
Individual Expression of Hepatitis A Virus 3C Protease Induces Ferroptosis in Human Cells In Vitro

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
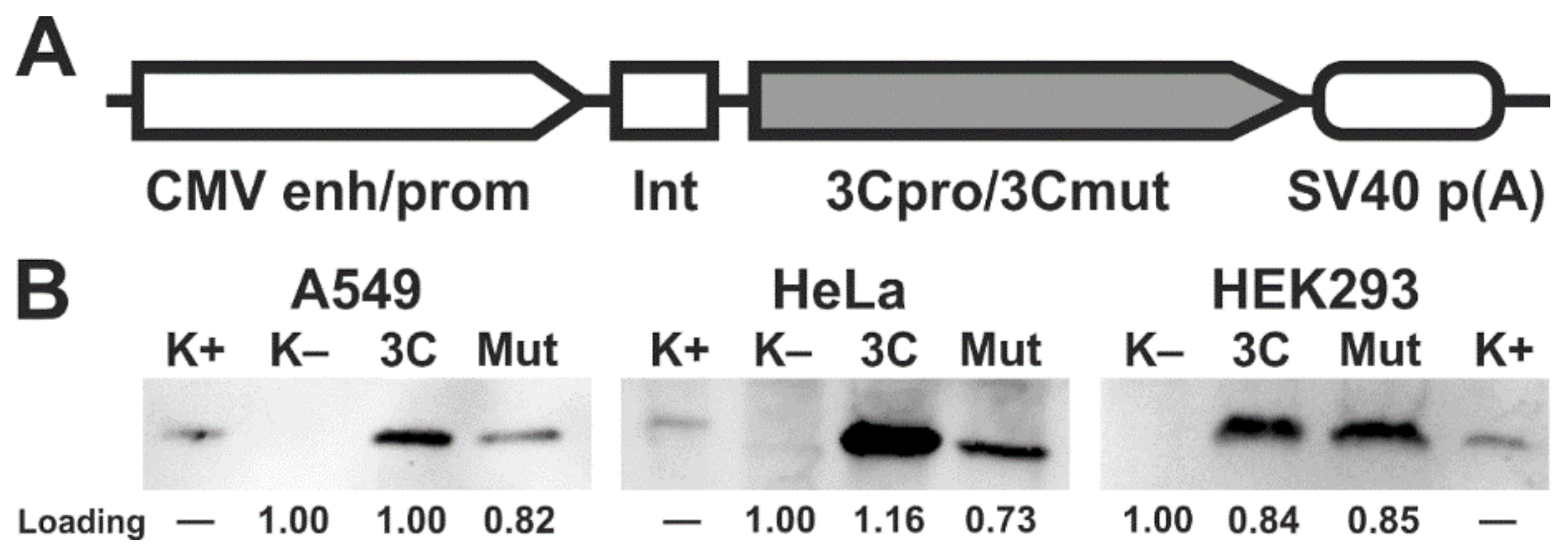
2.1. Ectopic Expression of 3Cpro and Its Inactive Form 3Cmut in Human Cells
2.2. Cpro Expression Induces Caspase-Independent Cell Death with Cytoplasmic Vacuolization
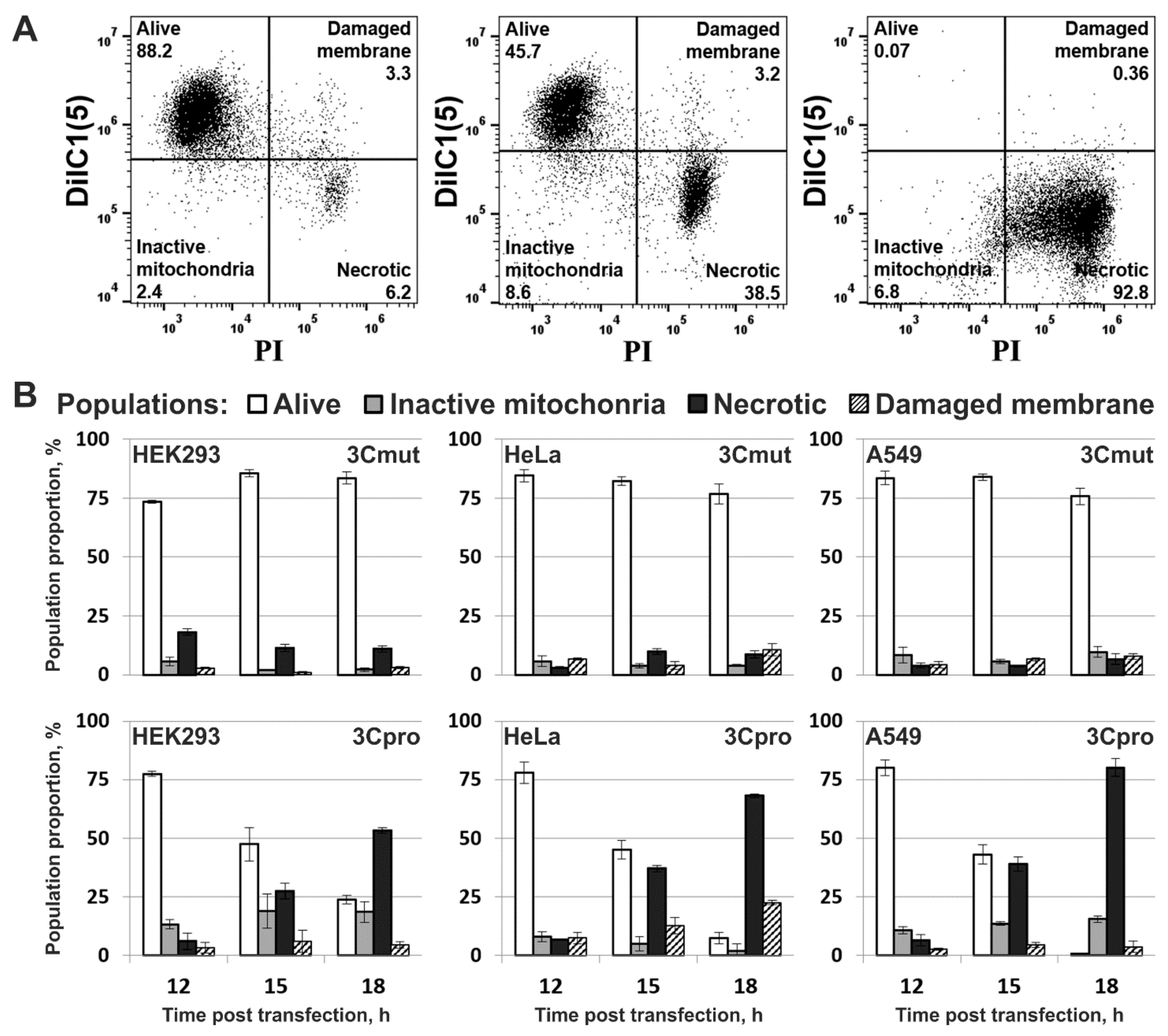
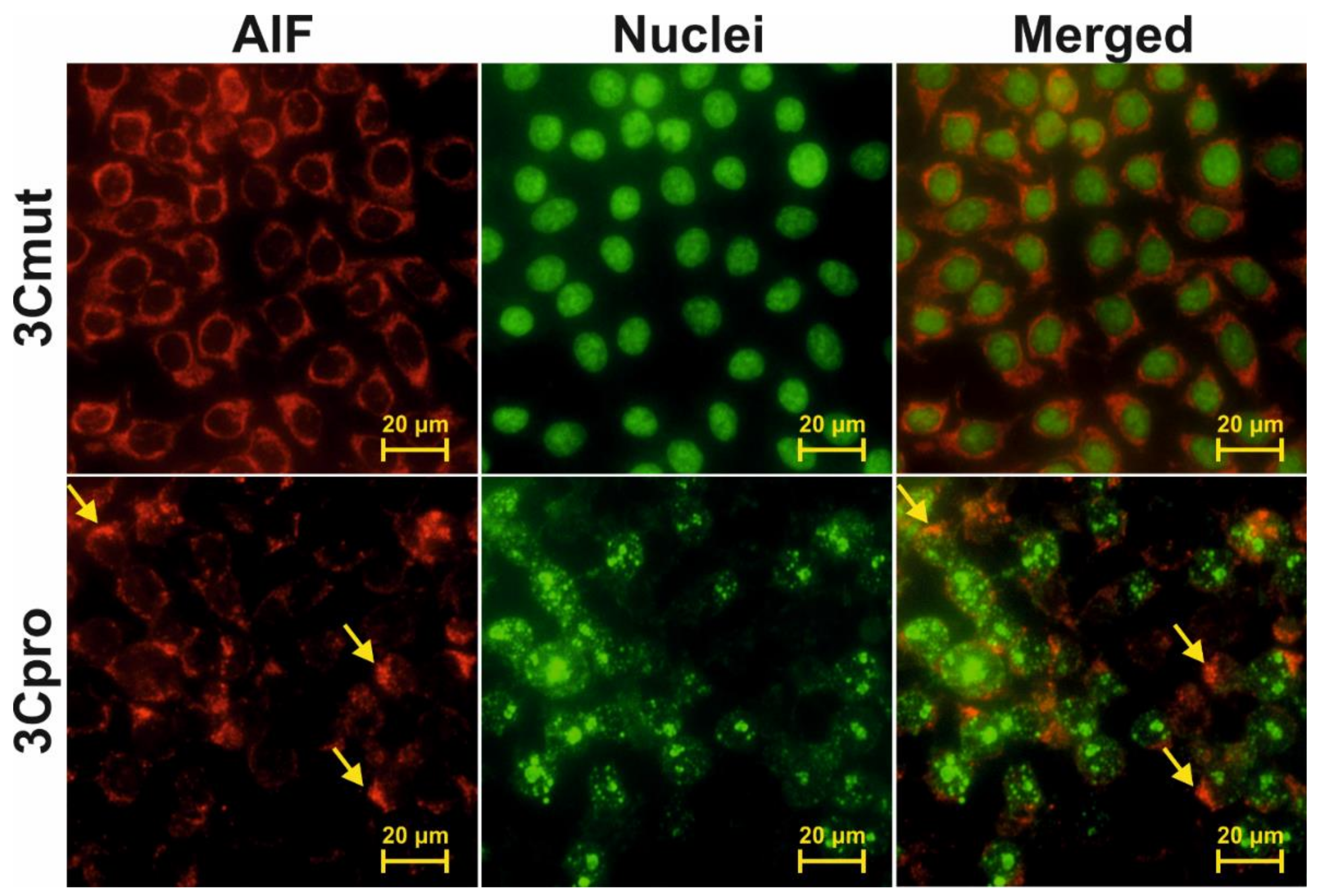
2.3. Cells Expressing 3Cpro Acquire Necrotic Morphology and Are Characterized by Nuclei and Mitochondria Swelling
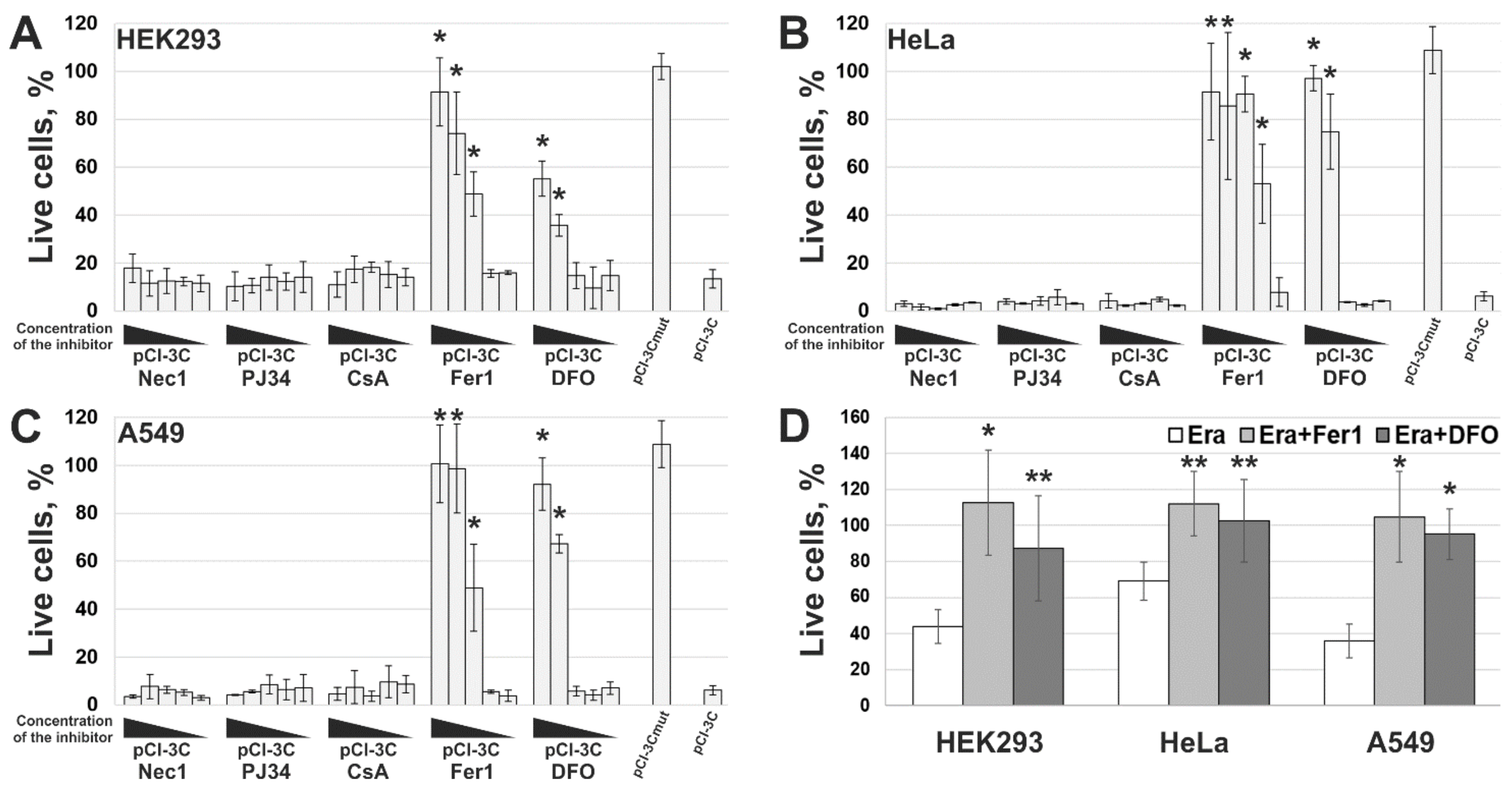
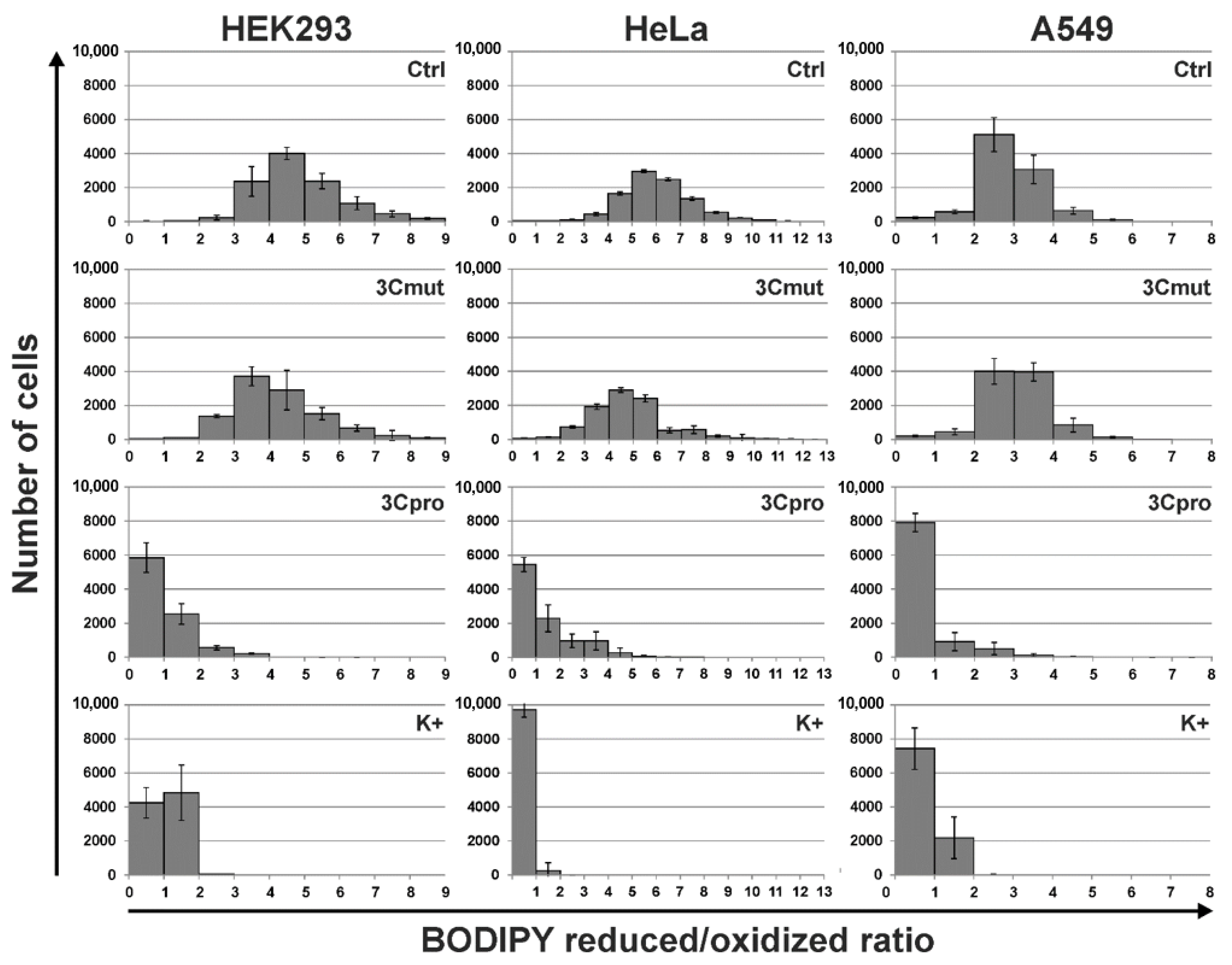
2.4. 3Cpro-Induced Cell Death Is Effectively Blocked by Ferroptosis Inhibitors and Is Accompinied by Lipid Peroxidation
3. Discussion
4. Materials and Methods
4.1. Plasmid Expression Constructs
4.2. Cell Cultures and Transfection
4.3. Western Blotting
4.4. Cell Viability Assay
4.5. Lipid Peroxidation Assay
4.6. Flow Cytometry
4.7. Fluorescence Microscopy
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allocati, N.; Masulli, M.; Di Ilio, C.; De Laurenzi, V. Die for the community: An overview of programmed cell death in bacteria. Cell Death Dis. 2015, 6, e1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, Y.; Steller, H. Programmed Cell Death in Animal Development and Disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, O.; Svedin, E.; Kapell, S.; Nurminen, A.; Hytönen, V.; Flodström-Tullberg, M. Enteroviral proteases: Structure, host interactions and pathogenicity. Rev. Med. Virol. 2016, 26, 251–267. [Google Scholar] [CrossRef]
