
Novel Therapeutic Approaches for Alzheimer’s Disease: An Updated Review


Abstract
:1. Introduction
2. Novel Therapeutic Approach
2.1. Anti-Amyloid Therapy
2.1.1. Secretase Inhibitors
2.1.2. Aβ aggregation Inhibitors
2.1.3. Aβ Immunotherapy
2.2. Anti-tau Therapy
2.2.1. Phosphatase Modifiers
2.2.2. Kinase Inhibitors
2.2.3. Tau Aggregation Inhibitors
2.2.4. Microtubule Stabilizers
2.2.5. Tau Immunotherapy
2.3. Anti-neuroinflammatory Therapy
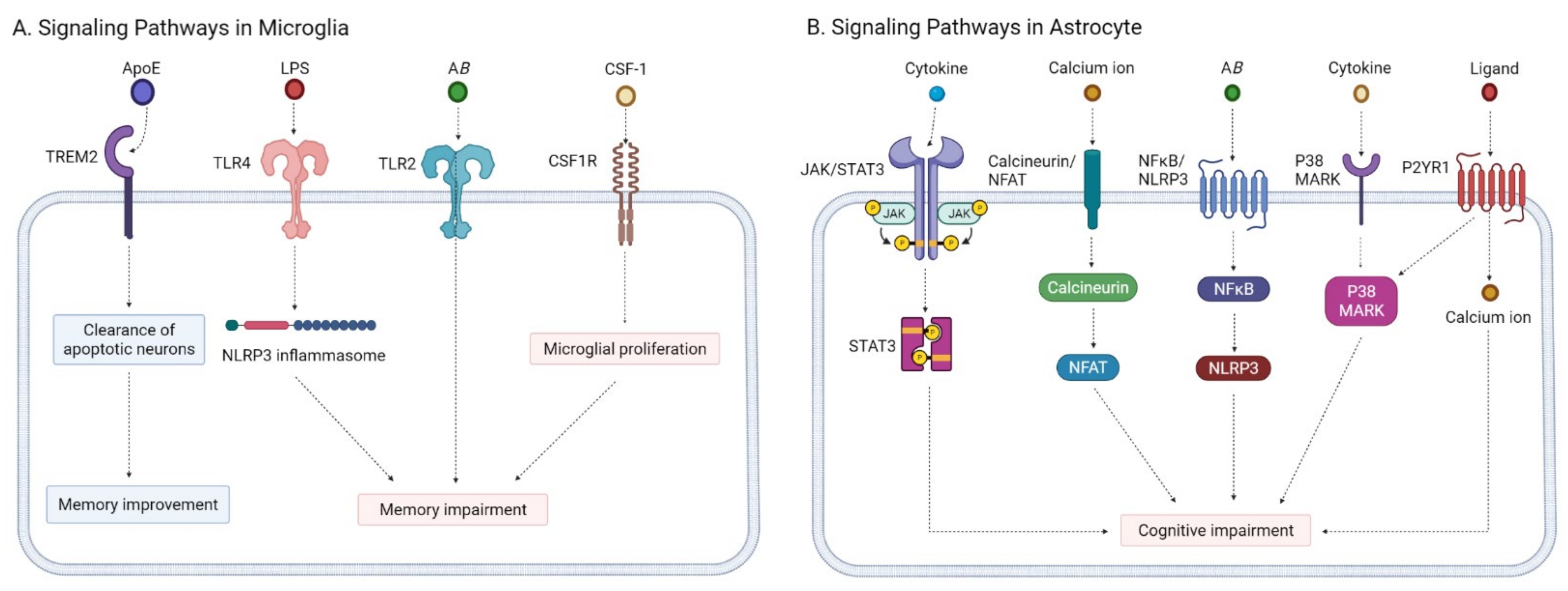
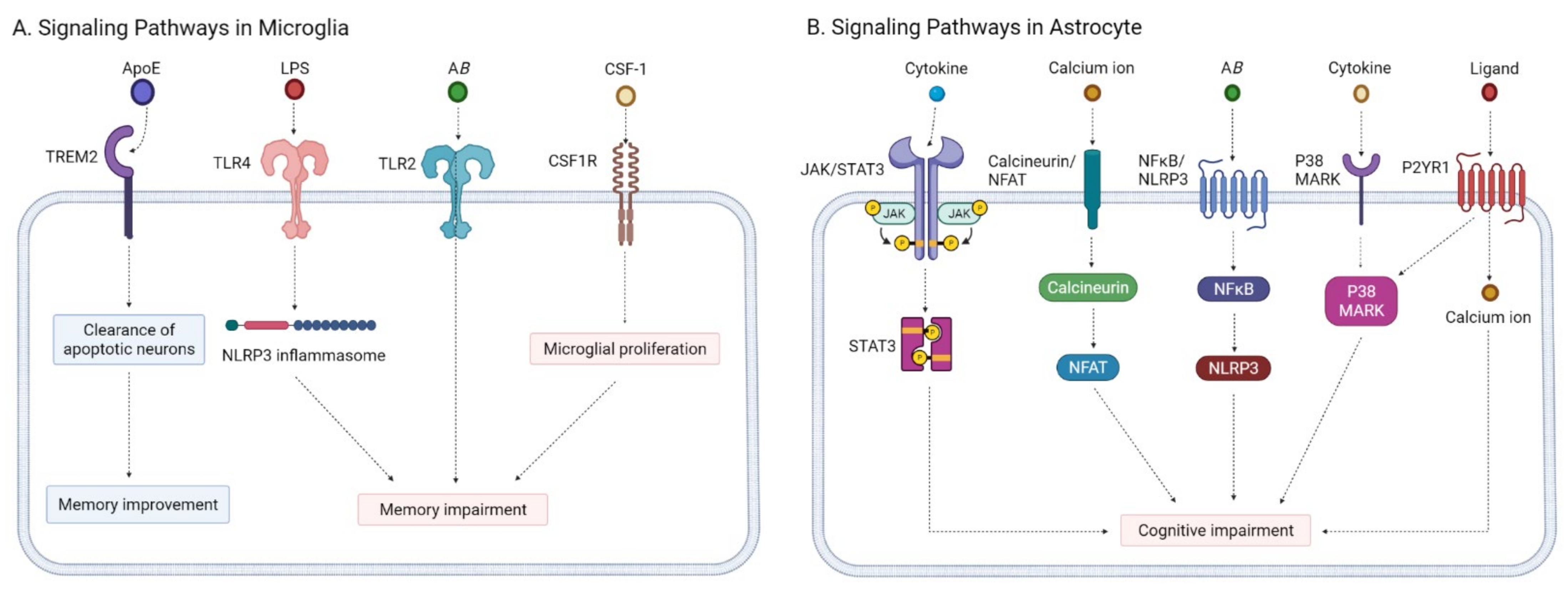
2.3.1. Microglia Modulators
2.3.2. Astrocyte Modulators
2.3.3. Insulin Resistance Management
2.3.4. Microbiome Therapy
2.4. Neuroprotective Agents
2.4.1. Antiepileptic Drugs
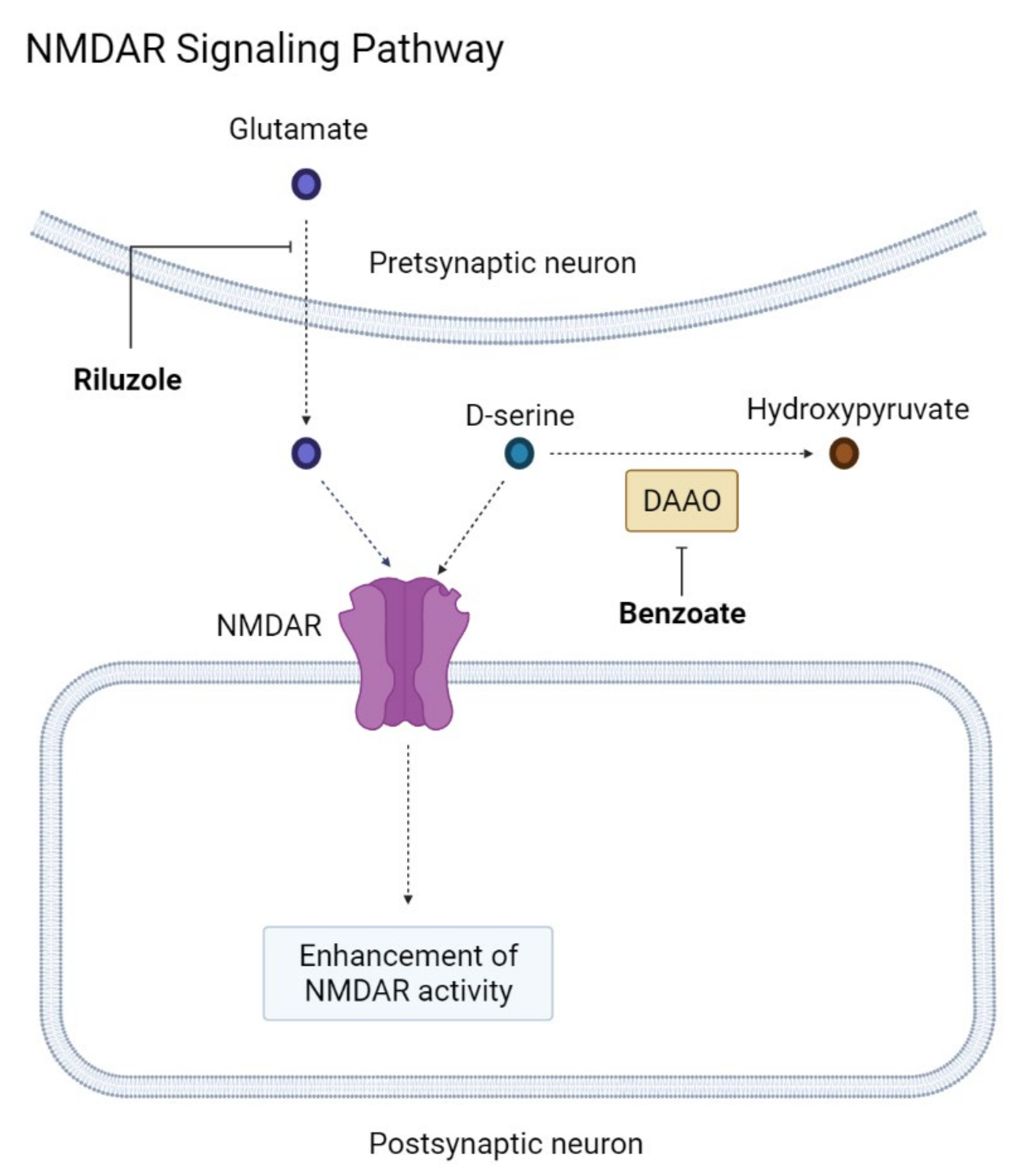
2.4.2. NMDAR Modification
2.4.3. Omega 3 Polyunsaturated Fatty Acid Supplements
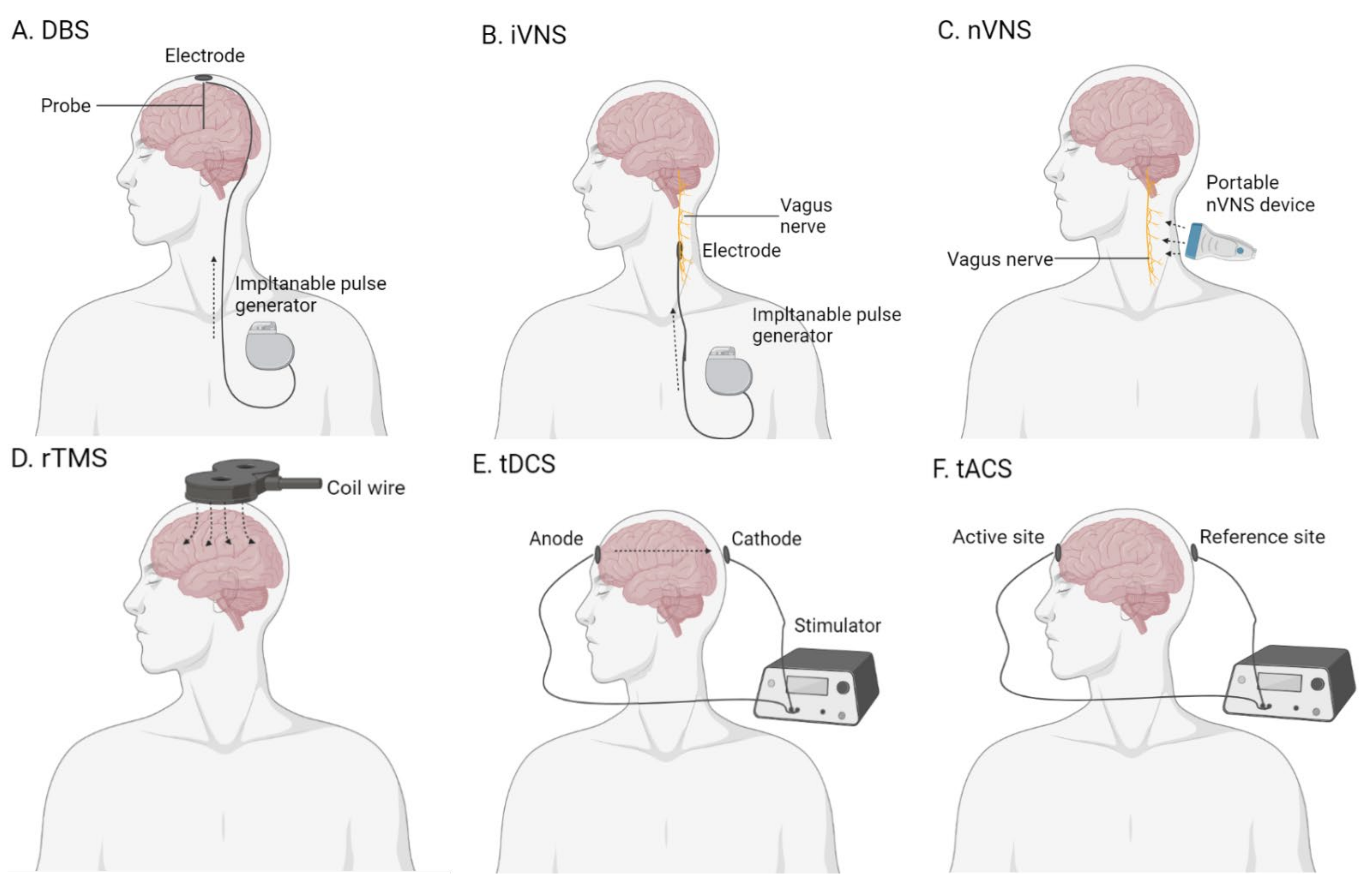
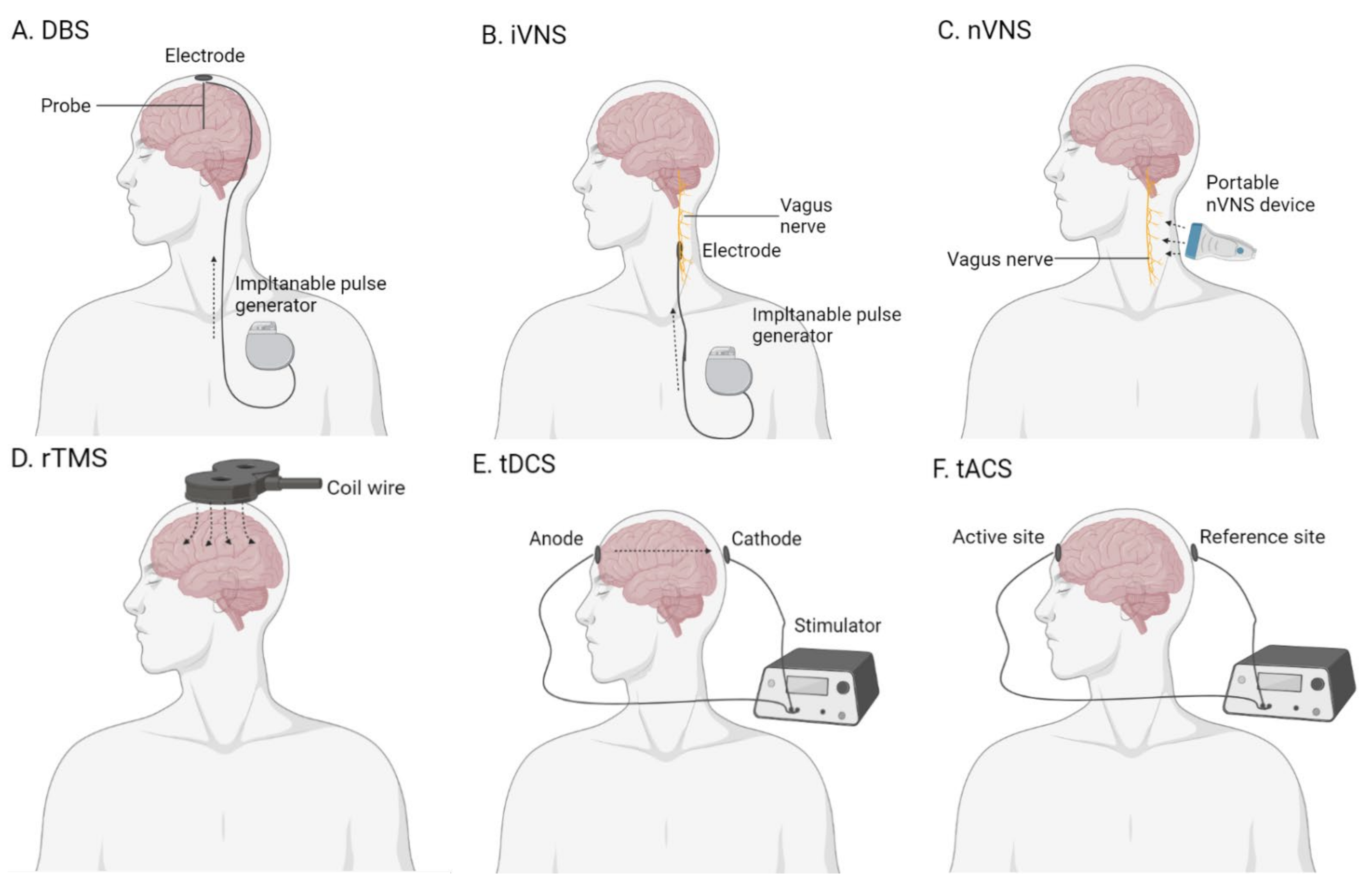
2.5. Brain Stimulation
2.5.1. Deep-Brain Stimulation
2.5.2. Vagus Nerve Stimulation
2.5.3. Transcranial Magnetic Stimulation
2.5.4. Transcranial Electrical Stimulation
3. Discussion
4. Future Research Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scheltens, P.; Blennow, K. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- GBD 2016 Dementia Collaborators. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Niu, H.; Álvarez-Álvarez, I. Prevalence and incidence of Alzheimer’s disease in Europe: A meta-analysis. Neurologia 2017, 32, 523–532. [Google Scholar] [CrossRef] [PubMed]
- United Nations Department of Economic and Social Affairs, Population Division. World Population Ageing 2020 Highlights: Living Arrangements of Older Persons. 2020. (ST/ESA/SER.A/451). Available online: https://www.un.org/development/desa/pd/sites/www.un.org.development.desa.pd/files/undesa_pd-2020_world_population_ageing_highlights.pdf (accessed on 4 April 2021).
- Hebert, L.E.; Weuve, J. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [Green Version]
- Eratne, D.; Loi, S.M. Alzheimer’s disease: Clinical update on epidemiology, pathophysiology and diagnosis. Australas Psychiatry 2018, 26, 347–357. [Google Scholar] [CrossRef]
- 2020 Alzheimer’s Disease Facts and Figures. Alzheimers Dementia. 2020. Available online: https://alz-journals.onlinelibrary.wiley.com/doi/full/10.1002/alz.12068 (accessed on 4 April 2021).
- Mokdad, A.H.; Ballestros, K. The State of US Health, 1990–2016: Burden of Diseases, Injuries, and Risk Factors among US States. JAMA 2018, 319, 1444–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.H.; Lane, H.Y. The Role of N-Methyl-D-Aspartate Receptor Neurotransmission and Precision Medicine in Behavioral and Psychological Symptoms of Dementia. Front. Pharmacol. 2019, 10, 540. [Google Scholar] [CrossRef] [Green Version]
- Baharudin, A.D.; Din, N.C. The associations between behavioral-psychological symptoms of dementia (BPSD) and coping strategy, burden of care and personality style among low-income caregivers of patients with dementia. BMC Public Health 2019, 19 (Suppl. 4), 447. [Google Scholar] [CrossRef] [PubMed]
