
Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer


Abstract
1. Introduction
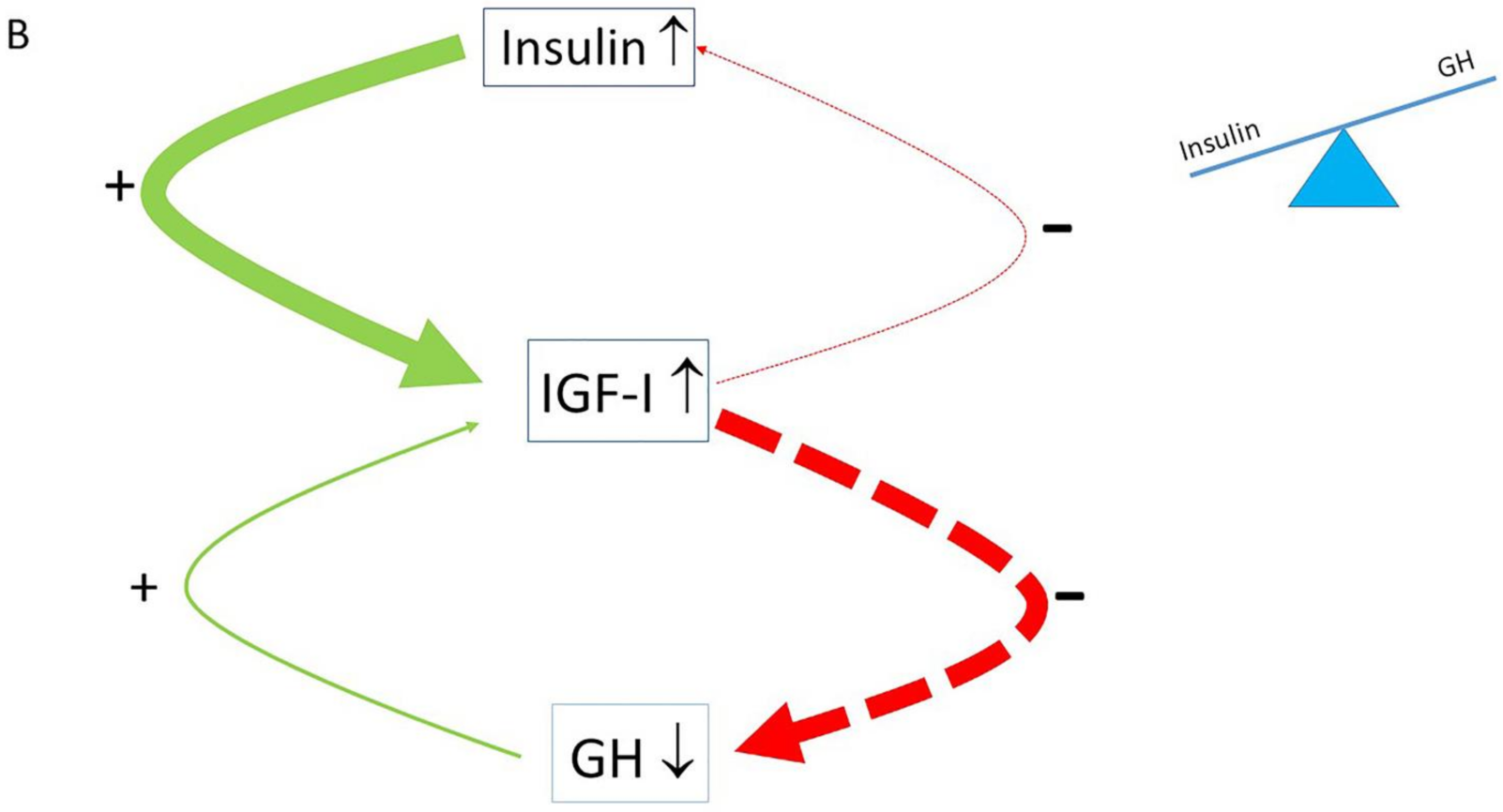
2. The Role of the Insulin–GH–IGF-I Axis in Healthy Subjects
3. The Role of the Insulin–GH–IGF-I Axis in Metabolism
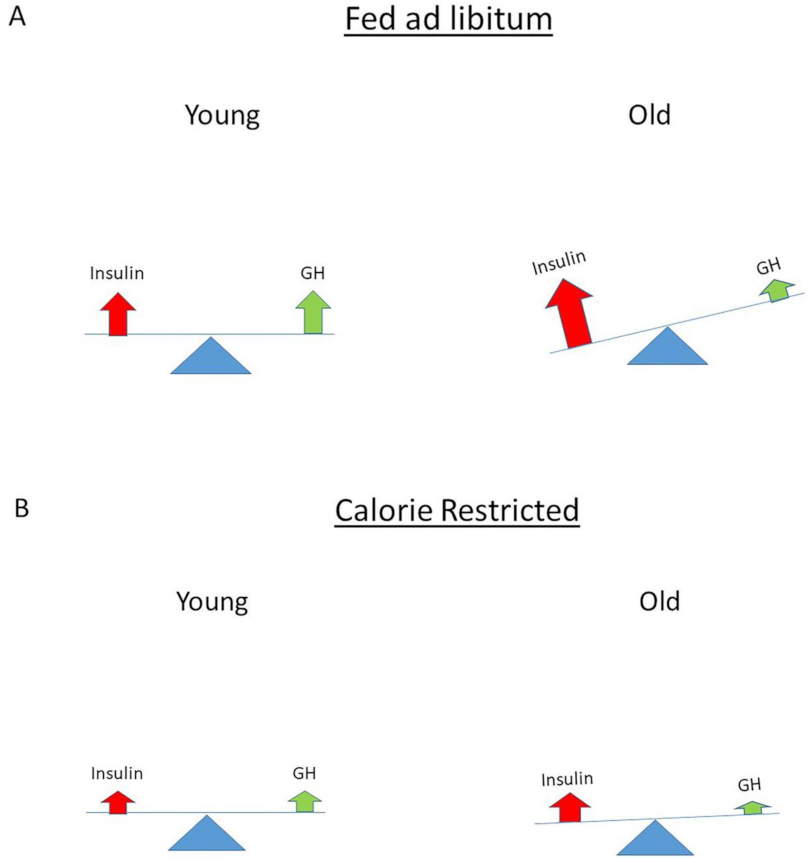
4. The Normal Balance between Insulin, IGF-I and GH Is Disturbed in Modern Societies
5. Methods to Measure Insulin
6. How to Define Circulating Hyperinsulinemia
7. Loss of Pulsatile Insulin Secretion Contributes to Insulin Resistance
8. Hyperinsulinemia Precedes Insulin Resistance
9. Which Factors Cause Hyperinsulinemia?
10. Hyperinsulinemia Is a Common Etiological Factor in Many Diseases
11. The Role of Insulin (Signaling) in Longevity
12. How Can Hyperinsulinemia Be Modified?
13. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lakka, H.M.; Lakka, T.A.; Tuomilehto, J.; Sivenius, J.; Salonen, J.T. Hyperinsulinemia and the risk of cardiovascular death and acute coronary and cerebrovascular events in men: The Kuopio Ischaemic Heart Disease Risk Factor Study. Arch. Int. Med. 2000, 160, 1160–1168. [Google Scholar] [CrossRef]
- Despres, J.P.; Lamarche, B.; Mauriege, P.; Cantin, B.; Dagenais, G.R.; Moorjani, S.; Lupien, P.J. Hyperinsulinemia as an independent risk factor for ischemic heart disease. N. Engl. J. Med. 1996, 334, 952–957. [Google Scholar] [CrossRef]
- Pyorala, M.; Miettinen, H.; Laakso, M.; Pyorala, K. Hyperinsulinemia predicts coronary heart disease risk in healthy middle-aged men: The 22-year follow-up results of the Helsinki Policemen Study. Circulation 1998, 98, 398–404. [Google Scholar] [CrossRef]
- Sigal, R.J.; El-Hashimy, M.; Martin, B.C.; Soeldner, J.S.; Krolewski, A.S.; Warram, J.H. Acute postchallenge hyperinsulinemia predicts weight gain: A prospective study. Diabetes 1997, 46, 1025–1029. [Google Scholar] [CrossRef]
- Odeleye, O.E.; de Courten, M.; Pettitt, D.J.; Ravussin, E. Fasting hyperinsulinemia is a predictor of increased body weight gain and obesity in Pima Indian children. Diabetes 1997, 46, 1341–1345. [Google Scholar] [CrossRef]
- Balkau, B.; Kahn, H.S.; Courbon, D.; Eschwege, E.; Ducimetiere, P.; Paris Prospective, S. Hyperinsulinemia predicts fatal liver cancer but is inversely associated with fatal cancer at some other sites: The Paris Prospective Study. Diabetes Care 2001, 24, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, T.; Kajio, H.; Sugiyama, T. Association between hyperinsulinemia and increased risk of cancer death in nonobese and obese people: A population-based observational study. Int. J. Cancer 2017, 141, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Giovannucci, E.; Pollak, M.; Leavitt, A.; Tao, Y.; Gaziano, J.M.; Stampfer, M.J. A prospective study of plasma C-peptide and colorectal cancer risk in men. J. Natl. Cancer Inst. 2004, 96, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Kekalainen, P.; Sarlund, H.; Pyorala, K.; Laakso, M. Hyperinsulinemia cluster predicts the development of type 2 diabetes independently of family history of diabetes. Diabetes Care 1999, 22, 86–92. [Google Scholar] [CrossRef]
- Mehran, A.E.; Templeman, N.M.; Brigidi, G.S.; Lim, G.E.; Chu, K.Y.; Hu, X.; Botezelli, J.D.; Asadi, A.; Hoffman, B.G.; Kieffer, T.J.; et al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 2012, 16, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Sbraccia, P.; D’Adamo, M.; Guglielmi, V. Is type 2 diabetes an adiposity-based metabolic disease? From the origin of insulin resistance to the concept of dysfunctional adipose tissue. Eat Weight Disord. 2021. [Google Scholar] [CrossRef]
- Shanik, M.H.; Xu, Y.; Skrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin resistance and hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31 (Suppl. 2), S262–S268. [Google Scholar] [CrossRef] [PubMed]
- Corkey, B.E. Banting lecture 2011: Hyperinsulinemia: Cause or consequence? Diabetes 2012, 61, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Dankner, R.; Chetrit, A.; Shanik, M.H.; Raz, I.; Roth, J. Basal-state hyperinsulinemia in healthy normoglycemic adults is predictive of type 2 diabetes over a 24-year follow-up: A preliminary report. Diabetes Care 2009, 32, 1464–1466. [Google Scholar] [CrossRef] [PubMed]
- Nolan, C.J.; Ruderman, N.B.; Kahn, S.E.; Pedersen, O.; Prentki, M. Insulin resistance as a physiological defense against metabolic stress: Implications for the management of subsets of type 2 diabetes. Diabetes 2015, 64, 673–686. [Google Scholar] [CrossRef]
- Schofield, C.J.; Sutherland, C. Disordered insulin secretion in the development of insulin resistance and Type 2 diabetes. Diabet. Med. 2012, 29, 972–979. [Google Scholar] [CrossRef]
- Aspinwall, C.A.; Lakey, J.R.; Kennedy, R.T. Insulin-stimulated insulin secretion in single pancreatic beta cells. J. Biol. Chem. 1999, 274, 6360–6365. [Google Scholar] [CrossRef]
- Froesch, E.R.; Zapf, J. Insulin-like growth factors and insulin: Comparative aspects. Diabetologia 1985, 28, 485–493. [Google Scholar] [CrossRef]
- De Meyts, P.; Sajid, W.; Palsgaard, J.; Theede, A.-M.; Gaugain, L.; Aladdin, H.; Whittaker, J. Insulin and IGF-I Receptor Structure and Binding Mechanism. In Mechanism of Insulin Action; Pessin, A.R.S.A.J.E., Ed.; Landes Bioscience: Austin, TX, USA, 2007; pp. 1–32. [Google Scholar]
- Clemmons, D.R. Involvement of insulin-like growth factor-I in the control of glucose homeostasis. Curr. Opin. Pharmacol. 2006, 6, 620–625. [Google Scholar] [CrossRef]
- Giustina, A.; Berardelli, R.; Gazzaruso, C.; Mazziotti, G. Insulin and GH-IGF-I axis: Endocrine pacer or endocrine disruptor? Acta Diabetol. 2015, 52, 433–443. [Google Scholar] [CrossRef]
