414 | Effective | [159] |
| Pembrolizumab | Second-line for advanced HCC | Versus placebo | NCT02702401 | Negative | [160] |
| Pembrolizumab | Adjuvant after curative treatment | Versus placebo | NCT03867084 | Ongoing |
| Regimen | Target Population | Design | Registration Number | Result | Reference |
|---|---|---|---|---|---|
| Anti-CTLA-4 antibody-based Combination therapy | |||||
| Tremelimumab + TACE | First-line for advanced HCC | Single group assignment | NCT01853618 | Effective | [168] |
| Anti-PD-1 antibody-based Combination therapy | |||||
| Nivolumab + cabozantinib | Neoadjuvant before resection | Single group assignment | NCT03299946 | Ongoing | |
| Nivolumab + ipilimumab | First-line for advanced HCC | Versus lenvatinib or sorafenib | NCT04039607 | Ongoing | |
| Nivolumab following SIRT | Intermediate HCC | Single group assignment | NCT03380130 | Ongoing | |
| Nivolumab following resection | First diagnosis of HCC | Versus placebo | NCT03383458 | Ongoing | |
| Nivolumab + galunisertib | Second-line for advanced HCC | Single group assignment | NCT02423343 | Ongoing | |
| Nivolumab + lenvatinib | First-line for advanced HCC | Single group assignment | NCT03418922 | Ongoing | |
| Nivolumab + sorafenib | Advanced HCC | Single group assignment | NCT03439891 | Ongoing | |
| Nivolumab + DEB-TACE | Intermediate HCC | Parallel assignment | NCT03143270 | Ongoing | |
| Nivolumab + mogamulizumab | Second-line for advanced HCC | Single group assignment | NCT02705105 | Ongoing | |
| Nivolumab + relatlimab | Advanced HCC | Parallel assignment | NCT01968109 | Ongoing | |
| Anti-PD-L1 antibody-based Combination therapy | |||||
| Atezolizumab + bevacizumab | First-line for advanced HCC | Versus sorafenib | NCT03434379 | Positive | [12] |
| Atezolizumab + cabozantinib | First-line for advanced HCC | Versus sorafenib | MCT03755791 | Ongoing | |
| Durvalumab + tremelimumab | First-line for advanced HCC | Versus sorafenib | NCT03298451 | Ongoing | |
| Durvalumab + ramucirumab | Second-line for advanced HCC | Parallel assignment | NCT02572687 | Ongoing | |
| Regimen | Target Population | Design | Registration Number |
|---|---|---|---|
| CIK monotherapy | |||
| CIKs | HCC | Phase III clinical trial | NCT00769106 |
| CIKs | HCC, RCC, and lung cancer | Phase I clinical trial | NCT01914263 |
| DC-CIKs | HCC | Phase III clinical trial | NCT01821482 |
| CIK-based Combination therapy | |||
| CIKs + PD-1 antibodies | HCC, RCC, bladder cancer, Colorectal cancer, and NSCLC | Phase II clinical trial | NCT02886897 |
| CIKs + TACE | HCC | Phase III clinical trial | NCT02487017 |
| CIKs + RFA | HCC | Phase III clinical trial | NCT02678013 |
| CAR-T trials | |||
| Anti-GPC3 CAR-T | HCC | Phase I/II clinical trial | NCT03084380 |
| Anti-GPC3 CAR-T | HCC | Phase I/II clinical trial | NCT02723942 |
| Anti-GPC3 CAR-T | AFP-expressing HCC | Phase I clinical trial | NCT03349255 |
| Anti-GPC3 CAR-T | Advanced HCC | Phase I clinical trial | NCT03198546 |
| TAI-GP3-CAR-T | HCC | Phase I/II clinical trial | NCT02715362 |
| Anti-Mucin1 CAR-T | HCC, NSCLC, pancreatic cancer, and triple-negative breast cancer | Phase I/II clinical trial | NCT02587689 |
| CAR-T targeting TAAs | HCC, pancreatic cancer, and colorectal cancer | Phase I/II clinical trial | NCT02959151 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oura, K.; Morishita, A.; Tani, J.; Masaki, T. Tumor Immune Microenvironment and Immunosuppressive Therapy in Hepatocellular Carcinoma: A Review. Int. J. Mol. Sci. 2021, 22, 5801. https://doi.org/10.3390/ijms22115801
Oura K, Morishita A, Tani J, Masaki T. Tumor Immune Microenvironment and Immunosuppressive Therapy in Hepatocellular Carcinoma: A Review. International Journal of Molecular Sciences. 2021; 22(11):5801. https://doi.org/10.3390/ijms22115801
Chicago/Turabian StyleOura, Kyoko, Asahiro Morishita, Joji Tani, and Tsutomu Masaki. 2021. "Tumor Immune Microenvironment and Immunosuppressive Therapy in Hepatocellular Carcinoma: A Review" International Journal of Molecular Sciences 22, no. 11: 5801. https://doi.org/10.3390/ijms22115801
APA StyleOura, K., Morishita, A., Tani, J., & Masaki, T. (2021). Tumor Immune Microenvironment and Immunosuppressive Therapy in Hepatocellular Carcinoma: A Review. International Journal of Molecular Sciences, 22(11), 5801. https://doi.org/10.3390/ijms22115801