- Friedman, E.M.; Shih, R.A. US Prevalence and Predictors of Informal Caregiving for Dementia. Health Aff. 2015, 34, 1637–1641. [Google Scholar] [CrossRef]
- Kevadiya, B.D.; Ottemann, B.M. Neurotheranostics as personalized medicines. Adv. Drug Deliv. Rev. 2019, 148, 252–289. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Silvente, L.; Castells, X. Discontinuation, Efficacy, and Safety of Cholinesterase Inhibitors for Alzheimer’s Disease: A Meta-Analysis and Meta-Regression of 43 Randomized Clinical Trials Enrolling 16 106 Patients. Int. J. Neuropsychopharmacol. 2017, 20, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Silvente, L.; Capellà, D. Predictors of discontinuation, efficacy, and safety of memantine treatment for Alzheimer’s disease: Meta-analysis and meta-regression of 18 randomized clinical trials involving 5004 patients. BMC Geriatr. 2018, 18, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal, S.L.; Lockhart, S.N. Subthreshold Amyloid Predicts Tau Deposition in Aging. J. Neurosci. 2018, 38, 4482–4489. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.N.; Lockhart, S.N. Relationships between Tau and Glucose Metabolism Reflect Alzheimer’s Disease Pathology in Cognitively Normal Older Adults. Cereb. Cortex 2019, 29, 1997–2009. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Benzinger, T.L. Evaluation of Tau Imaging in Staging Alzheimer Disease and Revealing Interactions between β-Amyloid and Tauopathy. JAMA Neurol. 2016, 73, 1070–1077. [Google Scholar] [CrossRef]
- Jagust, W. Imaging the evolution and pathophysiology of Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 687–700. [Google Scholar] [CrossRef]
- Hossain, M.F.; Wang, N. Exploring the multifunctional role of melatonin in regulating autophagy and sleep to mitigate Alzheimer’s disease neuropathology. Ageing Res. Rev. 2021, 67, 101304. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J. Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 1691–1703. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J. Randomized Trial of Verubecestat for Prodromal Alzheimer’s Disease. N. Engl. J. Med. 2019, 380, 1408–1420. [Google Scholar] [CrossRef] [PubMed]
- Henley, D.; Raghavan, N. Preliminary Results of a Trial of Atabecestat in Preclinical Alzheimer’s Disease. N. Engl. J. Med. 2019, 380, 1483–1485. [Google Scholar] [CrossRef] [PubMed]
- Wessels, A.M.; Tariot, P.N. Efficacy and Safety of Lanabecestat for Treatment of Early and Mild Alzheimer Disease: The AMARANTH and DAYBREAK-ALZ Randomized Clinical Trials. JAMA Neurol. 2020, 77, 199–209. [Google Scholar] [CrossRef]
- Lo, A.C.; Evans, C.D. Phase II (NAVIGATE-AD study) Results of LY3202626 Effects on Patients with Mild Alzheimer’s Disease Dementia. J. Alzheimers Dis. Rep. 2021, 5, 321–336. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Study of CNP520 Versus Placebo in Participants at Risk for the Onset of Clinical Symptoms of Alzheimer’s Disease. NCT03131453. Available online: https://ClinicalTrials.gov/show/NCT03131453 (accessed on 4 April 2021).