- Hartman, M.L.; Clayton, P.E.; Johnson, M.L.; Celniker, A.; Perlman, A.J.; Alberti, K.G.; Thorner, M.O. A low dose euglycemic infusion of recombinant human insulin-like growth factor I rapidly suppresses fasting-enhanced pulsatile growth hormone secretion in humans. J. Clin. Investig. 1993, 91, 2453–2462. [Google Scholar] [CrossRef]
- Bondy, C.A.; Underwood, L.E.; Clemmons, D.R.; Guler, H.P.; Bach, M.A.; Skarulis, M. Clinical uses of insulin-like growth factor I. Ann. Intern. Med. 1994, 120, 593–601. [Google Scholar] [CrossRef]
- Janssen, J.A. Advantages and disadvantages of GH/IGF-I combination treatment. Rev. Endocr. Metab. Disord. 2009, 10, 157–162. [Google Scholar] [CrossRef]
- Obici, S.; Zhang, B.B.; Karkanias, G.; Rossetti, L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat. Med. 2002, 8, 1376–1382. [Google Scholar] [CrossRef]
- Scherer, T.; O’Hare, J.; Diggs-Andrews, K.; Schweiger, M.; Cheng, B.; Lindtner, C.; Zielinski, E.; Vempati, P.; Su, K.; Dighe, S.; et al. Brain insulin controls adipose tissue lipolysis and lipogenesis. Cell Metab. 2011, 13, 183–194. [Google Scholar] [CrossRef]
- Newsholme, P.; Gaudel, C.; McClenaghan, N.H. Nutrient regulation of insulin secretion and beta-cell functional integrity. Adv. Exp. Med. Biol. 2010, 654, 91–114. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, D.; Zierler, K.L. A Metabolic Regulating Device Based on the Actions of Human Growth Hormone and of Insulin, Singly and Together, on the Human Forearm. Nature 1963, 199, 913–915. [Google Scholar] [CrossRef] [PubMed]
- Froesch, E. Insulin-like Growth Factor: Endocrine and Autocrine/Paracrine Implications and Relations to Diabetes Mellitus. In Contributions of Physiology to the Understanding of Diabetes; GR Zand, C.W., Ed.; Springer: Berlin, Germany, 1997; pp. 127–147. [Google Scholar]
- Huang, Z.; Huang, L.; Waters, M.J.; Chen, C. Insulin and Growth Hormone Balance: Implications for Obesity. Trends Endocrinol. Metab. 2020, 31, 642–654. [Google Scholar] [CrossRef] [PubMed]
- Guyenet, S.J.; Schwartz, M.W. Clinical review: Regulation of food intake, energy balance, and body fat mass: Implications for the pathogenesis and treatment of obesity. J. Clin. Endocrinol. Metab. 2012, 97, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Frystyk, J.; Vestbo, E.; Skjaerbaek, C.; Mogensen, C.E.; Orskov, H. Free insulin-like growth factors in human obesity. Metabolism 1995, 44, 37–44. [Google Scholar] [CrossRef]
- Brugts, M.P.; van Duijn, C.M.; Hofland, L.J.; Witteman, J.C.; Lamberts, S.W.; Janssen, J.A. Igf-I bioactivity in an elderly population: Relation to insulin sensitivity, insulin levels, and the metabolic syndrome. Diabetes 2010, 59, 505–508. [Google Scholar] [CrossRef]
- Gahete, M.D.; Cordoba-Chacon, J.; Lin, Q.; Bruning, J.C.; Kahn, C.R.; Castano, J.P.; Christian, H.; Luque, R.M.; Kineman, R.D. Insulin and IGF-I inhibit GH synthesis and release in vitro and in vivo by separate mechanisms. Endocrinology 2013, 154, 2410–2420. [Google Scholar] [CrossRef]
- Yamashita, S.; Melmed, S. Effects of insulin on rat anterior pituitary cells. Inhibition of growth hormone secretion and mRNA levels. Diabetes 1986, 35, 440–447. [Google Scholar] [CrossRef]
- Cornford, A.S.; Barkan, A.L.; Horowitz, J.F. Rapid suppression of growth hormone concentration by overeating: Potential mediation by hyperinsulinemia. J. Clin. Endocrinol. Metab. 2011, 96, 824–830. [Google Scholar] [CrossRef]
- Ji, S.; Guan, R.; Frank, S.J.; Messina, J.L. Insulin inhibits growth hormone signaling via the growth hormone receptor/JAK2/STAT5B pathway. J. Biol. Chem. 1999, 274, 13434–13442. [Google Scholar] [CrossRef]
- Kreitschmann-Andermahr, I.; Suarez, P.; Jennings, R.; Evers, N.; Brabant, G. GH/IGF-I regulation in obesity--mechanisms and practical consequences in children and adults. Horm. Res. Paediatr. 2010, 73, 153–160. [Google Scholar] [CrossRef]
- Chevenne, D.; Trivin, F.; Porquet, D. Insulin assays and reference values. Diabetes Metab. 1999, 25, 459–476. [Google Scholar] [PubMed]
- Janssen, J.; Llaurado, G.; Varewijck, A.J.; Groop, P.H.; Forsblom, C.; Fernandez-Veledo, S.; van den Dungen, E.S.R.; Vendrell, J.; Hofland, L.J.; Yki-Jarvinen, H. Serum Insulin Bioassay Reflects Insulin Sensitivity and Requirements in Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2017, 102, 3814–3821. [Google Scholar] [CrossRef] [PubMed]
- Clark, P.M. Assays for insulin, proinsulin(s) and C-peptide. Ann. Clin. Biochem. 1999, 36 Pt 5, 541–564. [Google Scholar] [CrossRef]
- Yalow, R.S.; Berson, S.A. Immunoassay of endogenous plasma insulin in man. 1960. Obes. Res. 1996, 4, 583–600. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.W.; Clarke, N.J.; Chen, Z.; McPhaul, M.J. A high-throughput mass spectrometry assay to simultaneously measure intact insulin and C-peptide. Clin. Chim. Acta 2016, 455, 202–208. [Google Scholar] [CrossRef]
- Marcovina, S.; Bowsher, R.R.; Miller, W.G.; Staten, M.; Myers, G.; Caudill, S.P.; Campbell, S.E.; Steffes, M.W.; Insulin Standardization, W. Standardization of insulin immunoassays: Report of the American Diabetes Association Workgroup. Clin. Chem. 2007, 53, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Sapin, R. Insulin immunoassays: Fast approaching 50 years of existence and still calling for standardization. Clin. Chem. 2007, 53, 810–812. [Google Scholar] [CrossRef] [PubMed]
- Tohidi, M.; Arbab, P.; Ghasemi, A. Assay-dependent variability of serum insulin concentrations: A comparison of eight assays. Scand. J. Clin. Lab. Investig. 2017, 77, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Porksen, N.; Hollingdal, M.; Juhl, C.; Butler, P.; Veldhuis, J.D.; Schmitz, O. Pulsatile insulin secretion: Detection, regulation, and role in diabetes. Diabetes 2002, 51 (Suppl. 1), S245–S254. [Google Scholar] [CrossRef]
- Crofts, C.; Zinn, C.; Wheldon, M.; Schofield, G. Hyperinsulinemia: A unifying theory of chronic disease. Diabesity 2015, 4, 34–43. [Google Scholar] [CrossRef]