- Sun, D.; Chen, S.; Cheng, A.; Wang, M. Roles of the Picornaviral 3C Proteinase in the Viral Life Cycle and Host Cells. Viruses 2016, 8, 82. [Google Scholar] [CrossRef] [Green Version]
- Calandria, C.; Irurzun, A.; Barco, A.; Carrasco, L. Individual expression of poliovirus 2Apro and 3Cpro induces activation of caspase-3 and PARP cleavage in HeLa cells. Virus Res. 2004, 104, 39–49. [Google Scholar] [CrossRef]
- Li, M.-L.; Hsu, T.-A.; Chen, T.-C.; Chang, S.-C.; Lee, J.-C.; Chen, C.-C.; Stollar, V.; Shih, S.-R. The 3C Protease Activity of Enterovirus 71 Induces Human Neural Cell Apoptosis. Virology 2002, 293, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Chau, D.H.W.; Yuan, J.; Zhang, H.; Cheung, P.; Lim, T.; Liu, Z.; Sall, A.; Yang, D. Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis 2006, 12, 513–524. [Google Scholar] [CrossRef]
- Lötzerich, M.; Roulin, P.S.; Boucke, K.; Witte, R.; Georgiev, O.; Greber, U.F. Rhinovirus 3C protease suppresses apoptosis and triggers caspase-independent cell death. Cell Death Dis. 2018, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Shubin, A.V.; Lunina, N.A.; Shedova, E.N.; Roshina, M.P.; Demidyuk, I.V.; Vinogradova, T.V.; Kopantsev, E.P.; Chernov, I.P.; Kostrov, S.V. Evaluation of the toxic effects evoked by the transient expression of protease genes from human pathogens in HEK293 cells. Appl. Biochem. Microbiol. 2013, 49, 750–755. [Google Scholar] [CrossRef]