- Imbimbo, B.P.; Watling, M. Investigational BACE inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2019, 28, 967–975. [Google Scholar] [CrossRef]
- Iraji, A.; Khoshneviszadeh, M. Novel small molecule therapeutic agents for Alzheimer disease: Focusing on BACE1 and multi-target directed ligands. Bioorg. Chem. 2020, 97, 103649. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Raman, R. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Coric, V.; Salloway, S. Targeting Prodromal Alzheimer Disease with Avagacestat: A Randomized Clinical Trial. JAMA Neurol. 2015, 72, 1324–1333. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C.; Schneider, L.S. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: A randomized controlled trial. JAMA 2009, 302, 2557–2564. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, C.W.; Bush, A.I. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: A pilot phase 2 clinical trial. Arch. Neurol. 2003, 60, 1685–1691. [Google Scholar] [CrossRef]
- Lannfelt, L.; Blennow, K. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: A phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008, 7, 779–786. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Rowe, C.C. A randomized, exploratory molecular imaging study targeting amyloid β with a novel 8-OH quinoline in Alzheimer’s disease: The PBT2-204 IMAGINE study. Alzheimers Dement. 2017, 3, 622–635. [Google Scholar] [CrossRef]
- Mantile, F.; Prisco, A. Vaccination against β-Amyloid as a Strategy for the Prevention of Alzheimer’s Disease. Biology 2020, 9, 425. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S. Aducanumab, gantenerumab, BAN2401, and ALZ-801-the first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res. Ther. 2020, 12, 95. [Google Scholar] [CrossRef]
- Ostrowitzki, S.; Lasser, R.A. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res. Ther. 2017, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Alexander, G.C.; Emerson, S. Evaluation of Aducanumab for Alzheimer Disease: Scientific Evidence and Regulatory Review Involving Efficacy, Safety, and Futility. JAMA 2021, 325, 1717–1718. [Google Scholar] [CrossRef]
- Howard, R.; Liu, K.Y. Questions EMERGE as Biogen claims aducanumab turnaround. Nat. Rev. Neurol. 2020, 16, 63–64. [Google Scholar] [CrossRef]
- Malpas, C.B.; Vivash, L. A Phase IIa Randomized Control Trial of VEL015 (Sodium Selenate) in Mild-Moderate Alzheimer’s Disease. J. Alzheimers Dis. 2016, 54, 223–232. [Google Scholar] [CrossRef]
- Cardoso, B.R.; Roberts, B.R. Supranutritional Sodium Selenate Supplementation Delivers Selenium to the Central Nervous System: Results from a Randomized Controlled Pilot Trial in Alzheimer’s Disease. Neurotherapeutics 2019, 16, 192–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, H.S.; Mitev, V. Discovery and development of Seliciclib. How systems biology approaches can lead to better drug performance. J. Biotechnol. 2015, 202, 40–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leggio, G.M.; Catania, M.V. The antineoplastic drug flavopiridol reverses memory impairment induced by Amyloid-ß1-42 oligomers in mice. Pharmacol. Res. 2016, 106, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Lovestone, S.; Boada, M. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef]
- Nunes, M.A.; Viel, T.A. Microdose lithium treatment stabilized cognitive impairment in patients with Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, S.; Kishi, T. Lithium as a Treatment for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2015, 48, 403–410. [Google Scholar] [CrossRef]
- Matsunaga, S.; Fujishiro, H. Efficacy and Safety of Glycogen Synthase Kinase 3 Inhibitors for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2019, 69, 1031–1039. [Google Scholar] [CrossRef]
- Wischik, C.M.; Staff, R.T. Tau aggregation inhibitor therapy: An exploratory phase 2 study in mild or moderate Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 705–720. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, S.; Feldman, H.H. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: A randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 2016, 388, 2873–2884. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.S.; Wahlqvist, M.L. Turmeric improves post-prandial working memory in pre-diabetes independent of insulin. Asia Pac. J. Clin. Nutr. 2014, 23, 581–591. [Google Scholar] [CrossRef]
- Cox, K.H.; Pipingas, A. Investigation of the effects of solid lipid curcumin on cognition and mood in a healthy older population. J. Psychopharmacol. 2015, 29, 642–651. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Study to Evaluate the Safety, Tolerability and the Effect of BMS-241027 on Cerebrospinal Fluid Biomarkers in Subjects with Mild Alzheimer’s Disease. NCT01492374. Available online: https://ClinicalTrials.gov/show/NCT01492374 (accessed on 4 April 2021).
- Morimoto, B.H.; Schmechel, D. A double-blind, placebo-controlled, ascending-dose, randomized study to evaluate the safety, tolerability and effects on cognition of AL-108 after 12 weeks of intranasal administration in subjects with mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 2013, 35, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Gozes, I.; Stewart, A. Addressing Alzheimer’s disease tangles: From NAP to AL-108. Curr. Alzheimer Res. 2009, 6, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Tsai, R.M.; Miller, Z. Reactions to Multiple Ascending Doses of the Microtubule Stabilizer TPI-287 in Patients with Alzheimer Disease, Progressive Supranuclear Palsy, and Corticobasal Syndrome: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 215–224. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. 24 Months Safety and Efficacy Study of AADvac1 in Patients with Mild Alzheimer’s Disease. NCT02579252. Available online: https://ClinicalTrials.gov/show/NCT02579252 (accessed on 4 April 2021).
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Zhu, K. Early active immunization with Aβ(3-10)-KLH vaccine reduces tau phosphorylation in the hippocampus and protects cognition of mice. Neural Regen. Res. 2020, 15, 519–527. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Phase 2 Study of BIIB092 in Participants with Early Alzheimer’s Disease. NCT03352557. Available online: https://ClinicalTrials.gov/show/NCT03352557 (accessed on 4 April 2021).
- ClinicalTrials.gov. A Study to Evaluate the Efficacy and Safety of ABBV-8E12 in Subjects with Early Alzheimer’s Disease. NCT02880956. Available online: https://ClinicalTrials.gov/show/NCT02880956 (accessed on 4 April 2021).
- ClinicalTrials.gov. An Extension Study of ABBV-8E12 in Early Alzheimer’s Disease (AD). NCT03712787. Available online: https://ClinicalTrials.gov/show/NCT03712787 (accessed on 4 April 2021).
- ClinicalTrials.gov. A Study to Evaluate the Efficacy and Safety of Semorinemab in Patients with Prodromal to Mild Alzheimer’s Disease. NCT03289143. Available online: https://ClinicalTrials.gov/show/NCT03289143 (accessed on 4 April 2021).
- ClinicalTrials.gov. A Study of Semorinemab in Patients with Moderate Alzheimer’s Disease. NCT03828747. Available online: https://ClinicalTrials.gov/show/NCT03828747 (accessed on 4 April 2021).
- ClinicalTrials.gov. Single-Ascending-Dose Study of BIIB076 in Healthy Volunteers and Participants with Alzheimer’s Disease. NCT03056729. Available online: https://ClinicalTrials.gov/show/NCT03056729 (accessed on 4 April 2021).
- ClinicalTrials.gov. A Study of LY3303560 in Participants with Early Symptomatic Alzheimer’s Disease. NCT03518073. Available online: https://ClinicalTrials.gov/show/NCT03518073 (accessed on 4 April 2021).
- ClinicalTrials.gov. A Study of JNJ-63733657 in Participants with Early Alzheimer’s Disease. NCT04619420. Available online: https://ClinicalTrials.gov/show/NCT04619420 (accessed on 4 April 2021).
- ClinicalTrials.gov. A Study to Test the Safety and Tolerability and Pharmacokinetics of Single Doses of UCB0107 in Healthy Japanese Subjects. NCT03605082. Available online: https://ClinicalTrials.gov/show/NCT03605082 (accessed on 4 April 2021).
- Courade, J.P.; Angers, R. Epitope determines efficacy of therapeutic anti-Tau antibodies in a functional assay with human Alzheimer Tau. Acta Neuropathol. 2018, 136, 729–745. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Chen, Y. TLR4 Targeting as a Promising Therapeutic Strategy for Alzheimer Disease Treatment. Front. Neurosci. 2020, 14, 602508. [Google Scholar] [CrossRef]
- Olmos-Alonso, A.; Schetters, S.T. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain 2016, 139 Pt 3, 891–907. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, R.; Fryatt, G. CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain 2019, 142, 3243–3264. [Google Scholar] [CrossRef]
- Sosna, J.; Philipp, S. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Kim, H. Inhibition of STAT3 phosphorylation attenuates impairments in learning and memory in 5XFAD mice, an animal model of Alzheimer’s disease. J. Pharmacol. Sci. 2020, 143, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Millot, P.; San, C. STAT3 inhibition protects against neuroinflammation and BACE1 upregulation induced by systemic inflammation. Immunol. Lett. 2020, 228, 129–134. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Pilot Open Labeled Study of Tacrolimus in Alzheimer’s Disease. NCT04263519. Available online: https://ClinicalTrials.gov/show/NCT04263519 (accessed on 18 April 2021).