- Wallace, T.M.; Levy, J.C.; Matthews, D.R. Use and abuse of HOMA modeling. Diabetes Care 2004, 27, 1487–1495. [Google Scholar] [CrossRef]
- De Leon, D.D.; Stanley, C.A. Determination of insulin for the diagnosis of hyperinsulinemic hypoglycemia. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 763–769. [Google Scholar] [CrossRef]
- Najjar, S.M.; Perdomo, G. Hepatic Insulin Clearance: Mechanism and Physiology. Physiology 2019, 34, 198–215. [Google Scholar] [CrossRef] [PubMed]
- Eaton, R.P.; Allen, R.C.; Schade, D.S. Hepatic removal of insulin in normal man: Dose response to endogenous insulin secretion. J. Clin. Endocrinol. Metab. 1983, 56, 1294–1300. [Google Scholar] [CrossRef]
- Tokarz, V.L.; MacDonald, P.E.; Klip, A. The cell biology of systemic insulin function. J. Cell Biol. 2018, 217, 2273–2289. [Google Scholar] [CrossRef]
- Horwitz, D.L.; Starr, J.I.; Mako, M.E.; Blackard, W.G.; Rubenstein, A.H. Proinsulin, insulin, and C-peptide concentrations in human portal and peripheral blood. J. Clin. Investig. 1975, 55, 1278–1283. [Google Scholar] [CrossRef]
- Bergman, R.N.; Piccinini, F.; Kabir, M.; Kolka, C.M.; Ader, M. Hypothesis: Role of Reduced Hepatic Insulin Clearance in the Pathogenesis of Type 2 Diabetes. Diabetes 2019, 68, 1709–1716. [Google Scholar] [CrossRef]
- Polonsky, K.S.; Rubenstein, A.H. C-peptide as a measure of the secretion and hepatic extraction of insulin. Pitfalls and limitations. Diabetes 1984, 33, 486–494. [Google Scholar] [CrossRef]
- Van Cauter, E.; Mestrez, F.; Sturis, J.; Polonsky, K.S. Estimation of insulin secretion rates from C-peptide levels. Comparison of individual and standard kinetic parameters for C-peptide clearance. Diabetes 1992, 41, 368–377. [Google Scholar] [CrossRef]
- Porksen, N.; Nyholm, B.; Veldhuis, J.D.; Butler, P.C.; Schmitz, O. In humans at least 75% of insulin secretion arises from punctuated insulin secretory bursts. Am. J. Physiol. 1997, 273, E908–E914. [Google Scholar] [CrossRef] [PubMed]
- Marfella, R.; D’Amico, M.; Di Filippo, C.; Siniscalchi, M.; Sasso, F.C.; Ferraraccio, F.; Rossi, F.; Paolisso, G. The possible role of the ubiquitin proteasome system in the development of atherosclerosis in diabetes. Cardiovasc. Diabetol. 2007, 6, 35. [Google Scholar] [CrossRef]
- Li, C.; Ford, E.S.; McGuire, L.C.; Mokdad, A.H.; Little, R.R.; Reaven, G.M. Trends in hyperinsulinemia among nondiabetic adults in the U.S. Diabetes Care 2006, 29, 2396–2402. [Google Scholar] [CrossRef] [PubMed]
- Lan-Pidhainy, X.; Wolever, T.M. Are the glycemic and insulinemic index values of carbohydrate foods similar in healthy control, hyperinsulinemic and type 2 diabetic patients? Eur. J. Clin. Nutr. 2011, 65, 727–734. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nilsson, P.; Nilsson, J.A.; Hedblad, B.; Eriksson, K.F.; Berglund, G. Hyperinsulinaemia as long-term predictor of death and ischaemic heart disease in nondiabetic men: The Malmo Preventive Project. J. Intern. Med. 2003, 253, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.R. Physiological implications of pulsatile hormone secretion. Ann. N. Y. Acad. Sci. 1991, 618, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Ward, G.M.; Walters, J.M.; Aitken, P.M.; Best, J.D.; Alford, F.P. Effects of prolonged pulsatile hyperinsulinemia in humans. Enhancement of insulin sensitivity. Diabetes 1990, 39, 501–507. [Google Scholar] [CrossRef]
- Rizza, R.A.; Mandarino, L.J.; Genest, J.; Baker, B.A.; Gerich, J.E. Production of insulin resistance by hyperinsulinaemia in man. Diabetologia 1985, 28, 70–75. [Google Scholar] [CrossRef] [PubMed]
- O’Rahilly, S.; Turner, R.C.; Matthews, D.R. Impaired pulsatile secretion of insulin in relatives of patients with non-insulin-dependent diabetes. N. Engl. J. Med. 1988, 318, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Matveyenko, A.V.; Liuwantara, D.; Gurlo, T.; Kirakossian, D.; Dalla Man, C.; Cobelli, C.; White, M.F.; Copps, K.D.; Volpi, E.; Fujita, S.; et al. Pulsatile portal vein insulin delivery enhances hepatic insulin action and signaling. Diabetes 2012, 61, 2269–2279. [Google Scholar] [CrossRef]
- Meier, J.J.; Veldhuis, J.D.; Butler, P.C. Pulsatile insulin secretion dictates systemic insulin delivery by regulating hepatic insulin extraction in humans. Diabetes 2005, 54, 1649–1656. [Google Scholar] [CrossRef]
- Gavin, J.R., 3rd; Roth, J.; Neville, D.M., Jr.; de Meyts, P.; Buell, D.N. Insulin-dependent regulation of insulin receptor concentrations: A direct demonstration in cell culture. Proc. Natl. Acad. Sci. USA 1974, 71, 84–88. [Google Scholar] [CrossRef]
- Le Marchand, Y.; Loten, E.G.; Assimacopoulos-Jeannet, F.; Forgue, M.E.; Freychet, P.; Jeanrenaud, B. Effect of fasting and streptozotocin in the obese-hyperglycemic (ob/ob) mouse. Apparent lack of a direct relationship between insulin binding and insulin effects. Diabetes 1977, 26, 582–590. [Google Scholar] [CrossRef]
- Zick, Y. Role of Ser/Thr kinases in the uncoupling of insulin signaling. Int. J. Obes. Relat. Metab. Disord. 2003, 27 (Suppl. 3), S56–S60. [Google Scholar] [CrossRef]
- Zick, Y.; Grunberger, G.; Podskalny, J.M.; Moncada, V.; Taylor, S.I.; Gorden, P.; Roth, J. Insulin stimulates phosphorylation of serine residues in soluble insulin receptors. Biochem. Biophys. Res. Commun. 1983, 116, 1129–1135. [Google Scholar] [CrossRef]
- Kusari, J.; Kenner, K.A.; Suh, K.I.; Hill, D.E.; Henry, R.R. Skeletal muscle protein tyrosine phosphatase activity and tyrosine phosphatase 1B protein content are associated with insulin action and resistance. J. Clin. Investig. 1994, 93, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Krebs, D.L.; Hilton, D.J. A new role for SOCS in insulin action. Suppressor of cytokine signaling. Sci. STKE 2003, 2003, PE6. [Google Scholar] [CrossRef] [PubMed]