- Shubin, A.V.; Demidyuk, I.V.; Lunina, N.A.; Komissarov, A.A.; Roschina, M.P.; Leonova, O.G.; Kostrov, S.V. Protease 3C of hepatitis A virus induces vacuolization of lysosomal/endosomal organelles and caspase-independent cell death. BMC Cell Biol. 2015, 16, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shubin, A.V.; Komissarov, A.A.; Karaseva, M.A.; Padman, B.S.; Kostrov, S.V.; Demidyuk, I.V. Human hepatitis A virus 3C protease exerts a cytostatic effect on Saccharomyces cerevisiae and affects the vacuolar compartment. Biologia 2021, 76, 321–327. [Google Scholar] [CrossRef]
- Komissarov, A.; Karaseva, M.A.; Safina, D.R.; Roschina, M.P.; Bednova, O.P.; Kazakov, A.A.; Demkin, V.V.; Demidyuk, I.V. Comparative evaluation of the transgene expression efficiency provided by the model genetic constructs of different structure. Mol. Genet. Microbiol. Virol. 2016, 31, 156–162. [Google Scholar] [CrossRef]
- Komissarov, A.; Demidyuk, I.; Safina, D.; Roschina, M.; Shubin, A.; Lunina, N.; Karaseva, M.; Kostrov, S. Cytotoxic effect of co-expression of human hepatitis A virus 3C protease and bifunctional suicide protein FCU1 genes in a bicistronic vector. Mol. Biol. Rep. 2017, 44, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Western Blot Normalization Using Image Lab Software, Quick Start Guide. Bio-Rad Bulletin 6434; Bio-Rad Laboratories: Hercules, CA, USA, 2015.