- Kheiri, G.; Dolatshahi, M. Role of p38/MAPKs in Alzheimer’s disease: Implications for amyloid beta toxicity targeted therapy. Rev. Neurosci. 2018, 30, 9–30. [Google Scholar] [CrossRef]
- Maphis, N.; Jiang, S. Selective suppression of the α isoform of p38 MAPK rescues late-stage tau pathology. Alzheimers Res. Ther. 2016, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Gee, M.S.; Son, S.H. A selective p38α/β MAPK inhibitor alleviates neuropathology and cognitive impairment, and modulates microglia function in 5XFAD mouse. Alzheimers Res. Ther. 2020, 12, 45. [Google Scholar] [CrossRef] [Green Version]
- Reichenbach, N.; Delekate, A. P2Y1 receptor blockade normalizes network dysfunction and cognition in an Alzheimer’s disease model. J. Exp. Med. 2018, 215, 1649–1663. [Google Scholar] [CrossRef]
- Craft, S.; Claxton, A. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimers Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef] [Green Version]
- Avgerinos, K.I.; Kalaitzidis, G. Intranasal insulin in Alzheimer’s dementia or mild cognitive impairment: A systematic review. J. Neurol. 2018, 265, 1497–1510. [Google Scholar] [CrossRef]
- Craft, S.; Raman, R. Safety, Efficacy, and Feasibility of Intranasal Insulin for the Treatment of Mild Cognitive Impairment and Alzheimer Disease Dementia: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Salameh, T.S.; Rhea, E.M. Brain uptake pharmacokinetics of incretin receptor agonists showing promise as Alzheimer’s and Parkinson’s disease therapeutics. Biochem. Pharmacol. 2020, 180, 114187. [Google Scholar] [CrossRef]
- Gejl, M.; Gjedde, A. In Alzheimer’s Disease, 6-Month Treatment with GLP-1 Analog Prevents Decline of Brain Glucose Metabolism: Randomized, Placebo-Controlled, Double-Blind Clinical Trial. Front. Aging Neurosci. 2016, 8, 108. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Perez, T. Metformin in Amnestic Mild Cognitive Impairment: Results of a Pilot Randomized Placebo Controlled Clinical Trial. J. Alzheimers Dis. 2016, 51, 501–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenig, A.M.; Mechanic-Hamilton, D. Effects of the Insulin Sensitizer Metformin in Alzheimer Disease: Pilot Data From a Randomized Placebo-controlled Crossover Study. Alzheimer Dis. Assoc. Disord. 2017, 31, 107–113. [Google Scholar] [CrossRef]
- Wójtowicz, S.; Strosznajder, A.K. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Hanyu, H. Efficacy of PPAR-γ agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging 2011, 32, 1626–1633. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. AD-4833/TOMM40_303 Extension Study of the Safety and Efficacy of Pioglitazone to Slow Cognitive Decline in Participants with Mild Cognitive Impairment Due to Alzheimer Disease. NCT02284906. Available online: https://ClinicalTrials.gov/show/NCT02284906 (accessed on 22 April 2021).
- ClinicalTrials.gov. Biomarker Qualification for Risk of Mild Cognitive Impairment (MCI) Due to Alzheimer’s Disease (AD) and Safety and Efficacy Evaluation of Pioglitazone in Delaying Its Onset. NCT01931566. Available online: https://ClinicalTrials.gov/show/NCT01931566 (accessed on 22 April 2021).
- Reich, D.; Gallucci, G. Therapeutic Advantages of Dual Targeting of PPAR-δ and PPAR-γ in an Experimental Model of Sporadic Alzheimer’s Disease. J. Parkinsons Dis. Alzheimers Dis. 2018, 5. [Google Scholar] [CrossRef]
- Wang, T.; Kuang, W. A phase II randomized trial of sodium oligomannate in Alzheimer’s dementia. Alzheimers Res. Ther. 2020, 12, 110. [Google Scholar] [CrossRef]
- Xiao, S.; Chan, P. A 36-week multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 3 clinical trial of sodium oligomannate for mild-to-moderate Alzheimer’s dementia. Alzheimers Res. Ther. 2021, 13, 62. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Study of AGB101 in Mild Cognitive Impairment Due to Alzheimer’s Disease. NCT03486938. Available online: https://ClinicalTrials.gov/show/NCT03486938 (accessed on 4 April 2021).
- ClinicalTrials.gov. Nighttime Agitation and Restless Legs Syndrome in People with Alzheimer’s Disease. NCT03082755. Available online: https://ClinicalTrials.gov/show/NCT03082755 (accessed on 1 May 2021).
- Lin, C.H.; Chen, P.K. Benzoate, a D-amino acid oxidase inhibitor, for the treatment of early-phase Alzheimer disease: A randomized, double-blind, placebo-controlled trial. Biol. Psychiatry 2014, 75, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Lane, H.Y.; Tu, C.H. Brain Activity of Benzoate, a D-Amino Acid Oxidase Inhibitor, in Patients with Mild Cognitive Impairment in a Randomized, Double-Blind, Placebo Controlled Clinical Trial. Int. J. Neuropsychopharmacol. 2021, 24, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Chen, P.K. Effect of Sodium Benzoate on Cognitive Function among Patients with Behavioral and Psychological Symptoms of Dementia: Secondary Analysis of a Randomized Clinical Trial. JAMA Netw. Open 2021, 4, e216156. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Riluzole in Mild Alzheimer’s Disease. NCT01703117. Available online: https://ClinicalTrials.gov/show/NCT01703117 (accessed on 28 April 2021).
- ClinicalTrials.gov. Study of BHV-4157 in Alzheimer’s Disease. NCT03605667. Available online: https://ClinicalTrials.gov/show/NCT03605667 (accessed on 28 April 2021).
- Quinn, J.F.; Raman, R. Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: A randomized trial. JAMA 2010, 304, 1903–1911. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. DHA Brain Delivery Trial (PreventE4). NCT03613844. Available online: https://ClinicalTrials.gov/show/NCT03613844 (accessed on 19 July 2021).
- Bhatt, D.L.; Hull, M.A. Beyond cardiovascular medicine: Potential future uses of icosapent ethyl. Eur. Heart J. Suppl. 2020, 22 (Suppl. J), J54–J64. [Google Scholar] [CrossRef]
- Lozano, A.M.; Fosdick, L. A Phase II Study of Fornix Deep Brain Stimulation in Mild Alzheimer’s Disease. J. Alzheimers Dis. 2016, 54, 777–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoutsakos, J.S.; Yan, H. Deep Brain Stimulation Targeting the Fornix for Mild Alzheimer Dementia (the ADvance Trial): A Two Year Follow-up Including Results of Delayed Activation. J. Alzheimers Dis. 2018, 64, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.J.; Lozano, A.M. Current Status of Deep Brain Stimulation for Alzheimer’s Disease: From Chance Observation to Clinical Trials. Cold Spring Harb. Symp. Quant. Biol. 2018, 83, 201–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, J.; Hardenacke, K. Deep brain stimulation of the nucleus basalis of Meynert in Alzheimer’s dementia. Mol. Psychiatry 2015, 20, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Sjögren, M.J.; Hellström, P.T. Cognition-enhancing effect of vagus nerve stimulation in patients with Alzheimer’s disease: A pilot study. J. Clin. Psychiatry 2002, 63, 972–980. [Google Scholar] [CrossRef]
- Merrill, C.A.; Jonsson, M.A. Vagus nerve stimulation in patients with Alzheimer’s disease: Additional follow-up results of a pilot study through 1 year. J. Clin. Psychiatry 2006, 67, 1171–1178. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Treatment of Mild Cognitive Impairment with Transcutaneous Vagal Nerve Stimulation. NCT03359902. Available online: https://ClinicalTrials.gov/show/NCT03359902 (accessed on 3 May 2021).
- Padala, P.R.; Padala, K.P. Repetitive transcranial magnetic stimulation for apathy in mild cognitive impairment: A double-blind, randomized, sham-controlled, cross-over pilot study. Psychiatry Res. 2018, 261, 312–318. [Google Scholar] [CrossRef]
- Taylor, J.L.; Hambro, B.C. The effects of repetitive transcranial magnetic stimulation in older adults with mild cognitive impairment: A protocol for a randomized, controlled three-arm trial. BMC Neurol. 2019, 19, 326. [Google Scholar] [CrossRef]
- Cotelli, M.; Calabria, M. Improved language performance in Alzheimer disease following brain stimulation. J. Neurol. Neurosurg. Psychiatry 2011, 82, 794–797. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, W. Adjunctive treatment with high frequency repetitive transcranial magnetic stimulation for the behavioral and psychological symptoms of patients with Alzheimer’s disease: A randomized, double-blind, sham-controlled study. Shanghai Arch. Psychiatry 2015, 27, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Bagattini, C.; Zanni, M. Enhancing cognitive training effects in Alzheimer’s disease: rTMS as an add-on treatment. Brain Stimul. 2020, 13, 1655–1664. [Google Scholar] [CrossRef]
- Zhang, F.; Qin, Y. High-frequency repetitive transcranial magnetic stimulation combined with cognitive training improves cognitive function and cortical metabolic ratios in Alzheimer’s disease. J. Neural Transm. (Vienna) 2019, 126, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Alcalá-Lozano, R.; Morelos-Santana, E. Similar clinical improvement and maintenance after rTMS at 5 Hz using a simple vs. complex protocol in Alzheimer’s disease. Brain Stimul. 2018, 11, 625–627. [Google Scholar] [CrossRef]
- Ahmed, M.A.; Darwish, E.S. Effects of low versus high frequencies of repetitive transcranial magnetic stimulation on cognitive function and cortical excitability in Alzheimer’s dementia. J. Neurol. 2012, 259, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, G.; Lithgow, B. Short and Long-term Effects of rTMS Treatment on Alzheimer’s Disease at Different Stages: A Pilot Study. J. Exp. Neurosci. 2015, 9, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Eliasova, I.; Anderkova, L. Non-invasive brain stimulation of the right inferior frontal gyrus may improve attention in early Alzheimer’s disease: A pilot study. J. Neurol. Sci. 2014, 346, 318–322. [Google Scholar] [CrossRef]
- Anderkova, L.; Eliasova, I. Distinct Pattern of Gray Matter Atrophy in Mild Alzheimer’s Disease Impacts on Cognitive Outcomes of Noninvasive Brain Stimulation. J. Alzheimers Dis. 2015, 48, 251–260. [Google Scholar] [CrossRef]
- Koch, G.; Bonnì, S. Transcranial magnetic stimulation of the precuneus enhances memory and neural activity in prodromal Alzheimer’s disease. Neuroimage 2018, 169, 302–311. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Li, Z. Repetitive transcranial magnetic stimulation improves cognitive function of Alzheimer’s disease patients. Oncotarget 2017, 8, 33864–33871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turriziani, P.; Smirni, D. Enhancing memory performance with rTMS in healthy subjects and individuals with Mild Cognitive Impairment: The role of the right dorsolateral prefrontal cortex. Front. Hum. Neurosci. 2012, 6, 62. [Google Scholar] [CrossRef] [Green Version]
- Boggio, P.S.; Khoury, L.P. Temporal cortex direct current stimulation enhances performance on a visual recognition memory task in Alzheimer disease. J. Neurol. Neurosurg. Psychiatry 2009, 80, 444–447. [Google Scholar] [CrossRef]
- Khedr, E.M.; Gamal, N.F. A double-blind randomized clinical trial on the efficacy of cortical direct current stimulation for the treatment of Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 275. [Google Scholar] [CrossRef] [PubMed]
- Suemoto, C.K.; Apolinario, D. Effects of a non-focal plasticity protocol on apathy in moderate Alzheimer’s disease: A randomized, double-blind, sham-controlled trial. Brain Stimul. 2014, 7, 308–313. [Google Scholar] [CrossRef]
- Im, J.J.; Jeong, H. Effects of 6-month at-home transcranial direct current stimulation on cognition and cerebral glucose metabolism in Alzheimer’s disease. Brain Stimul. 2019, 12, 1222–1228. [Google Scholar] [CrossRef] [PubMed]
- Manenti, R.; Sandrini, M. Effects of Transcranial Direct Current Stimulation on Episodic Memory in Amnestic Mild Cognitive Impairment: A Pilot Study. J. Gerontol. B Psychol. Sci. Soc. Sci. 2020, 75, 1403–1413. [Google Scholar] [CrossRef]
- Cruz Gonzalez, P.; Fong, K.N.K. The Effects of Transcranial Direct Current Stimulation on the Cognitive Functions in Older Adults with Mild Cognitive Impairment: A Pilot Study. Behav. Neurol. 2018, 2018, 5971385. [Google Scholar] [CrossRef] [Green Version]
- Bystad, M.; Grønli, O. Transcranial direct current stimulation as a memory enhancer in patients with Alzheimer’s disease: A randomized, placebo-controlled trial. Alzheimers Res. Ther. 2016, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, R.; Mameli, F. Transcranial direct current stimulation improves recognition memory in Alzheimer disease. Neurology 2008, 71, 493–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boggio, P.S.; Ferrucci, R. Prolonged visual memory enhancement after direct current stimulation in Alzheimer’s disease. Brain Stimul. 2012, 5, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Khedr, E.M.; Salama, R.H. Therapeutic Role of Transcranial Direct Current Stimulation in Alzheimer Disease Patients: Double-Blind, Placebo-Controlled Clinical Trial. Neurorehabil. Neural Repair 2019, 33, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Gangemi, A.; Colombo, B. Effects of short- and long-term neurostimulation (tDCS) on Alzheimer’s disease patients: Two randomized studies. Aging Clin. Exp. Res. 2021, 33, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Kehler, L.; Francisco, C.O. The effect of transcranial alternating current stimulation (tACS) on cognitive function in older adults with dementia. In Proceedings of the 2020 42nd Annual International Conference of the IEEE Engineering in Medicine & Biology Society (EMBC), Montreal, QC, Canada, 20–24 July 2020; Volume 2020, pp. 3649–3653. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Gamma tACS in Alzheimer’s Disease. NCT04515433. Available online: https://ClinicalTrials.gov/show/NCT04515433 (accessed on 7 May 2021).