- Zick, Y. Insulin resistance: A phosphorylation-based uncoupling of insulin signaling. Trends Cell Biol. 2001, 11, 437–441. [Google Scholar] [CrossRef]
- Ha, J.; Satin, L.S.; Sherman, A.S. A Mathematical Model of the Pathogenesis, Prevention, and Reversal of Type 2 Diabetes. Endocrinology 2016, 157, 624–635. [Google Scholar] [CrossRef]
- Corkey, B.E. Diabetes: Have we got it all wrong? Insulin hypersecretion and food additives: Cause of obesity and diabetes? Diabetes Care 2012, 35, 2432–2437. [Google Scholar] [CrossRef]
- Ramlo-Halsted, B.A.; Edelman, S.V. The natural history of type 2 diabetes. Implications for clinical practice. Prim. Care 1999, 26, 771–789. [Google Scholar] [CrossRef]
- Billings, L.K.; Florez, J.C. The genetics of type 2 diabetes: What have we learned from GWAS? Ann. N. Y. Acad. Sci. 2010, 1212, 59–77. [Google Scholar] [CrossRef]
- Ferrannini, E.; Gastaldelli, A.; Miyazaki, Y.; Matsuda, M.; Mari, A.; DeFronzo, R.A. beta-Cell function in subjects spanning the range from normal glucose tolerance to overt diabetes: A new analysis. J. Clin. Endocrinol. Metab. 2005, 90, 493–500. [Google Scholar] [CrossRef]
- Pories, W.J.; Dohm, G.L. Diabetes: Have we got it all wrong? Hyperinsulinism as the culprit: Surgery provides the evidence. Diabetes Care 2012, 35, 2438–2442. [Google Scholar] [CrossRef]
- Lustig, R.H. Which comes first? The obesity or the insulin? The behavior or the biochemistry? J. Pediatrics 2008, 152, 601–602. [Google Scholar] [CrossRef]
- Hansen, B.C.; Bodkin, N.L. Beta-cell hyperresponsiveness: Earliest event in development of diabetes in monkeys. Am. J. Physiol. 1990, 259, R612–R617. [Google Scholar] [CrossRef]
- Sokooti, S.; Kieneker, L.M.; Borst, M.H.; Muller Kobold, A.; Kootstra-Ros, J.E.; Gloerich, J.; van Gool, A.J.; Heerspink, H.J.L.; Gansevoort, R.T.; Dullaart, R.P.F.; et al. Plasma C-Peptide and Risk of Developing Type 2 Diabetes in the General Population. J. Clin. Med. 2020, 9, 1. [Google Scholar] [CrossRef]
- Crofts, C.; Schofield, G.; Zinn, C.; Wheldon, M.; Kraft, J. Identifying hyperinsulinaemia in the absence of impaired glucose tolerance: An examination of the Kraft database. Diabetes Res. Clin. Pract. 2016, 118, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Weyer, C.; Hanson, R.L.; Tataranni, P.A.; Bogardus, C.; Pratley, R.E. A high fasting plasma insulin concentration predicts type 2 diabetes independent of insulin resistance: Evidence for a pathogenic role of relative hyperinsulinemia. Diabetes 2000, 49, 2094–2101. [Google Scholar] [CrossRef]
- Ferrannini, E.; Natali, A.; Bell, P.; Cavallo-Perin, P.; Lalic, N.; Mingrone, G. Insulin resistance and hypersecretion in obesity. European Group for the Study of Insulin Resistance (EGIR). J. Clin. Investig. 1997, 100, 1166–1173. [Google Scholar] [CrossRef]
- Mari, A.; Tura, A.; Natali, A.; Anderwald, C.; Balkau, B.; Lalic, N.; Walker, M.; Ferrannini, E.; Investigators, R. Influence of hyperinsulinemia and insulin resistance on in vivo beta-cell function: Their role in human beta-cell dysfunction. Diabetes 2011, 60, 3141–3147. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, D.J.; Guilherme, A.; Danai, L.V.; Heyda, L.; Matevossian, A.; Cohen, J.; Nicoloro, S.M.; Straubhaar, J.; Noh, H.L.; Jung, D.; et al. A major role of insulin in promoting obesity-associated adipose tissue inflammation. Mol. Metab. 2015, 4, 507–518. [Google Scholar] [CrossRef]
- Nolan, C.J.; Prentki, M. Insulin resistance and insulin hypersecretion in the metabolic syndrome and type 2 diabetes: Time for a conceptual framework shift. Diab. Vasc. Dis. Res. 2019, 16, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; Corkey, B.E.; Istfan, N.W.; Apovian, C.M. Hyperinsulinemia: An Early Indicator of Metabolic Dysfunction. J. Endocr. Soc. 2019, 3, 1727–1747. [Google Scholar] [CrossRef]
- Baier, L.J.; Hanson, R.L. Genetic studies of the etiology of type 2 diabetes in Pima Indians: Hunting for pieces to a complicated puzzle. Diabetes 2004, 53, 1181–1186. [Google Scholar] [CrossRef]
- Schousboe, K.; Visscher, P.M.; Henriksen, J.E.; Hopper, J.L.; Sorensen, T.I.; Kyvik, K.O. Twin study of genetic and environmental influences on glucose tolerance and indices of insulin sensitivity and secretion. Diabetologia 2003, 46, 1276–1283. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Chen, X. Effects of fatty acids and ketone bodies on basal insulin secretion in type 2 diabetes. Diabetes 1999, 48, 577–583. [Google Scholar] [CrossRef]
- Saadeh, M.; Ferrante, T.C.; Kane, A.; Shirihai, O.; Corkey, B.E.; Deeney, J.T. Reactive oxygen species stimulate insulin secretion in rat pancreatic islets: Studies using mono-oleoyl-glycerol. PLoS ONE 2012, 7, e30200. [Google Scholar] [CrossRef]
- Malaisse, W.J.; Vanonderbergen, A.; Louchami, K.; Jijakli, H.; Malaisse-Lagae, F. Effects of artificial sweeteners on insulin release and cationic fluxes in rat pancreatic islets. Cell Signal. 1998, 10, 727–733. [Google Scholar] [CrossRef]
- Pi, J.; Bai, Y.; Zhang, Q.; Wong, V.; Floering, L.M.; Daniel, K.; Reece, J.M.; Deeney, J.T.; Andersen, M.E.; Corkey, B.E.; et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 2007, 56, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Nair, K.S.; Short, K.R. Hormonal and signaling role of branched-chain amino acids. J. Nutr. 2005, 135, 1547S–1552S. [Google Scholar] [CrossRef]
- Van Loon, L.J.; Kruijshoop, M.; Menheere, P.P.; Wagenmakers, A.J.; Saris, W.H.; Keizer, H.A. Amino acid ingestion strongly enhances insulin secretion in patients with long-term type 2 diabetes. Diabetes Care 2003, 26, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P. Estimation of beta-cell mass by metabolic tests: Necessary, but how sufficient? Diabetes 2007, 56, 2420–2424. [Google Scholar] [CrossRef][Green Version]
