
TDP-43 and Inflammation: Implications for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia


Abstract
1. Introduction
1.1. Neuroinflammation
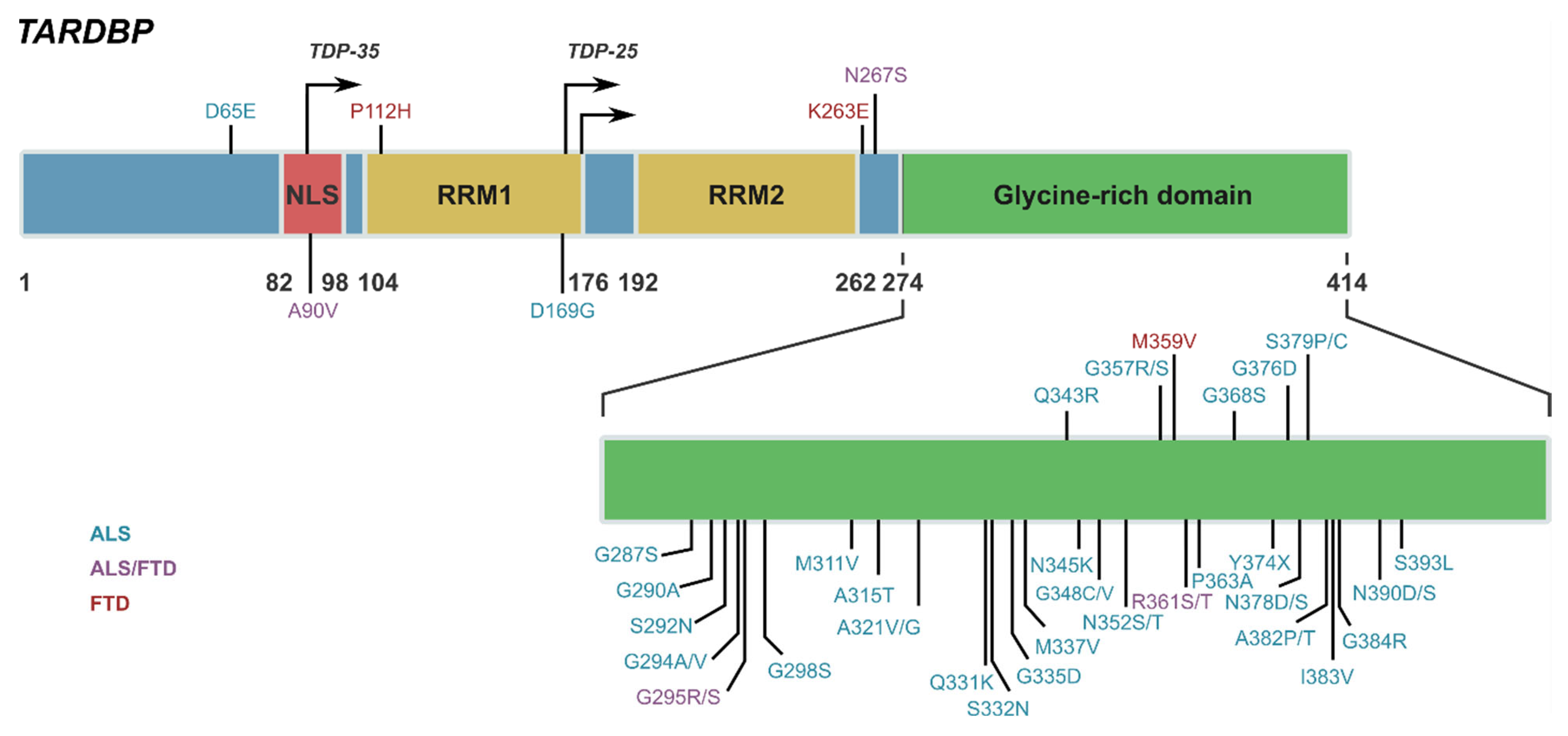
1.2. TAR DNA Binding Protein 43 (TDP-43)
2. ALS and FTD Causative and Susceptibility Genes Associated with TDP-43 Implicated in Immunity and Inflammation
3. Chromosome Open Reading Frame 72 (C9orf72)
4. Granulin (GRN)
5. TANK Binding Kinase 1 (TBK1)
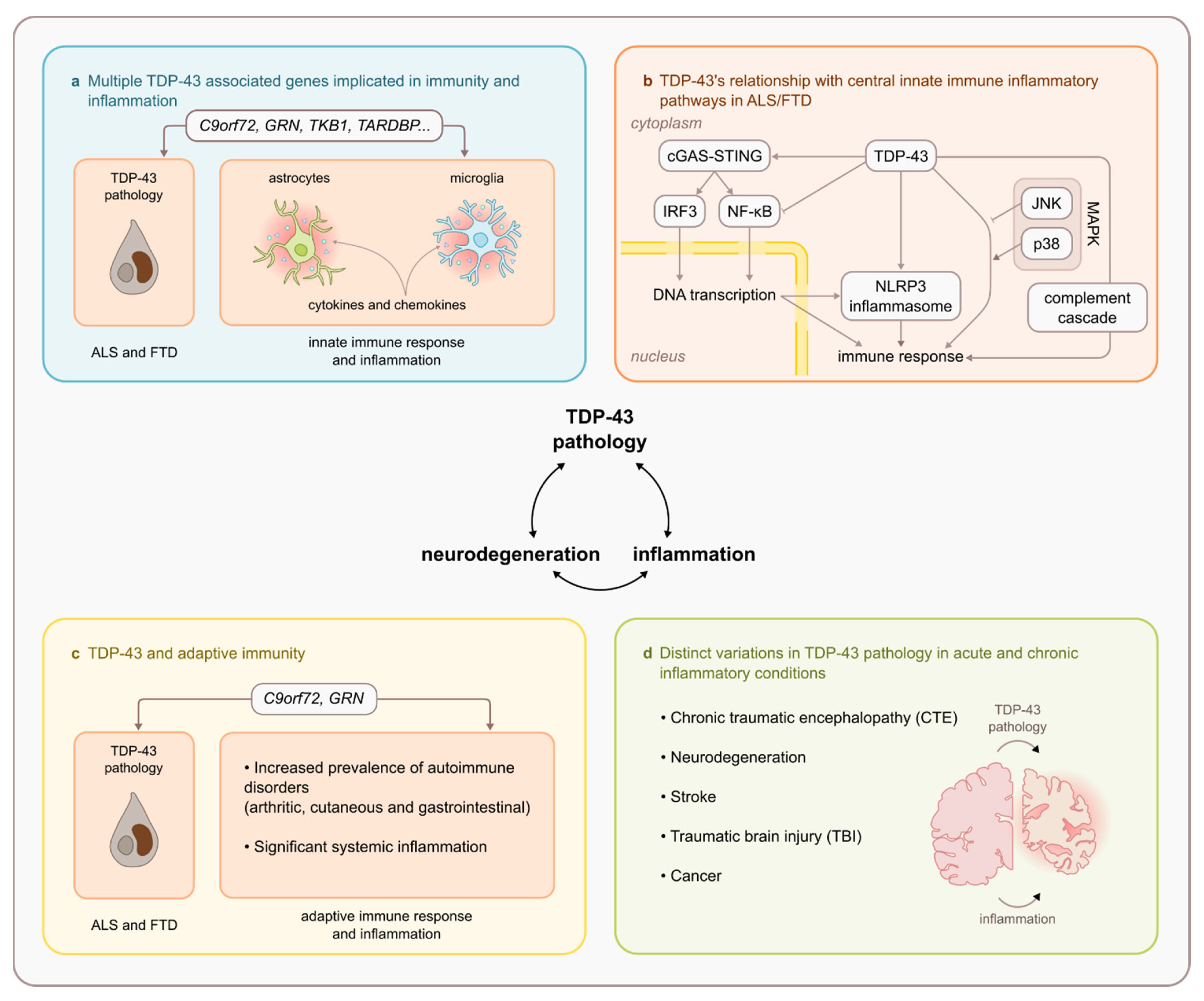
6. Relationship between TDP-43 and Key Innate Immune Inflammatory Pathways
6.1. TDP-43 and NF-κβ/p65
6.2. TDP-43 and cGAS/STING Pathway
6.3. TDP43 and NLRP3 Inflammasome
6.4. TDP43 and MAPK Pathway
6.5. TDP-43 and Complement Cascade
7. TDP-43 and Adaptive Immunity
8. Presence of TDP-43 in Other Acute and Chronic Neuroinflammatory Conditions
8.1. TDP43 and Stroke
8.2. TDP-43, Traumatic Brain Injury and Chronic Traumatic Encephalopathy
9. Conclusions
- The involvement of various ALS and FTD causative and susceptibility genes (notably C9orf72, GRN, and TBK1) in immunity and inflammation.