- ClinicalTrials.gov. Memory Functions in Mild Alzheimer’s Disease. NCT04785053. Available online: https://ClinicalTrials.gov/show/NCT04785053 (accessed on 7 May 2021).
- Ono, K. Alzheimer’s disease as oligomeropathy. Neurochem. Int. 2018, 119, 57–70. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Morris, J.C. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamelin, L.; Lagarde, J. Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18F-DPA-714 PET imaging. Brain 2016, 139 Pt 4, 1252–1264. [Google Scholar] [CrossRef] [Green Version]
- Aisen, P.S. The development of anti-amyloid therapy for Alzheimer’s disease: From secretase modulators to polymerisation inhibitors. CNS Drugs 2005, 19, 989–996. [Google Scholar] [CrossRef]
- Lynch, S.Y.; Kaplow, J. Elenbecestat, e2609, a bace inhibitor: Results from a phase-2 study in subjects with mild cognitive impairment and mild-to-moderate dementia due to alzheimer’s disease. Alzheimer’s Dement. 2018, 14 (Suppl. 7), P1623. [Google Scholar] [CrossRef]
- Huang, L.K.; Chao, S.P. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Lozupone, M. Do BACE inhibitor failures in Alzheimer patients challenge the amyloid hypothesis of the disease? Expert Rev. Neurother. 2019, 19, 599–602. [Google Scholar] [CrossRef]
- Miranda, A.; Montiel, E. Selective Secretase Targeting for Alzheimer’s Disease Therapy. J. Alzheimers Dis. 2021. [Google Scholar] [CrossRef]
- Uddin, M.S.; Hossain, M.F. Exploring the multimodal role of phytochemicals in the modulation of cellular signaling pathways to combat age-related neurodegeneration. Sci. Total Environ. 2020, 725, 138313. [Google Scholar] [CrossRef] [PubMed]
- Pagano, K.; Tomaselli, S. Natural Compounds as Inhibitors of Aβ Peptide Aggregation: Chemical Requirements and Molecular Mechanisms. Front. Neurosci. 2020, 14, 619667. [Google Scholar] [CrossRef] [PubMed]
- Nie, Q.; Du, X.G. Small molecule inhibitors of amyloid β peptide aggregation as a potential therapeutic strategy for Alzheimer’s disease. Acta Pharmacol. Sin. 2011, 32, 545–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampson, E.L.; Jenagaratnam, L. Metal protein attenuating compounds for the treatment of Alzheimer’s dementia. Cochrane Database Syst. Rev. 2014, 2, Cd005380. [Google Scholar] [CrossRef]
- Makin, S. The amyloid hypothesis on trial. Nature 2018, 559, S4–S7. [Google Scholar] [CrossRef] [Green Version]
- Cheignon, C.; Tomas, M. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox. Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Schenk, D. Amyloid-beta immunotherapy for Alzheimer’s disease: The end of the beginning. Nat. Rev. Neurosci. 2002, 3, 824–828. [Google Scholar] [CrossRef]
- Loureiro, J.C.; Pais, M.V. Passive antiamyloid immunotherapy for Alzheimer’s disease. Curr. Opin. Psychiatry 2020, 33, 284–291. [Google Scholar] [CrossRef]
- Mahase, E. Three FDA advisory panel members resign over approval of Alzheimer’s drug. BMJ 2021, 373, n1503. [Google Scholar] [CrossRef] [PubMed]
- Naseri, N.N.; Wang, H. The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 2019, 705, 183–194. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2016, 12, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Alafuzoff, I. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef]
- Soeda, Y.; Takashima, A. New Insights Into Drug Discovery Targeting Tau Protein. Front. Mol. Neurosci. 2020, 13, 590896. [Google Scholar] [CrossRef]
- Pillai, R.; Uyehara-Lock, J.H. Selenium and selenoprotein function in brain disorders. IUBMB Life 2014, 66, 229–239. [Google Scholar] [CrossRef]
- Shultz, S.R.; Wright, D.K. Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain 2015, 138 Pt 5, 1297–1313. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.L.; Wang, C. The Role of Cdk5 in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 4328–4342. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, F.; Lucas, J.J. GSK3 and tau: Two convergence points in Alzheimer’s disease. J. Alzheimers Dis. 2013, 33 (Suppl. 1), S141–S144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicenas, J.; Kalyan, K. Roscovitine in cancer and other diseases. Ann. Transl. Med. 2015, 3, 135. [Google Scholar] [CrossRef]
- Zeidner, J.F.; Karp, J.E. Clinical activity of alvocidib (flavopiridol) in acute myeloid leukemia. Leuk. Res. 2015, 39, 1312–1318. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Edwards, P.C. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. USA 1996, 93, 11213–11218. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, S.; Suzuki, N. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 2005, 280, 7614–7623. [Google Scholar] [CrossRef] [Green Version]
- Soeda, Y.; Saito, M. Methylene Blue Inhibits Formation of Tau Fibrils but not of Granular Tau Oligomers: A Plausible Key to Understanding Failure of a Clinical Trial for Alzheimer’s Disease. J. Alzheimers Dis. 2019, 68, 1677–1686. [Google Scholar] [CrossRef]
- Rane, J.S.; Bhaumik, P. Curcumin Inhibits Tau Aggregation and Disintegrates Preformed Tau Filaments in vitro. J. Alzheimers Dis. 2017, 60, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Baum, L.; Lam, C.W. Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J. Clin. Psychopharmacol. 2008, 28, 110–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringman, J.M.; Frautschy, S.A. Oral curcumin for Alzheimer’s disease: Tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimers Res. Ther. 2012, 4, 43. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Taghibiglou, C. The Mechanisms of Action of Curcumin in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Rainey-Smith, S.R.; Brown, B.M. Curcumin and cognition: A randomised, placebo-controlled, double-blind study of community-dwelling older adults. Br. J. Nutr. 2016, 115, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.N.; Mei, X. Curcumin intervention for cognitive function in different types of people: A systematic review and meta-analysis. Phytother. Res. 2019, 33, 524–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollag, D.M.; McQueney, P.A. Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action. Cancer Res. 1995, 55, 2325–2333. [Google Scholar] [PubMed]
- Zhang, B.; Carroll, J. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J. Neurosci. 2012, 32, 3601–3611. [Google Scholar] [CrossRef]
- Barten, D.M.; Fanara, P. Hyperdynamic microtubules, cognitive deficits, and pathology are improved in tau transgenic mice with low doses of the microtubule-stabilizing agent BMS-241027. J. Neurosci. 2012, 32, 7137–7145. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Valenzuela, J.J.; Sanchez-Varo, R. Enhancing microtubule stabilization rescues cognitive deficits and ameliorates pathological phenotype in an amyloidogenic Alzheimer’s disease model. Sci. Rep. 2020, 10, 14776. [Google Scholar] [CrossRef]
- Gozes, I. Microtubules (tau) as an emerging therapeutic target: NAP (davunetide). Curr. Pharm. Des. 2011, 17, 3413–3417. [Google Scholar] [CrossRef]
- Gozes, I. NAP (davunetide) provides functional and structural neuroprotection. Curr. Pharm. Des. 2011, 17, 1040–1044. [Google Scholar] [CrossRef]
- Morimoto, B.H.; Fox, A.W. Davunetide: A review of safety and efficacy data with a focus on neurodegenerative diseases. Expert Rev. Clin. Pharmacol. 2013, 6, 483–502. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, D.P.; Emerson, D.L. TPI-287, a new taxane family member, reduces the brain metastatic colonization of breast cancer cells. Mol Cancer Ther 2012, 11, 1959–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumbar, C.T.; Usubalieva, A. The CNS penetrating taxane TPI 287 and the AURKA inhibitor alisertib induce synergistic apoptosis in glioblastoma cells. J. Neurooncol. 2018, 137, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Mondal, P.; Das, G. Crafting of Neuroprotective Octapeptide from Taxol-Binding Pocket of β-Tubulin. ACS Chem. Neurosci. 2018, 9, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Solfrizzi, V. Tau-based therapeutics for Alzheimer’s disease: Active and passive immunotherapy. Immunotherapy 2016, 8, 1119–1134. [Google Scholar] [CrossRef]
- Novak, P.; Zilka, N. AADvac1, an Active Immunotherapy for Alzheimer’s Disease and Non Alzheimer Tauopathies: An Overview of Preclinical and Clinical Development. J. Prev. Alzheimers Dis. 2019, 6, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Schmidt, R. Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer’s disease: A randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol. 2017, 16, 123–134. [Google Scholar] [CrossRef]
- Novak, P.; Schmidt, R. Fundamant: An interventional 72-week phase 1 follow-up study of AADvac1, an active immunotherapy against tau protein pathology in Alzheimer’s disease. Alzheimers Res. Ther. 2018, 10, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theunis, C.; Crespo-Biel, N. Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in tau.P301L mice that model tauopathy. PLoS ONE 2013, 8, e72301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoskin, J.L.; Sabbagh, M.N. Tau immunotherapies for Alzheimer’s disease. Expert Opin. Investig. Drugs 2019, 28, 545–554. [Google Scholar] [CrossRef]
- Qureshi, I.A.; Tirucherai, G. A randomized, single ascending dose study of intravenous BIIB092 in healthy participants. Alzheimers Dement. 2018, 4, 746–755. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Safety, Tolerability, and Pharmacokinetics of C2N-8E12 in Subjects with Progressive Supranuclear Palsy. NCT02494024. Available online: https://ClinicalTrials.gov/show/NCT02494024 (accessed on 4 April 2021).
- ClinicalTrials.gov. A Study of LY3303560 in Healthy Participants and Participants with Alzheimer’s Disease (AD). NCT02754830. Available online: https://ClinicalTrials.gov/show/NCT02754830 (accessed on 4 April 2021).