- Chmurzynska, A. Fetal programming: Link between early nutrition, DNA methylation, and complex diseases. Nutr. Rev. 2010, 68, 87–98. [Google Scholar] [CrossRef]
- Portha, B.; Chavey, A.; Movassat, J. Early-life origins of type 2 diabetes: Fetal programming of the beta-cell mass. Exp. Diabetes Res. 2011, 2011, 105076. [Google Scholar] [CrossRef] [PubMed]
- Mitrani, P.; Srinivasan, M.; Dodds, C.; Patel, M.S. Autonomic involvement in the permanent metabolic programming of hyperinsulinemia in the high-carbohydrate rat model. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1364–E1377. [Google Scholar] [CrossRef][Green Version]
- Fenichel, P.; Chevalier, N. Environmental endocrine disruptors: New diabetogens? Comptes Rendus Biol. 2017, 340, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Indumathi, D.; Jayashree, S.; Selvaraj, J.; Sathish, S.; Mayilvanan, C.; Akilavalli, N.; Balasubramanian, K. Effect of bisphenol-A on insulin signal transduction and glucose oxidation in skeletal muscle of adult male albino rat. Hum. Exp. Toxicol. 2013, 32, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Templeman, N.M.; Skovso, S.; Page, M.M.; Lim, G.E.; Johnson, J.D. A causal role for hyperinsulinemia in obesity. J. Endocrinol. 2017, 232, R173–R183. [Google Scholar] [CrossRef]
- Polidori, D.C.; Bergman, R.N.; Chung, S.T.; Sumner, A.E. Hepatic and Extrahepatic Insulin Clearance Are Differentially Regulated: Results From a Novel Model-Based Analysis of Intravenous Glucose Tolerance Data. Diabetes 2016, 65, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Piccinini, F.; Polidori, D.C.; Gower, B.A.; Bergman, R.N. Hepatic but Not Extrahepatic Insulin Clearance Is Lower in African American Than in European American Women. Diabetes 2017, 66, 2564–2570. [Google Scholar] [CrossRef]
- Bojsen-Moller, K.N.; Lundsgaard, A.M.; Madsbad, S.; Kiens, B.; Holst, J.J. Hepatic Insulin Clearance in Regulation of Systemic Insulin Concentrations-Role of Carbohydrate and Energy Availability. Diabetes 2018, 67, 2129–2136. [Google Scholar] [CrossRef]
- Guo, X.; Cui, J.; Jones, M.R.; Haritunians, T.; Xiang, A.H.; Chen, Y.D.; Taylor, K.D.; Buchanan, T.A.; Davis, R.C.; Hsueh, W.A.; et al. Insulin clearance: Confirmation as a highly heritable trait, and genome-wide linkage analysis. Diabetologia 2012, 55, 2183–2192. [Google Scholar] [CrossRef]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [PubMed]
- Fosam, A.; Sikder, S.; Abel, B.S.; Tella, S.H.; Walter, M.F.; Mari, A.; Muniyappa, R. Reduced Insulin Clearance and Insulin-Degrading Enzyme Activity Contribute to Hyperinsulinemia in African Americans. J. Clin. Endocrinol. Metab. 2020, 105. [Google Scholar] [CrossRef] [PubMed]
- Borges, D.O.; Patarrao, R.S.; Ribeiro, R.T.; de Oliveira, R.M.; Duarte, N.; Belew, G.D.; Martins, M.; Andrade, R.; Costa, J.; Correia, I.; et al. Loss of postprandial insulin clearance control by Insulin-degrading enzyme drives dysmetabolism traits. Metabolism 2021, 118, 154735. [Google Scholar] [CrossRef] [PubMed]
- Preeyasombat, C.; Bacchetti, P.; Lazar, A.A.; Lustig, R.H. Racial and etiopathologic dichotomies in insulin hypersecretion and resistance in obese children. J. Pediatrics 2005, 146, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Astley, C.M.; Todd, J.N.; Salem, R.M.; Vedantam, S.; Ebbeling, C.B.; Huang, P.L.; Ludwig, D.S.; Hirschhorn, J.N.; Florez, J.C. Genetic Evidence That Carbohydrate-Stimulated Insulin Secretion Leads to Obesity. Clin. Chem. 2018, 64, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Le Stunff, C.; Bougneres, P. Early changes in postprandial insulin secretion, not in insulin sensitivity, characterize juvenile obesity. Diabetes 1994, 43, 696–702. [Google Scholar] [CrossRef]
- Templeman, N.M.; Clee, S.M.; Johnson, J.D. Suppression of hyperinsulinaemia in growing female mice provides long-term protection against obesity. Diabetologia 2015, 58, 2392–2402. [Google Scholar] [CrossRef]
- Rajan, S.; Shankar, K.; Beg, M.; Varshney, S.; Gupta, A.; Srivastava, A.; Kumar, D.; Mishra, R.K.; Hussain, Z.; Gayen, J.R.; et al. Chronic hyperinsulinemia reduces insulin sensitivity and metabolic functions of brown adipocyte. J. Endocrinol. 2016, 230, 275–290. [Google Scholar] [CrossRef]
- Trico, D.; Natali, A.; Arslanian, S.; Mari, A.; Ferrannini, E. Identification, pathophysiology, and clinical implications of primary insulin hypersecretion in nondiabetic adults and adolescents. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Sicree, R.A.; Zimmet, P.Z.; King, H.O.; Coventry, J.S. Plasma insulin response among Nauruans. Prediction of deterioration in glucose tolerance over 6 yr. Diabetes 1987, 36, 179–186. [Google Scholar] [CrossRef]
- Saad, M.F.; Knowler, W.C.; Pettitt, D.J.; Nelson, R.G.; Charles, M.A.; Bennett, P.H. A two-step model for development of non-insulin-dependent diabetes. Am. J. Med. 1991, 90, 229–235. [Google Scholar] [CrossRef]
- Haffner, S.M.; Stern, M.P.; Mitchell, B.D.; Hazuda, H.P.; Patterson, J.K. Incidence of type II diabetes in Mexican Americans predicted by fasting insulin and glucose levels, obesity, and body-fat distribution. Diabetes 1990, 39, 283–288. [Google Scholar] [CrossRef]
- Zimmet, P.Z.; Collins, V.R.; Dowse, G.K.; Knight, L.T. Hyperinsulinaemia in youth is a predictor of type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia 1992, 35, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Charles, M.A.; Fontbonne, A.; Thibult, N.; Warnet, J.M.; Rosselin, G.E.; Eschwege, E. Risk factors for NIDDM in white population. Paris prospective study. Diabetes 1991, 40, 796–799. [Google Scholar] [CrossRef]