- A demonstrated relationship between TDP-43 and central innate immune inflammatory pathways including NF-κβ/p65, cGAS/STING, NLRP3 inflammasome, MAPK/JNK/p38, and the innate immune complement cascade.
- Altered adaptive immunity in ALS and FTD that is intrinsically linked to TDP-43 pathophysiology (notably in C9orf72 repeat expansion and GRN mutation carriers)
- TDP-43 proteinopathy is observed in other acute and chronic inflammatory CNS conditions (notably stroke, TBI and CTE).
- Investigation into the substantial amount of evidence supporting TDP-43’s relationship with immunity and inflammation that centres around microglia.
- Determining the manner and involvement of TDP-43 structural and functional sites (e.g., NLS and RRM1 domain) with key inflammatory pathways (e.g., NF-κβ/p65)
- Investigation of the mechanisms underlying TDP-43’s role in triggering cytoplasmic mitochondrial DNA release and activating central inflammatory pathways (i.e., cGAS/STING)
- Deciphering the association between TDP-43 and autoimmunity to determine whether systemic inflammation is a risk factor for TDP-43 proteinopathy or the possibility of common shared mechanisms between TDP-43 proteinopathies and adaptive immune dysregulation.
- Explore the mechanisms underlying TDP-43’s presence and involvement in strokes, TBI, and CTE to determine whether shared mechanisms exist in the context of neuroinflammation.
Author Contributions
Funding
Conflicts of Interest
References
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease—A double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Prather, E.R.; Stetskiv, M.; Garrison, D.E.; Meade, J.R.; Peace, T.I.; Zhou, T. Inflammaging and Oxidative Stress in Human Diseases: From Molecular Mechanisms to Novel Treatments. Int. J. Mol. Sci. 2019, 20, 4472. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236. [Google Scholar] [CrossRef]
- McCauley, M.E.; Baloh, R.H. Inflammation in ALS/FTD pathogenesis. Acta. Neuropathol. 2019, 137, 715–730. [Google Scholar] [CrossRef]
- Bright, F.; Werry, E.L.; Dobson-Stone, C.; Piguet, O.; Ittner, L.M.; Halliday, G.M.; Hodges, J.R.; Kiernan, M.C.; Loy, C.T.; Kassiou, M.; et al. Neuroinflammation in frontotemporal dementia. Nat. Rev. Neurol. 2019, 15, 540–555. [Google Scholar] [CrossRef]
- Calabrese, V.; Santoro, A.; Monti, D.; Crupi, R.; Di Paola, R.; Latteri, S.; Cuzzocrea, S.; Zappia, M.; Giordano, J.; Calabrese, E.J.; et al. Aging and Parkinson’s Disease: Inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic Biol. Med. 2018, 115, 80–91. [Google Scholar] [CrossRef]
- Ou, S.H.; Wu, F.; Harrich, D.; Garcia-Martinez, L.F.; Gaynor, R.B. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 1995, 69, 3584–3596. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Conicella, A.E.; Schmidt, H.B.; Martin, E.W.; Rhoads, S.N.; Reeb, A.N.; Nourse, A.; Ramirez Montero, D.; Ryan, V.H.; Rohatgi, R.; et al. A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation, and RNA splicing. EMBO J. 2018, 37, e97452. [Google Scholar] [CrossRef]
- Mompean, M.; Romano, V.; Pantoja-Uceda, D.; Stuani, C.; Baralle, F.E.; Buratti, E.; Laurents, D.V. Point mutations in the N-terminal domain of transactive response DNA-binding protein 43 kDa (TDP-43) compromise its stability, dimerization, and functions. J. Biol. Chem. 2017, 292, 11992–12006. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front. Biosci. 2008, 13, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. TDP-43: Gumming up neurons through protein-protein and protein-RNA interactions. Trends Biochem. Sci. 2012, 37, 237–247. [Google Scholar] [CrossRef]
- Mercado, P.A.; Ayala, Y.M.; Romano, M.; Buratti, E.; Baralle, F.E. Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic Acids Res. 2005, 33, 6000–6010. [Google Scholar] [CrossRef]