- ClinicalTrials.gov. A Study of LY3303560 in Participants with Mild Cognitive Impairment or Alzheimer’s Disease. NCT03019536. Available online: https://ClinicalTrials.gov/show/NCT03019536 (accessed on 4 April 2021).
- ClinicalTrials.gov. A Study of JNJ-63733657 in Healthy Japanese Participants. NCT03689153. Available online: https://ClinicalTrials.gov/show/NCT03689153 (accessed on 4 April 2021).
- ClinicalTrials.gov. A Study to Investigate Safety and Tolerability, Pharmacokinetics and Pharmacodynamics of JNJ-63733657 in Healthy Subjects and Subjects with Alzheimer’s Disease. NCT03375697. Available online: https://ClinicalTrials.gov/show/NCT03375697 (accessed on 4 April 2021).
- Kreisl, W.C.; Lyoo, C.H. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain 2013, 136 Pt 7, 2228–2238. [Google Scholar] [CrossRef] [Green Version]
- Kaur, D.; Sharma, V. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef] [PubMed]
- Regen, F.; Hellmann-Regen, J. Neuroinflammation and Alzheimer’s Disease: Implications for Microglial Activation. Curr. Alzheimer Res. 2017, 14, 1140–1148. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Atagi, Y.; Liu, C.C. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J. Biol. Chem. 2015, 290, 26043–26050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, C.M.; Fitz, N.F. The Role of APOE and TREM2 in Alzheimer’s Disease-Current Understanding and Perspectives. Int. J. Mol. Sci. 2018, 20, 81. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.Y.D.; Daggett, A. Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron 2018, 97, 1032–1048.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jay, T.R.; Hirsch, A.M. Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2017, 37, 637–647. [Google Scholar] [CrossRef]
- Yang, J.; Wise, L. TLR4 Cross-Talk with NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease. Front. Immunol. 2020, 11, 724. [Google Scholar] [CrossRef]
- Zakaria, R.; Wan Yaacob, W.M. Lipopolysaccharide-induced memory impairment in rats: A model of Alzheimer’s disease. Physiol. Res. 2017, 66, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Lax, N.; Fainstein, N. Systemic microbial TLR2 agonists induce neurodegeneration in Alzheimer’s disease mice. J. Neuroinflamm. 2020, 17, 55. [Google Scholar] [CrossRef]
- Richard, K.L.; Filali, M. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid beta 1-42 and delay the cognitive decline in a mouse model of Alzheimer’s disease. J. Neurosci. 2008, 28, 5784–5793. [Google Scholar] [CrossRef] [Green Version]
- McDonald, C.L.; Hennessy, E. Inhibiting TLR2 activation attenuates amyloid accumulation and glial activation in a mouse model of Alzheimer’s disease. Brain Behav. Immun. 2016, 58, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Sun, X. Genomic deletion of TLR2 induces aggravated white matter damage and deteriorated neurobehavioral functions in mouse models of Alzheimer’s disease. Aging (Albany NY) 2019, 11, 7257–7273. [Google Scholar] [CrossRef]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef] [Green Version]
- Ben Haim, L.; Ceyzériat, K. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J. Neurosci. 2015, 35, 2817–2829. [Google Scholar] [CrossRef] [PubMed]
- Nagamoto-Combs, K.; Combs, C.K. Microglial phenotype is regulated by activity of the transcription factor, NFAT (nuclear factor of activated T cells). J. Neurosci. 2010, 30, 9641–9646. [Google Scholar] [CrossRef] [Green Version]
- Hudry, E.; Wu, H.Y. Inhibition of the NFAT pathway alleviates amyloid β neurotoxicity in a mouse model of Alzheimer’s disease. J. Neurosci. 2012, 32, 3176–3192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojanathammanee, L.; Floden, A.M. Attenuation of microglial activation in a mouse model of Alzheimer’s disease via NFAT inhibition. J. Neuroinflamm. 2015, 12, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugan, L.L.; Ali, S.S. IL-6 mediated degeneration of forebrain GABAergic interneurons and cognitive impairment in aged mice through activation of neuronal NADPH oxidase. PLoS ONE 2009, 4, e5518. [Google Scholar] [CrossRef] [PubMed]
- François, A.; Rioux Bilan, A. Longitudinal follow-up of autophagy and inflammation in brain of APPswePS1dE9 transgenic mice. J. Neuroinflamm. 2014, 11, 139. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.S.; Tan, Z.X. The involvement of NLRP3 inflammasome in the treatment of Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101192. [Google Scholar] [CrossRef]
- Lee, J.K.; Kim, N.J. Recent Advances in the Inhibition of p38 MAPK as a Potential Strategy for the Treatment of Alzheimer’s Disease. Molecules 2017, 22, 1287. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Li, G. p38 MAPK mediates glial P2 × 7R-neuronal P2Y1R inhibitory control of P2 × 3R expression in dorsal root ganglion neurons. Mol. Pain 2015, 11, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simões, A.P.; Silva, C.G. Glutamate-induced and NMDA receptor-mediated neurodegeneration entails P2Y1 receptor activation. Cell Death Dis. 2018, 9, 297. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Thirumala, V. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M. Insulin Resistance and Neurodegeneration: Progress Towards the Development of New Therapeutics for Alzheimer’s Disease. Drugs 2017, 77, 47–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Femminella, G.D.; Frangou, E. Evaluating the effects of the novel GLP-1 analogue liraglutide in Alzheimer’s disease: Study protocol for a randomised controlled trial (ELAD study). Trials 2019, 20, 191. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.M.; Stephenson, M.D. Metformin Use Associated with Reduced Risk of Dementia in Patients with Diabetes: A Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2018, 65, 1225–1236. [Google Scholar] [CrossRef] [Green Version]
- Samaras, K.; Makkar, S. Metformin Use Is Associated With Slowed Cognitive Decline and Reduced Incident Dementia in Older Adults With Type 2 Diabetes: The Sydney Memory and Ageing Study. Diabetes Care 2020, 43, 2691–2701. [Google Scholar] [CrossRef]
- Iglesias, J.; Morales, L. Metabolic and Inflammatory Adaptation of Reactive Astrocytes: Role of PPARs. Mol. Neurobiol. 2017, 54, 2518–2538. [Google Scholar] [CrossRef]
- Tufano, M.; Pinna, G. Is There a Future for PPARs in the Treatment of Neuropsychiatric Disorders? Molecules 2020, 25, 1062. [Google Scholar] [CrossRef] [Green Version]
- Megur, A.; Baltriukienė, D. The Microbiota-Gut-Brain Axis and Alzheimer’s Disease: Neuroinflammation Is to Blame? Nutrients 2020, 13, 37. [Google Scholar] [CrossRef]
- Sochocka, M.; Donskow-Łysoniewska, K. The Gut Microbiome Alterations and Inflammation-Driven Pathogenesis of Alzheimer’s Disease-a Critical Review. Mol. Neurobiol. 2019, 56, 1841–1851. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Sun, G. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef]
- Syed, Y.Y. Sodium Oligomannate: First Approval. Drugs 2020, 80, 441–444. [Google Scholar] [CrossRef]
- Longo, F.M.; Massa, S.M. Neuroprotective strategies in Alzheimer’s disease. NeuroRx 2004, 1, 117–127. [Google Scholar] [CrossRef]
- Eddy, C.M.; Rickards, H.E. The cognitive impact of antiepileptic drugs. Ther. Adv. Neurol. Disord. 2011, 4, 385–407. [Google Scholar] [CrossRef]
- Daniels, V.; Wood, M. Modulation of the conformational state of the SV2A protein by an allosteric mechanism as evidenced by ligand binding assays. Br. J. Pharmacol. 2013, 169, 1091–1101. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Altamirano, J.L.; Olmos-Hernández, A. Levetiracetam as an antiepileptic, neuroprotective, and hyperalgesic drug. Neurol. India 2016, 64, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Sola, I.; Aso, E. Novel Levetiracetam Derivatives That Are Effective against the Alzheimer-like Phenotype in Mice: Synthesis, in Vitro, ex Vivo, and in Vivo Efficacy Studies. J. Med. Chem. 2015, 58, 6018–6032. [Google Scholar] [CrossRef] [PubMed]
- Schoenberg, M.R.; Rum, R.S. A randomized, double-blind, placebo-controlled crossover study of the effects of levetiracetam on cognition, mood, and balance in healthy older adults. Epilepsia 2017, 58, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.C.; Wang, J. Neuroprotective Effects of Gabapentin Against Cerebral Ischemia Reperfusion-Induced Neuronal Autophagic Injury via Regulation of the PI3K/Akt/mTOR Signaling Pathways. J. Neuropathol. Exp. Neurol. 2019, 78, 157–171. [Google Scholar] [CrossRef] [Green Version]
- Ortinski, P.; Meador, K.J. Cognitive side effects of antiepileptic drugs. Epilepsy Behav. 2004, 5 (Suppl. 1), S60–S65. [Google Scholar] [CrossRef]
- Supasitthumrong, T.; Bolea-Alamanac, B.M. Gabapentin and pregabalin to treat aggressivity in dementia: A systematic review and illustrative case report. Br. J. Clin. Pharmacol. 2019, 85, 690–703. [Google Scholar] [CrossRef]
- Liu, J.; Chang, L. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.J.; Lin, C.H. NMDA Neurotransmission Dysfunction in Behavioral and Psychological Symptoms of Alzheimer’s Disease. Curr. Neuropharmacol. 2012, 10, 272–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.H.; Huang, Y.J. NMDA neurotransmission dysfunction in mild cognitive impairment and Alzheimer’s disease. Curr. Pharm. Des. 2014, 20, 5169–5179. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Chen, Y.M. Novel Treatment for the Most Resistant Schizophrenia: Dual Activation of NMDA Receptor and Antioxidant. Curr. Drug Targets 2020, 21, 610–615. [Google Scholar] [CrossRef]
- Lin, C.H.; Yang, H.T. Precision Medicine of Sodium Benzoate for the Treatment of Behavioral and Psychological Symptoms of Dementia (BPSD). Neuropsychiatr. Dis. Treat. 2020, 16, 509–518. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.H.; Lin, C.H. Sodium Benzoate, a D-Amino Acid Oxidase Inhibitor, Added to Clozapine for the Treatment of Schizophrenia: A Randomized, Double-Blind, Placebo-Controlled Trial. Biol. Psychiatry 2018, 84, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Gray, J.D. Riluzole reduces amyloid beta pathology, improves memory, and restores gene expression changes in a transgenic mouse model of early-onset Alzheimer’s disease. Transl. Psychiatry 2018, 8, 153. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, N.V. Hippocampal hyperglutamatergic signaling matters: Early targeting glutamate neurotransmission as a preventive strategy in Alzheimer’s disease: An Editorial Highlight for “Riluzole attenuates glutamatergic tone and cognitive decline in AβPP/PS1 mice” on page 513. J. Neurochem. 2021, 156, 399–402. [Google Scholar] [CrossRef]