- Spadaro, L.; Alagona, C.; Palermo, F.; Piro, S.; Calanna, S.; Parrinello, G.; Purrello, F.; Rabuazzo, A.M. Early phase insulin secretion is increased in subjects with normal fasting glucose and metabolic syndrome: A premature feature of beta-cell dysfunction. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Dankner, R.; Chetrit, A.; Shanik, M.H.; Raz, I.; Roth, J. Basal state hyperinsulinemia in healthy normoglycemic adults heralds dysglycemia after more than two decades of follow up. Diabetes Metab. Res. Rev. 2012, 28, 618–624. [Google Scholar] [CrossRef]
- Zavaroni, I.; Bonini, L.; Gasparini, P.; Barilli, A.L.; Zuccarelli, A.; Dall’Aglio, E.; Delsignore, R.; Reaven, G.M. Hyperinsulinemia in a normal population as a predictor of non-insulin-dependent diabetes mellitus, hypertension, and coronary heart disease: The Barilla factory revisited. Metabolism 1999, 48, 989–994. [Google Scholar] [CrossRef]
- Yaghootkar, H.; Scott, R.A.; White, C.C.; Zhang, W.; Speliotes, E.; Munroe, P.B.; Ehret, G.B.; Bis, J.C.; Fox, C.S.; Walker, M.; et al. Genetic evidence for a normal-weight “metabolically obese” phenotype linking insulin resistance, hypertension, coronary artery disease, and type 2 diabetes. Diabetes 2014, 63, 4369–4377. [Google Scholar] [CrossRef] [PubMed]
- Zavaroni, I.; Bonora, E.; Pagliara, M.; Dall’Aglio, E.; Luchetti, L.; Buonanno, G.; Bonati, P.A.; Bergonzani, M.; Gnudi, L.; Passeri, M.; et al. Risk factors for coronary artery disease in healthy persons with hyperinsulinemia and normal glucose tolerance. N. Engl. J. Med. 1989, 320, 702–706. [Google Scholar] [CrossRef]
- Pyorala, K. Relationship of glucose tolerance and plasma insulin to the incidence of coronary heart disease: Results from two population studies in Finland. Diabetes Care 1979, 2, 131–141. [Google Scholar] [CrossRef]
- Welborn, T.A.; Wearne, K. Coronary heart disease incidence and cardiovascular mortality in Busselton with reference to glucose and insulin concentrations. Diabetes Care 1979, 2, 154–160. [Google Scholar] [CrossRef]
- Eschwege, E.; Richard, J.L.; Thibult, N.; Ducimetiere, P.; Warnet, J.M.; Claude, J.R.; Rosselin, G.E. Coronary heart disease mortality in relation with diabetes, blood glucose and plasma insulin levels. The Paris Prospective Study, ten years later. Horm. Metab. Res. Suppl. 1985, 15, 41–46. [Google Scholar]
- Stout, R.W. Hyperinsulinemia and atherosclerosis. Diabetes 1996, 45 (Suppl. 3), S45–S46. [Google Scholar] [CrossRef] [PubMed]
- Stout, R.W.; Bierman, E.L.; Ross, R. Effect of insulin on the proliferation of cultured primate arterial smooth muscle cells. Circ. Res. 1975, 36, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Pfeifle, B.; Ditschuneit, H. Effect of insulin on growth of cultured human arterial smooth muscle cells. Diabetologia 1981, 20, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Nakao, J.; Ito, H.; Kanayasu, T.; Murota, S. Stimulatory effect of insulin on aortic smooth muscle cell migration induced by 12-L-hydroxy-5,8,10,14-eicosatetraenoic acid and its modulation by elevated extracellular glucose levels. Diabetes 1985, 34, 185–191. [Google Scholar] [CrossRef]
- Young, I.R.; Stout, R.W. Effects of insulin and glucose on the cells of the arterial wall: Interaction of insulin with dibutyryl cyclic AMP and low density lipoprotein in arterial cells. Diabete Metab. 1987, 13, 301–306. [Google Scholar]
- Oppenheimer, M.J.; Sundquist, K.; Bierman, E.L. Downregulation of high-density lipoprotein receptor in human fibroblasts by insulin and IGF-I. Diabetes 1989, 38, 117–122. [Google Scholar] [CrossRef]
- Sasso, F.C.; Pafundi, P.C.; Marfella, R.; Calabro, P.; Piscione, F.; Furbatto, F.; Esposito, G.; Galiero, R.; Gragnano, F.; Rinaldi, L.; et al. Adiponectin and insulin resistance are related to restenosis and overall new PCI in subjects with normal glucose tolerance: The prospective AIRE Study. Cardiovasc. Diabetol. 2019, 18, 24. [Google Scholar] [CrossRef] [PubMed]
- Vollenweider, P.; Randin, D.; Tappy, L.; Jequier, E.; Nicod, P.; Scherrer, U. Impaired insulin-induced sympathetic neural activation and vasodilation in skeletal muscle in obese humans. J. Clin. Investig. 1994, 93, 2365–2371. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, P.; Weidmann, P. Insulin, insulin sensitivity and hypertension. J. Hypertens. 1990, 8, 491–500. [Google Scholar] [CrossRef]
- Medina-Santillan, R.; Lopez-Velazquez, J.A.; Chavez-Tapia, N.; Torres-Villalobos, G.; Uribe, M.; Mendez-Sanchez, N. Hepatic manifestations of metabolic syndrome. Diabetes Metab. Res. Rev. 2013. [Google Scholar] [CrossRef]
- Fruehwald-Schultes, B.; Kern, W.; Born, J.; Fehm, H.L.; Peters, A. Hyperinsulinemia causes activation of the hypothalamus-pituitary-adrenal axis in humans. Int. J. Obes. Relat. Metab. Disord. 2001, 25 (Suppl. 1), S38–S40. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Godsland, I.F. Insulin resistance and hyperinsulinaemia in the development and progression of cancer. Clin. Sci. 2009, 118, 315–332. [Google Scholar] [CrossRef] [PubMed]
- Pisani, P. Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Arch. Physiol. Biochem. 2008, 114, 63–70. [Google Scholar] [CrossRef]
- Kabat, G.C.; Kim, M.; Caan, B.J.; Chlebowski, R.T.; Gunter, M.J.; Ho, G.Y.; Rodriguez, B.L.; Shikany, J.M.; Strickler, H.D.; Vitolins, M.Z.; et al. Repeated measures of serum glucose and insulin in relation to postmenopausal breast cancer. Int. J. Cancer 2009, 125, 2704–2710. [Google Scholar] [CrossRef]
- Vigneri, R.; Goldfine, I.D.; Frittitta, L. Insulin, insulin receptors, and cancer. J. Endocrinol. Investig. 2016, 39, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, A.; Malaguarnera, R. Insulin receptor and cancer. Endocr. Relat. Cancer 2011, 18, R125–R147. [Google Scholar] [CrossRef] [PubMed]
- Frystyk, J. Free insulin-like growth factors—Measurements and relationships to growth hormone secretion and glucose homeostasis. Growth Horm. IGF Res. 2004, 14, 337–375. [Google Scholar] [CrossRef]