- Bertrand, R.; Solary, E.; O’Connor, P.; Kohn, K.W.; Pommier, Y. Induction of a Common Pathway of Apoptosis by Staurosporine. Exp. Cell Res. 1994, 211, 314–321. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, P.; Soriano, F.; Virág, L.; Liaudet, L.; Mabley, J.; Szabó, É.; Haskó, G.; Marton, A.; Lorigados, C.B.; Gallyas, F.; et al. Novel phenanthridinone inhibitors of poly(adenosine 5′-diphosphate-ribose) synthetase: Potent cytoprotective and antishock agents. Crit. Care Med. 2002, 30, 1071–1082. [Google Scholar] [CrossRef]
- Kajitani, K.; Fujihashi, M.; Kobayashi, Y.; Shimizu, S.; Tsujimoto, Y.; Miki, K. Crystal structure of human cyclophilin D in complex with its inhibitor, cyclosporin A at 0.96-Å resolution. Proteins Struct. Funct. Bioinform. 2007, 70, 1635–1639. [Google Scholar] [CrossRef]
- Zilka, O.; Shah, R.; Li, B.; Angeli, J.P.F.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Shah, R.; Pratt, D.A.; Conrad, M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol. Sci. 2017, 38, 489–498. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbasi, U.; Abbina, S.; Gill, A.; Bhagat, V.; Kizhakkedathu, J.N. A facile colorimetric method for the quantification of labile iron pool and total iron in cells and tissue specimens. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Bajbouj, K.; Shafarin, J.; Hamad, M. High-Dose Deferoxamine Treatment Disrupts Intracellular Iron Homeostasis, Reduces Growth, and Induces Apoptosis in Metastatic and Nonmetastatic Breast Cancer Cell Lines. Technol. Cancer Res. Treat. 