- Swanson, D.; Block, R. Omega-3 fatty acids EPA and DHA: Health benefits throughout life. Adv. Nutr. 2012, 3, 1–7. [Google Scholar] [CrossRef]
- Dyall, S.C. Long-chain omega-3 fatty acids and the brain: A review of the independent and shared effects of EPA, DPA and DHA. Front. Aging Neurosci. 2015, 7, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, J.; Thomas, C.J. Omega-3 Fatty Acids in Early Prevention of Inflammatory Neurodegenerative Disease: A Focus on Alzheimer’s Disease. Biomed. Res. Int. 2015, 2015, 172801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazereeuw, G.; Lanctôt, K.L. Effects of ω-3 fatty acids on cognitive performance: A meta-analysis. Neurobiol. Aging 2012, 33, 1482.e17–1482.e29. [Google Scholar] [CrossRef] [PubMed]
- Patrick, R.P. Role of phosphatidylcholine-DHA in preventing APOE4-associated Alzheimer’s disease. FASEB J. 2019, 33, 1554–1564. [Google Scholar] [CrossRef] [Green Version]
- Araya-Quintanilla, F.; Gutiérrez-Espinoza, H. Effectiveness of omega-3 fatty acid supplementation in patients with Alzheimer disease: A systematic review and meta-analysis. Neurologia 2020, 35, 105–114. [Google Scholar] [CrossRef]
- Hansen, N. Brain stimulation for combating Alzheimer’s disease. Front. Neurol. 2014, 5, 80. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Lane, H.Y. Brain Stimulation in Alzheimer’s Disease. Front. Psychiatry 2018, 9, 201. [Google Scholar] [CrossRef] [Green Version]
- McKinnon, C.; Gros, P. Deep brain stimulation: Potential for neuroprotection. Ann. Clin. Transl. Neurol. 2019, 6, 174–185. [Google Scholar] [CrossRef] [Green Version]
- Laxton, A.W.; Tang-Wai, D.F. A phase I trial of deep brain stimulation of memory circuits in Alzheimer’s disease. Ann. Neurol. 2010, 68, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Hardenacke, K.; Hashemiyoon, R. Deep Brain Stimulation of the Nucleus Basalis of Meynert in Alzheimer’s Dementia: Potential Predictors of Cognitive Change and Results of a Long-Term Follow-Up in Eight Patients. Brain Stimul. 2016, 9, 799–800. [Google Scholar] [CrossRef]
- Baldermann, J.C.; Hardenacke, K. Neuroanatomical Characteristics Associated with Response to Deep Brain Stimulation of the Nucleus Basalis of Meynert for Alzheimer’s Disease. Neuromodulation 2018, 21, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, S.D.; Yuan, H. Non-invasive vagus nerve stimulation for primary headache: A clinical update. Cephalalgia 2020, 40, 1370–1384. [Google Scholar] [CrossRef]
- Jacobs, H.I.; Riphagen, J.M. Transcutaneous vagus nerve stimulation boosts associative memory in older individuals. Neurobiol. Aging 2015, 36, 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Farmer, A.D.; Strzelczyk, A. International Consensus Based Review and Recommendations for Minimum Reporting Standards in Research on Transcutaneous Vagus Nerve Stimulation (Version 2020). Front. Hum. Neurosci. 2020, 14, 568051. [Google Scholar] [CrossRef] [PubMed]
- Luber, B.; McClintock, S.M. Applications of transcranial magnetic stimulation and magnetic seizure therapy in the study and treatment of disorders related to cerebral aging. Dialogues Clin. Neurosci. 2013, 15, 87–98. [Google Scholar] [CrossRef]
- Klomjai, W.; Katz, R. Basic principles of transcranial magnetic stimulation (TMS) and repetitive TMS (rTMS). Ann. Phys. Rehabil. Med. 2015, 58, 208–213. [Google Scholar] [CrossRef]
- Somani, A.; Kar, S.K. Efficacy of repetitive transcranial magnetic stimulation in treatment-resistant depression: The evidence thus far. Gen. Psychiatr. 2019, 32, e100074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drumond Marra, H.L.; Myczkowski, M.L. Transcranial Magnetic Stimulation to Address Mild Cognitive Impairment in the Elderly: A Randomized Controlled Study. Behav. Neurol. 2015, 2015, 287843. [Google Scholar] [CrossRef] [Green Version]
- Chou, Y.H.; Ton That, V. A systematic review and meta-analysis of rTMS effects on cognitive enhancement in mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 2020, 86, 1–10. [Google Scholar] [CrossRef]
- Rabey, J.M.; Dobronevsky, E. Repetitive transcranial magnetic stimulation combined with cognitive training is a safe and effective modality for the treatment of Alzheimer’s disease: A randomized, double-blind study. J. Neural Transm. (Vienna) 2013, 120, 813–819. [Google Scholar] [CrossRef]
- Woods, A.J.; Antal, A. A technical guide to tDCS, and related non-invasive brain stimulation tools. Clin. Neurophysiol. 2016, 127, 1031–1048. [Google Scholar] [CrossRef] [Green Version]
- Buss, S.S.; Fried, P.J. Therapeutic noninvasive brain stimulation in Alzheimer’s disease and related dementias. Curr. Opin. Neurol. 2019, 32, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Lefaucheur, J.P. A comprehensive database of published tDCS clinical trials (2005–2016). Neurophysiol. Clin. 2016, 46, 319–398. [Google Scholar] [CrossRef] [PubMed]
- Antonenko, D.; Faxel, M. Effects of Transcranial Alternating Current Stimulation on Cognitive Functions in Healthy Young and Older Adults. Neural Plast. 2016, 2016, 4274127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fröhlich, F.; Sellers, K.K. Targeting the neurophysiology of cognitive systems with transcranial alternating current stimulation. Expert Rev. Neurother. 2015, 15, 145–167. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Transcranial Alternating Current Stimulation for Patients with Mild Alzheimer’s Disease (TRANSFORM-AD). NCT03920826. Available online: https://ClinicalTrials.gov/show/NCT03920826 (accessed on 7 May 2021).

{kind=link}
{kind=link}
{kind=link}
| Class of Drugs | Compounds | Mechanism | Subjects | Status | Summary | [Ref] |
|---|---|---|---|---|---|---|
| 1. Anti-amyloid therapy | ||||||
| Secretase inhibitor | Verubecestat | BACE1 inhibitor | Prodromal to moderate AD | Phase II/III | Lack of efficacy | [20,21] |
| Atabecestat | BACE1 inhibitor | Prodromal AD | Phase II/III | Cognitive worsening, psychiatric disorder | [22] | |
| Lanabecestat | BACE1 inhibitor | MCI to mild AD | Phase III | Cognitive worsening, weight loss, psychiatric disorder | [23] | |
| LY3202626 | BACE1 inhibitor | Mild AD | Phase III | Lack of efficacy | [24] | |
| Umibecestat | BACE1 inhibitor | Cognitively healthy APOE4 carriers | Phase II/III | Completed, failed analysis due to small number of events | [25] | |
| Elenbecestat | BACE1 inhibitor | MCI to moderate AD | Phase III | Lack of efficacy, nightmare | [26,27] | |
| Semagacestat | γ-secretase inhibitor | Mild to moderate AD | Phase III | Lack of efficacy, skin cancer, weight loss, hematologic disorder, infection | [28] | |
| Avagacestat | γ-secretase inhibitor | MCI | Phase II | Lack of efficacy, non-melanoma cancer, gastrointestinal symptoms | [29] | |
| Tarenflurbil | γ-secretase modulator | Mild AD | Phase II | Lack of efficacy, anemia, infection | [30] | |
| Aβ aggregation inhibitor | PBT1 | MPAC | MCI to moderate AD | Phase II | Rescue of cognitive decline in severely affected patients (ADAS-cog ≥25), visual impairment | [31] |
| PBT2 | MPAC | Mild to moderate AD | Phase II | Lack of efficacy, large individual variance | [32,33] | |
| Aβ immunotherapy | ACI-24 | Aβ vaccine | Adults with Down syndrome | Phase II | Lack of immunogenicity | [34] |
| CAD106 | Aβ vaccine | Mild AD | Phase II | Lack of efficacy | [34] | |
| UB-311 | Aβ vaccine | Mild AD | Phase II | No published data | [34] | |
| ABVac40 | Aβ vaccine | MCI to mild AD | Phase II | Ongoing | [34] | |
| BAN2401 | Monoclonal antibody | MCI to mild AD | Phase III | Modest efficacy among APOE4 carriers | [35] | |
| Gantenerumab | Monoclonal antibody | Prodromal to mild AD | Phase III | Lack of efficacy | [36] | |
| Aducanumab | Monoclonal antibody | Monoclonal antibody | Phase III | Termination, little change in efficacy FDA approval for now | [37,38] | |
| 2. Anti-tau therapy | ||||||
| Phosphatase modifier | Selenate | PP2A activator | Mild to moderate AD | Phase II | Lack of efficacy | [39,40] |
| Kinase inhibitor | Roscovitine | CDK5 inhibitor | 5XFAD mice | In vivo | Prevention of tau phosphorylation | [41,42] |
| Flavopiridol | CDK5 inhibitor | CD1 mice | In vivo | Rescue of cognitive decline | [41,42] | |
| Tideglusib | GSK3β inhibitor | Mild to moderate AD | Phase II | Lack of efficacy, transaminase increase | [43] | |
| Lithium | GSK3β inhibitor | MCI | Phase II | Rescue of cognitive decline | [44,45,46] | |
| Tau aggregation inhibitor | MB | Disrupts polymerization | Mild to moderate AD | Phase II | Cognitive improvement | [47] |
| LMTX | Disrupts polymerization | Mild to moderate AD | Phase III | Lack of efficacy | [48] | |
| Curcumin | Decreases β-sheet formation in tau | Cognitively healthy elderly | Phase II | Improvement in working memory (short-term course) | [49,50] | |
| Microtubule stabilizer | EpoD | Enhances microtubule bundling | Mild AD | Phase I | Discontinuation, frequent adverse effects without published data | [51] |
| NAP | Protects microtubules from katanin disruption | MCI | Phase II | Cognitive and functional improvement | [52,53] | |
| TPI-287 | Stabilizes microtubules | Mild to moderate AD | Phase I | Rescue of cognitive decline, anaphylactoid reactions | [54] | |
| Tau immunotherapy | AADvac1 | Tau vaccine | Mild AD | Phase II | Completed, no published data | [55] |
| ACI-35 | Tau vaccine | Mild to moderate AD | Phase I | Safe and tolerated | [56] | |
| Aβ 3–10-KLH | Tau vaccine | 3×Tg-AD mice | In vivo | Cognitive improvement | [57] | |
| BIIB092 | Monoclonal antibody | Early AD | Phase II | Ongoing | [58] | |
| ABBV-8E12 | Monoclonal antibody | Early AD | Phase II | Ongoing | [59,60] | |
| RO7105705 | Monoclonal antibody | Prodromal to moderate AD | Phase II | Ongoing | [61,62] | |
| BIIB076 | Monoclonal antibody | Healthy volunteers, MCI | Phase I | Safe and tolerated | [63] | |
| LY3303560 | Monoclonal antibody | Early AD | Phase II | Completed, no available data | [64] | |
| JNJ-63733657 | Monoclonal antibody | Early AD | Phase II | Ongoing | [65] | |