- Arcidiacono, B.; Iiritano, S.; Nocera, A.; Possidente, K.; Nevolo, M.T.; Ventura, V.; Foti, D.; Chiefari, E.; Brunetti, A. Insulin resistance and cancer risk: An overview of the pathogenetic mechanisms. Exp. Diabetes Res. 2012, 2012, 789174. [Google Scholar] [CrossRef]
- Janssen, J.A.; Lamberts, S.W. Insulin-like growth factor-I and risk of breast cancer. Lancet 1998, 352, 490. [Google Scholar] [CrossRef]
- Gallagher, E.J.; LeRoith, D. Hyperinsulinaemia in cancer. Nat. Rev. Cancer 2020, 20, 629–644. [Google Scholar] [CrossRef]
- Janssen, J.A.; Lamberts, S.W. Igf-I and longevity. Horm. Res. 2004, 62 (Suppl. 3), 104–109. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Facchini, F.S.; Hua, N.W.; Reaven, G.M.; Stoohs, R.A. Hyperinsulinemia: The missing link among oxidative stress and age-related diseases? Free Radic. Biol. Med. 2000, 29, 1302–1306. [Google Scholar] [CrossRef]
- Broughton, S.J.; Piper, M.D.; Ikeya, T.; Bass, T.M.; Jacobson, J.; Driege, Y.; Martinez, P.; Hafen, E.; Withers, D.J.; Leevers, S.J.; et al. Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proc. Natl. Acad. Sci. USA 2005, 102, 3105–3110. [Google Scholar] [CrossRef]
- Parr, T. Insulin exposure and aging theory. Gerontology 1997, 43, 182–200. [Google Scholar] [CrossRef] [PubMed]
- Parr, T. Insulin exposure controls the rate of mammalian aging. Mech. Ageing Dev. 1996, 88, 75–82. [Google Scholar] [CrossRef]
- Vitale, G.; Brugts, M.P.; Ogliari, G.; Castaldi, D.; Fatti, L.M.; Varewijck, A.J.; Lamberts, S.W.; Monti, D.; Bucci, L.; Cevenini, E.; et al. Low circulating IGF-I bioactivity is associated with human longevity: Findings in centenarians’ offspring. Aging 2012, 4, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Masoro, E.J. Food restriction and the aging process. J. Am. Geriatr. Soc. 1984, 32, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.S.; Lane, M.A.; Ingram, D.K.; Mattison, J.A.; Elahi, D.; Tobin, J.D.; Muller, D.; Metter, E.J. Biomarkers of caloric restriction may predict longevity in humans. Science 2002, 297, 811. [Google Scholar] [CrossRef] [PubMed]
- Petruzzi, E.; Pinzani, P.; Orlando, C.; Poggesi, M.; Monami, M.; Pazzagli, M.; Masotti, G. Healthy centenarian subjects as living model of “successful aging”: A study on fasting glycemia, C-peptide, dehydroepiandrosterone sulphate (DHEAS) and insulin-like growth factor (IGF-1). Arch. Gerontol. Geriatr. Suppl. 2002, 8, 273–278. [Google Scholar] [CrossRef]
- Paolisso, G.; Gambardella, A.; Ammendola, S.; D’Amore, A.; Balbi, V.; Varricchio, M.; D’Onofrio, F. Glucose tolerance and insulin action in healthy centenarians. Am. J. Physiol. 1996, 270, E890–E894. [Google Scholar] [CrossRef]
- Crofts, C.; Zinn, C.; Wheldon, M.; Schofield, G. Hyperinsulinemia: Best management practice. Diabesity 2016, 2, 1–11. [Google Scholar] [CrossRef][Green Version]
- Bosello, O.; Zamboni, M.; Armellini, F.; Zocca, I.; Bergamo Andreis, I.A.; Smacchia, C.; Milani, M.P.; Cominacini, L. Modifications of abdominal fat and hepatic insulin clearance during severe caloric restriction. Ann. Nutr. Metab. 1990, 34, 359–365. [Google Scholar] [CrossRef]
- Bojsen-Moller, K.N.; Dirksen, C.; Jorgensen, N.B.; Jacobsen, S.H.; Serup, A.K.; Albers, P.H.; Hansen, D.L.; Worm, D.; Naver, L.; Kristiansen, V.B.; et al. Early enhancements of hepatic and later of peripheral insulin sensitivity combined with increased postprandial insulin secretion contribute to improved glycemic control after Roux-en-Y gastric bypass. Diabetes 2014, 63, 1725–1737. [Google Scholar] [CrossRef]
- Tiikkainen, M.; Hakkinen, A.M.; Korsheninnikova, E.; Nyman, T.; Makimattila, S.; Yki-Jarvinen, H. Effects of rosiglitazone and metformin on liver fat content, hepatic insulin resistance, insulin clearance, and gene expression in adipose tissue in patients with type 2 diabetes. Diabetes 2004, 53, 2169–2176. [Google Scholar] [CrossRef]
- Sutton, E.F.; Beyl, R.; Early, K.S.; Cefalu, W.T.; Ravussin, E.; Peterson, C.M. Early Time-Restricted Feeding Improves Insulin Sensitivity, Blood Pressure, and Oxidative Stress Even without Weight Loss in Men with Prediabetes. Cell Metab. 2018, 27, 1212–1221.e13. [Google Scholar] [CrossRef]
- Colberg, S.R.; Sigal, R.J.; Fernhall, B.; Regensteiner, J.G.; Blissmer, B.J.; Rubin, R.R.; Chasan-Taber, L.; Albright, A.L.; Braun, B.; American College of Sports, M.; et al. Exercise and type 2 diabetes: The American College of Sports Medicine and the American Diabetes Association: Joint position statement. Diabetes Care 2010, 33, e147–e167. [Google Scholar] [CrossRef]
- Richard, D.; Labrie, A.; Lupien, D.; Tremblay, A.; LeBlanc, J. Role of exercise-training in the prevention of hyperinsulinemia caused by high energy diet. J. Nutr. 1982, 112, 1756–1762. [Google Scholar] [CrossRef] [PubMed]
- Teich, T.; Zaharieva, D.P.; Riddell, M.C. Advances in Exercise, Physical Activity, and Diabetes Mellitus. Diabetes Technol. Ther. 2019, 21, S112–S122. [Google Scholar] [CrossRef] [PubMed]
- Wake, A.D. Antidiabetic Effects of Physical Activity: How It Helps to Control Type 2 Diabetes. Diabetes Metab. Syndr. Obes. 2020, 13, 2909–2923. [Google Scholar] [CrossRef] [PubMed]
- Heiskanen, M.A.; Motiani, K.K.; Mari, A.; Saunavaara, V.; Eskelinen, J.J.; Virtanen, K.A.; Koivumaki, M.; Loyttyniemi, E.; Nuutila, P.; Kalliokoski, K.K.; et al. Exercise training decreases pancreatic fat content and improves beta cell function regardless of baseline glucose tolerance: A randomised controlled trial. Diabetologia 2018, 61, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Leissring, M.A.; Gonzalez-Casimiro, C.M.; Merino, B.; Suire, C.N.; Perdomo, G. Targeting Insulin-Degrading Enzyme in Insulin Clearance. Int. J. Mol. Sci. 2021, 22, 2235. [Google Scholar] [CrossRef] [PubMed]