2018, 17, 1533033818764470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prus, E.; Fibach, E. Effect of Iron Chelators on Labile Iron and Oxidative Status of Thalassaemic Erythroid Cells. Acta Haematol. 2010, 123, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Glickstein, H.; Ben El, R.; Shvartsman, M.; Cabantchik, Z.I. Intracellular labile iron pools as direct targets of iron chelators: A fluorescence study of chelator action in living cells. Blood 2005, 106, 3242–3250. [Google Scholar] [CrossRef] [PubMed]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.-M.; Travain, V.B.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doll, S.; Conrad, M. Iron and ferroptosis: A still ill-defined liaison. IUBMB Life 2017, 69, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Liu, J.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Tang, D. Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 2020, 66, 89–100. [Google Scholar] [CrossRef]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Shubin, A.V.; Demidyuk, I.; Komissarov, A.; Rafieva, L.M.; Kostrov, S.V. Cytoplasmic vacuolization in cell death and survival. Oncotarget 2016, 7, 55863–55889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-Z.; Sui, C.-Y.; Chen, Q.; Chen, X.-P.; Zhang, H.; Zhou, X.-P. Promotion of autophagy at the maturation step by IL-6 is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Mol. Cell. Biochem. 2013, 380, 219–227. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death-apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Riegman, M.; Sagie, L.; Galed, C.; Levin, T.; Steinberg, N.; Dixon, S.J.; Wiesner, U.; Bradbury, M.S.; Niethammer, P.; Zaritsky, A.; et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat. Cell Biol. 2020, 22, 1–7. [Google Scholar] [CrossRef]
- Katikaneni, A.; Jelcic, M.; Gerlach, G.F.; Ma, Y.; Overholtzer, M.; Niethammer, P. Lipid peroxidation regulates long-range wound detection through 5-lipoxygenase in zebrafish. Nat. Cell Biol. 2020, 22, 1049–1055. [Google Scholar] [CrossRef]
- Lu, B.; Chen, X.B.; Ying, M.D.; He, Q.J.; Cao, J.; Yang, B. The Role of Ferroptosis in Cancer Development and Treatment Response. Front. Pharmacol. 2018, 8, 992. [Google Scholar] [CrossRef]
- Shen, Z.; Song, J.; Yung, B.C.; Zhou, Z.; Wu, A.; Chen, X. Emerging Strategies of Cancer Therapy Based on Ferroptosis. Adv. Mater. 2018, 30, e1704007. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, B.; Fleischer, S.; Maier, K.; Wiedmann, K.H.; Sacher, M.; Thaler, H.; Vallbracht, A. Clonal analysis of infiltrating T lymphocytes in liver tissue in viral hepatitis A. Immunology 1990, 69, 14–19. [Google Scholar] [PubMed]