| UCB0107 | Monoclonal antibody | Healthy volunteers | Phase I | Ongoing | [66,67] | |
| 3. Anti-neuroinflammatory therapy | ||||||
| Microglia modulator | Thymoquinone | TLR4 inhibitor | AD mice induced by AlCl3 | In vivo | Rescue of cognitive impairment | [68] |
| Ethyl pyruvate | TLR4 inhibitor | AD mice induced by AlCl3 | In vivo | Rescue of cognitive impairment | [68] | |
| TAK-242 | TLR4 inhibitor | APP/PS1 mice | In vivo | Cognitive improvement | [68] | |
| GW2580 | CSF1R inhibitor | APP/PS1 mice | In vivo | Recovery of short-term memory and behavioral deficit | [69] | |
| JN-J527 | CSF1R inhibitor | P301S mice | In vivo | Functional improvement | [70] | |
| PLX3397 | CSF1R inhibitor | 5XFAD mice | In vivo | Recovery of spatial and emotional memory deficit | [71] | |
| Astrocyte modulator | Stattic | STAT3 inhibitor | 5XFAD mice | In vivo | Rescue of learning and memory impairment | [72,73] |
| FK506 | Calcineurin/NFAT inhibitor | MCI to AD | Phase II | Not yet recruiting | [74] | |
| SB202190 | P38 MAPK inhibitor | Wip1-deficient mice | In vivo | Rescue of learning and memory impairment | [75] | |
| PD169316 | P38 MAPK inhibitor | Aβ-injected mice | In vivo | Rescue of spatial memory and learning impairment | [75] | |
| MW108 | P38 MAPK inhibitor | hTau mice | In vivo | Rescue of cognitive impairment | [76] | |
| NJK14047 | P38 MAPK inhibitor | 5XFAD mice | In vivo | Cognitive improvement | [77] | |
| MRS2179 | P2Y1R inhibitor | APPPS1 mice | In vivo | Spatial learning improvement | [78] | |
| BPTU | P2Y1R inhibitor | APPPS1 mice | In vivo | Spatial learning improvement | [78] | |
| Insulin resistance management | Intranasal insulin therapy | Intranasal supplement | MCI to moderate AD | Phase II | Cognitive improvement, modulation by APOE4 genotype | [79,80] |
| MCI to AD | Phase II/III | Lack of efficacy | [81] | |||
| Liraglutide | Incretin receptor agonist | Mild AD | Phase II | Delay of cognitive impairment | [82,83] | |
| Metformin | Biguanide | MCI | Phase II | Reduction in recall memory decline | [84] | |
| MCI to early AD | Phase II | Executive functional improvement | [85] | |||
| Gemfibrozil | PPAR-α agonist | MCI | Phase I | Completed, no published data | [86] | |
| Pioglitazone | PPAR-γ agonist | Mild AD | Phase II | Cognitive improvement | [87] | |
| MCI | Phase III | Lack of efficacy | [88,89] | |||
| T3D-959 | Hybrid PPAR-δ/γ agonist | STZ-induced AD mice | In vivo | Reduction in neuroinflammation | [90] | |
| Microbiome therapy | Sodium oligomannate | Dysbiosis of gut microbiota | Mild to moderate AD | Phase III | Cognitive improvement | [91,92] |
| 4. Neuroprotective agents | ||||||
| Antiepileptic drug | Levetiracetam | SV2A receptor | MCI | Phase III | Ongoing | [93] |
| Gabapentin | VGCCs inhibitor | Moderate to severe AD | Phase IV | Ongoing | [94] | |
| NMDAR modification | Sodium benzoate | DAAO inhibitor | MCI to mild AD | Phase II | Cognitive improvement | [95] |
| MCI | Phase II | Cognitive and functional improvement | [96] | |||
| Moderate to severe AD with BPSD | Phase II | Cognitive benefit in female gender | [97] | |||
| Riluzole | Glutamate modulator | Mild AD | Phase II | Completed, no published data | [98] | |
| Troriruzole | Glutamate modulator | Mild to moderate AD | Phase II | Ongoing | [99] | |
| Omega 3 polyunsaturated fatty acid supplements | DHA | Anti-oxidative effect | Mild to moderate AD | Phase III | Lack of efficacy | [100] |
| Cognitively healthy elderly | Phase II | Ongoing | [101] | |||
| Icosapent ethyl | Anti-oxidative effect | Cognitively healthy elderly | Phase III | Ongoing | [102] | |
| Methods | Targeted Region | Protocol | Subjects | Status | Summary | [Ref] |
|---|---|---|---|---|---|---|
| 1. Deep-brain stimulation | ||||||
| DBS | Fornix | Forneceal DBS | Mild AD | Phase II | Slight cognitive benefit in the elderly | [103,104] |
| Mild AD | Phase III | Ongoing | [105] | |||
| NBM | NBM-DBS | Mild to moderate AD | Phase I | Cognitive stabilization and improvement, response rate 67% | [106] | |
| 2. Vagus nerve stimulation | ||||||
| VNS | Tenth cranial nerve | Invasive VNS | Probable AD | Phase I | Cognitive stabilization and improvement, response rate 70% | [107,108] |
| Tenth cranial nerve | Non-invasive VNS | MCI | Not Applicable | Ongoing | [109] | |
| 3. Transcranial magnetic stimulation | ||||||
| High-frequency rTMS | Left DLPFC | 10 Hz/120% MT/3000 pulses per session/10 sessions/2 weeks * | MCI | Phase IV | Executive functional improvement | [110] |
| 10 Hz/120% MT/2000 pulses per session/20 sessions/4 weeks * | MCI | Not Applicable | Ongoing | [111] | ||
| 20 Hz/100% MT/2000 pulses per session/20 sessions/4 weeks * | Moderate AD | Not Applicable | Improved language performance | [112] | ||
| 20 Hz/80% MT/1200 pulses per session/20 sessions/4 weeks * | AD patients with BPSD | Not Applicable | Cognitive and functional improvement | [113] | ||
| 20 Hz/100% MT/2000 pulses per session/20 sessions/4 weeks * | Mild to moderate AD | Not Applicable | Improvement in trained associative memory, add-on effect | [114] | ||
| 20 Hz/80–100% MT/1000 pulses per session/20 sessions/4 weeks * | Mild to moderate AD | Not Applicable | Cognitive and functional improvement, add-on effect | [115] | ||
| 5 Hz/100% MT/1500 pulses per session/15 sessions/3 weeks * | Probable AD | Not Applicable | Cognitive and functional improvement | [116] | ||
| Bilateral DLPFCs | 20 Hz/90% MT/2000 pulses per session/5 sessions/5 days * | Mild to severe AD | Not Applicable | Cognitive and functional improvement in mild to moderate AD | [117] | |
| 20 Hz/90–100% MT/2000 pulses per session/13 sessions/4 weeks * | Mild to moderate AD | Not Applicable | Cognitive improvement | [118] | ||
| Right IFG | 10 Hz/90% MT/2250 pulses per session/single session * | MCI | Not Applicable | Improvement in attention and psychomotor speed | [119] | |
| 10 Hz/90% MT/2250 pulses per session/single session * | MCI to moderate AD | Not Applicable | Cognitive improvement | [120] | ||
| Right STG | 10 Hz/90% MT/2250 pulses per session/single session * | MCI to moderate AD | Not Applicable | Cognitive improvement | [120] | |
| Left parietal lobe | 20 Hz/100% MT/1600 pulses per session/10 sessions/1 week * | Early AD | Not Applicable | Improvement in episodic memory | [121] | |
| Bilateral parietal lobes | 20 Hz/Unavailable MT/1 h per session/30 sessions/6 weeks * | Mild to moderate AD | Not Applicable | Better performance in memory and language in mild AD | [122] | |
| Low-frequency rTMS | Bilateral DLPFCs | 1 Hz/90% MT/600 pulses per session/2 sessions/1 day * | Healthy individuals-MCI | Not Applicable | Improvement in recognition memory | [123] |
| 1 Hz/100% MT/2000 pulses per session/5 sessions/5 days * | Mild to severe AD | Not Applicable | Less cognitive efficacy than high-frequency rTMS | [117] | ||
| 4. Transcranial electrical stimulation | ||||||
| Transcranial direct current stimulation | Left DLPFC | 2 mA/30 min per session/single session ** | Mild to moderate AD | Not Applicable | Improved recognition memory | [124] |
| 2 mA/25 min per session/10 sessions/2 weeks ** | Mild to moderate AD | Not Applicable | Cognitive improvement | [125] | ||
| 2 mA/20 min per session/6 sessions/2 weeks ** | Moderate AD | Phase II | No cognitive or behavioral improvement, no change in apathy symptoms Requirement of more than 6 sessions | [126] | ||
| 2 mA/30 min per session/daily session/6 months ** | Early AD | Not Applicable | Cognitive and functional improvement, rescue of executive function | [127] | ||
| 1.5 mA/15 min per session/single session ** | MCI | Not Applicable | Enhanced free recall and recognition of memory | [128] | ||
| 2 mA/30 min per session/1-5 sessions ** | MCI | Not Applicable | Improvement in selective attention, processing speed, and planning ability tasks Optimal frequency of 3 sessions/week | [129] | ||
| Left parietal lobe | 2 mA/30 min per session/single session ** | Mild to moderate AD | Not Applicable | Improvement in recognition memory | [124] | |
| 2 mA/30 min per session/6 sessions/10 days ** | Mild to moderate AD | Not Applicable | No improved verbal memory function | [130] | ||
| Bilateral temporoparietal lobe | 1.5 mA/15 min per minute/2 sessions (anodal and cathodal) ** | Mild AD | Not Applicable | Improved word recognition in anodal group, cognitive worsening in cathodal group | [131] | |
| 2 mA/30 min per session/5 sessions/1 week ** | Mild to moderate AD | Not Applicable | Improvement in visual recognition memory | [132] | ||
| 2 mA/20 min per session/10 sessions/2 weeks ** | Early AD | Not Applicable | Improved cognitive performance | [133] | ||
| Left temporoparietal lobe | 2 mA/20 min per session/10 sessions/2 weeks ** | Advanced AD | Not Applicable | Stabilized neuropsychological performance, long-term protective effect | [134] | |
| Transcranial alternating current stimulation | Left DLPFC | 40 Hz/1.5 mA/30 min per session/40 sessions/4 weeks *** | MCI to moderate AD | Not Applicable | Improved cognitive performance | [135] |
| Superior parietal cortex | 40 Hz/3 mA/30 min per session/single session *** | AD patients | Not Applicable | Completed, no published data | [136] | |
| Left angular gyrus | 40 Hz/unavailable intensity/20 min per session/3 sessions *** | Healthy individuals to mild AD | Not Applicable | Ongoing | [137] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, T.-W.; Lane, H.-Y.; Lin, C.-H. Novel Therapeutic Approaches for Alzheimer’s Disease: An Updated Review. Int. J. Mol. Sci. 2021, 22, 8208. https://doi.org/10.3390/ijms22158208
Yu T-W, Lane H-Y, Lin C-H. Novel Therapeutic Approaches for Alzheimer’s Disease: An Updated Review. International Journal of Molecular Sciences. 2021; 22(15):8208. https://doi.org/10.3390/ijms22158208
Chicago/Turabian StyleYu, Tien-Wei, Hsien-Yuan Lane, and Chieh-Hsin Lin. 2021. "Novel Therapeutic Approaches for Alzheimer’s Disease: An Updated Review" International Journal of Molecular Sciences 22, no. 15: 8208. https://doi.org/10.3390/ijms22158208
APA StyleYu, T.-W., Lane, H.-Y., & Lin, C.-H. (2021). Novel Therapeutic Approaches for Alzheimer’s Disease: An Updated Review. International Journal of Molecular Sciences, 22(15), 8208. https://doi.org/10.3390/ijms22158208