- Alemzadeh, R.; Slonim, A.E.; Zdanowicz, M.M.; Maturo, J. Modification of insulin resistance by diazoxide in obese Zucker rats. Endocrinology 1993, 133, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, T.; Taguchi, N.; Sato, Y.; Nakabayashi, T.; Kobuchi, H.; Hidaka, H.; Nagasawa, T.; Ishihara, F.; Itoh, N.; Hashizume, K. Prophylaxis of genetically determined diabetes by diazoxide: A study in a rat model of naturally occurring obese diabetes. J. Pharmacol. Exp. Ther. 1995, 275, 194–199. [Google Scholar] [PubMed]
- Alemzadeh, R.; Langley, G.; Upchurch, L.; Smith, P.; Slonim, A.E. Beneficial effect of diazoxide in obese hyperinsulinemic adults. J. Clin. Endocrinol. Metab. 1998, 83, 1911–1915. [Google Scholar] [CrossRef]
- Velasquez-Mieyer, P.A.; Cowan, P.A.; Arheart, K.L.; Buffington, C.K.; Spencer, K.A.; Connelly, B.E.; Cowan, G.W.; Lustig, R.H. Suppression of insulin secretion is associated with weight loss and altered macronutrient intake and preference in a subset of obese adults. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 219–226. [Google Scholar] [CrossRef]
- Hundal, R.S.; Krssak, M.; Dufour, S.; Laurent, D.; Lebon, V.; Chandramouli, V.; Inzucchi, S.E.; Schumann, W.C.; Petersen, K.F.; Landau, B.R.; et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes 2000, 49, 2063–2069. [Google Scholar] [CrossRef]
- Morgillo, F.; Fasano, M.; Della Corte, C.M.; Sasso, F.C.; Papaccio, F.; Viscardi, G.; Esposito, G.; Di Liello, R.; Normanno, N.; Capuano, A.; et al. Results of the safety run-in part of the METAL (METformin in Advanced Lung cancer) study: A multicentre, open-label phase I-II study of metformin with erlotinib in second-line therapy of patients with stage IV non-small-cell lung cancer. ESMO Open 2017, 2, e000132. [Google Scholar] [CrossRef]
- Kalra, S.; Gupta, Y.; Patil, S. Sodium-glucose cotransporter-2 inhibition and the insulin: Glucagon ratio: Unexplored dimensions. Indian J. Endocrinol. Metab. 2015, 19, 426–429. [Google Scholar] [CrossRef]
- Ferrannini, E. Sodium-Glucose Co-transporters and Their Inhibition: Clinical Physiology. Cell Metab. 2017, 26, 27–38. [Google Scholar] [CrossRef]
- Ritzel, R.; Schulte, M.; Porksen, N.; Nauck, M.S.; Holst, J.J.; Juhl, C.; Marz, W.; Schmitz, O.; Schmiegel, W.H.; Nauck, M.A. Glucagon-like peptide 1 increases secretory burst mass of pulsatile insulin secretion in patients with type 2 diabetes and impaired glucose tolerance. Diabetes 2001, 50, 776–784. [Google Scholar] [CrossRef]
- Nagahisa, T.; Saisho, Y. Cardiorenal Protection: Potential of SGLT2 Inhibitors and GLP-1 Receptor Agonists in the Treatment of Type 2 Diabetes. Diabetes Ther. 2019, 10, 1733–1752. [Google Scholar] [CrossRef] [PubMed]
- Wahren, J.; Kallas, A. Loss of pulsatile insulin secretion: A factor in the pathogenesis of type 2 diabetes? Diabetes 2012, 61, 2228–2229. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mirbolooki, M.R.; Taylor, G.E.; Knutzen, V.K.; Scharp, D.W.; Willcourt, R.; Lakey, J.R. Pulsatile intravenous insulin therapy: The best practice to reverse diabetes complications? Med. Hypotheses 2009, 73, 363–369. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time Course | Major Fuel Used | Hormonal Control |
|---|---|---|
| Immediate Postprandial Period | Stimulation of glucose uptake Stimulation of glycogenesis Stimulation of amino acid uptake Stimulation of free fatty acid uptake Stimulation of lipogenesis | Insulin |
| Early Postprandial Period | Stimulation of amino acid uptake Stimulation of protein synthesis Reduction in insulin resistance Suppression of proteolysis | Insulin, GH + IGF-I |
| Late Postprandial Period | Reduction in insulin resistance Inhibiting of insulin release Stimulating of lipolysis Stimulating lipid oxidation Reduction in protein oxidation | IGF-I IGF-I IGF-I (indirectly) by reducing insulin secretion GH + IGF-I |
| Reference No | Type of Study | |
|---|---|---|
| Obesity | 4 | Prospective, observational |
| 5 | Prospective, observational | |
| 116 | Prospective, observational | |
| Impaired Glucose Tolerance | 119 | Prospective, observational |
| 126 | Prospective, observational | |
| 127 | Prospective, observational | |
| Type 2 Diabetes | 9 | Prospective, observational |
| 84 | Prospective, observational | |
| 86 | Prospective, observational | |
| 120 | Prospective, observational | |
| 124 | Prospective, observational | |
| 127 | Prospective, observational | |
| Cardiovascular Disease | 1 | Prospective, observational |
| 2 | Prospective, observational | |
| 3 | Prospective, observational | |
| 130 | Prospective, observational | |
| 131 | Prospective, observational | |
| 132 | Prospective, observational | |
| Cancer | ||
| Liver Cancer | 6 | Prospective, observational |
| Colorectal Cancer | 8 | Prospective, observational |
| Colorectal Cancer | 145 | Prospective, observational |
| Pancreas | 145 | Prospective, observational |
| Breast Cancer | 146 | Prospective, observational |
| Cancer Mortality | 7 | Prospective, observational |
| Decreased Survival | 164 | Prospective, observational |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janssen, J.A.M.J.L. Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. Int. J. Mol. Sci. 2021, 22, 7797. https://doi.org/10.3390/ijms22157797
Janssen JAMJL. Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. International Journal of Molecular Sciences. 2021; 22(15):7797. https://doi.org/10.3390/ijms22157797
Chicago/Turabian StyleJanssen, Joseph A. M. J. L. 2021. "Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer" International Journal of Molecular Sciences 22, no. 15: 7797. https://doi.org/10.3390/ijms22157797
APA StyleJanssen, J. A. M. J. L. (2021). Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. International Journal of Molecular Sciences, 22(15), 7797. https://doi.org/10.3390/ijms22157797