- Baba, M.; Hasegawa, H.; Nakayabu, M.; Fukai, K.; Suzuki, S. Cytolytic activity of natural killer cells and lymphokine activated killer cells against hepatitis A virus infected fibroblasts. J. Clin. Lab. Immunol. 1993, 40, 47–60. [Google Scholar] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komissarov, A.A.; Karaseva, M.A.; Roschina, M.P.; Shubin, A.V.; Lunina, N.A.; Kostrov, S.V.; Demidyuk, I.V. Individual Expression of Hepatitis A Virus 3C Protease Induces Ferroptosis in Human Cells In Vitro. Int. J. Mol. Sci. 2021, 22, 7906. https://doi.org/10.3390/ijms22157906
Komissarov AA, Karaseva MA, Roschina MP, Shubin AV, Lunina NA, Kostrov SV, Demidyuk IV. Individual Expression of Hepatitis A Virus 3C Protease Induces Ferroptosis in Human Cells In Vitro. International Journal of Molecular Sciences. 2021; 22(15):7906. https://doi.org/10.3390/ijms22157906
Chicago/Turabian StyleKomissarov, Alexey A., Maria A. Karaseva, Marina P. Roschina, Andrey V. Shubin, Nataliya A. Lunina, Sergey V. Kostrov, and Ilya V. Demidyuk. 2021. "Individual Expression of Hepatitis A Virus 3C Protease Induces Ferroptosis in Human Cells In Vitro" International Journal of Molecular Sciences 22, no. 15: 7906. https://doi.org/10.3390/ijms22157906
APA StyleKomissarov, A. A., Karaseva, M. A., Roschina, M. P., Shubin, A. V., Lunina, N. A., Kostrov, S. V., & Demidyuk, I. V. (2021). Individual Expression of Hepatitis A Virus 3C Protease Induces Ferroptosis in Human Cells In Vitro. International Journal of Molecular Sciences, 22(15), 7906. https://doi.org/10.3390/ijms22157906