- Hefferon, T.W.; Groman, J.D.; Yurk, C.E.; Cutting, G.R. A variable dinucleotide repeat in the CFTR gene contributes to phenotype diversity by forming RNA secondary structures that alter splicing. Proc. Natl. Acad. Sci. USA 2004, 101, 3504–3509. [Google Scholar] [CrossRef] [PubMed]
- Ederle, H.; Funk, C.; Abou-Ajram, C.; Hutten, S.; Funk, E.B.E.; Kehlenbach, R.H.; Bailer, S.M.; Dormann, D. Nuclear egress of TDP-43 and FUS occurs independently of Exportin-1/CRM1. Sci. Rep. 2018, 8, 7084. [Google Scholar] [CrossRef]
- Pinarbasi, E.S.; Cagatay, T.; Fung, H.Y.J.; Li, Y.C.; Chook, Y.M.; Thomas, P.J. Active nuclear import and passive nuclear export are the primary determinants of TDP-43 localization. Sci. Rep. 2018, 8, 7083. [Google Scholar] [CrossRef]
- Archbold, H.C.; Jackson, K.L.; Arora, A.; Weskamp, K.; Tank, E.M.; Li, X.; Miguez, R.; Dayton, R.D.; Tamir, S.; Klein, R.L.; et al. TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci. Rep. 2018, 8, 4606. [Google Scholar] [CrossRef] [PubMed]
- Sephton, C.F.; Good, S.K.; Atkin, S.; Dewey, C.M.; Mayer, P., 3rd; Herz, J.; Yu, G. TDP-43 is a developmentally regulated protein essential for early embryonic development. J. Biol. Chem. 2010, 285, 6826–6834. [Google Scholar] [CrossRef]
- Chu, J.F.; Majumder, P.; Chatterjee, B.; Huang, S.L.; Shen, C.J. TDP-43 Regulates Coupled Dendritic mRNA Transport-Translation Processes in Co-operation with FMRP and Staufen1. Cell Rep. 2019, 29, 3118–3133. [Google Scholar] [CrossRef]
- Ayala, Y.M.; De Conti, L.; Avendano-Vazquez, S.E.; Dhir, A.; Romano, M.; D’Ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E.; et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011, 30, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.S.; McCaffery, J.M.; Lindquist, S.; Gitler, A.D. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 6439–6444. [Google Scholar] [CrossRef]
- Kraemer, B.C.; Schuck, T.; Wheeler, J.M.; Robinson, L.C.; Trojanowski, J.Q.; Lee, V.M.; Schellenberg, G.D. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta. Neuropathol. 2010, 119, 409–419. [Google Scholar] [CrossRef]
- Voigt, A.; Herholz, D.; Fiesel, F.C.; Kaur, K.; Muller, D.; Karsten, P.; Weber, S.S.; Kahle, P.J.; Marquardt, T.; Schulz, J.B. TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PLoS ONE 2010, 5, e12247. [Google Scholar] [CrossRef]
- Leibiger, C.; Deisel, J.; Aufschnaiter, A.; Ambros, S.; Tereshchenko, M.; Verheijen, B.M.; Buttner, S.; Braun, R.J. TDP-43 controls lysosomal pathways thereby determining its own clearance and cytotoxicity. Hum. Mol. Genet. 2018, 27, 1593–1607. [Google Scholar] [CrossRef]
- Chang, C.K.; Chiang, M.H.; Toh, E.K.; Chang, C.F.; Huang, T.H. Molecular mechanism of oxidation-induced TDP-43 RRM1 aggregation and loss of function. FEBS Lett. 2013, 587, 575–582. [Google Scholar] [CrossRef]
- Shodai, A.; Morimura, T.; Ido, A.; Uchida, T.; Ayaki, T.; Takahashi, R.; Kitazawa, S.; Suzuki, S.; Shirouzu, M.; Kigawa, T.; et al. Aberrant assembly of RNA recognition motif 1 links to pathogenic conversion of TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 2013, 288, 14886–14905. [Google Scholar] [CrossRef] [PubMed]
- Garnier, C.; Devred, F.; Byrne, D.; Puppo, R.; Roman, A.Y.; Malesinski, S.; Golovin, A.V.; Lebrun, R.; Ninkina, N.N.; Tsvetkov, P.O. Zinc binding to RNA recognition motif of TDP-43 induces the formation of amyloid-like aggregates. Sci. Rep. 2017, 7, 6812. [Google Scholar] [CrossRef] [PubMed]
- Zacco, E.; Grana-Montes, R.; Martin, S.R.; de Groot, N.S.; Alfano, C.; Tartaglia, G.G.; Pastore, A. RNA as a key factor in driving or preventing self-assembly of the TAR DNA-binding protein 43. J. Mol. Biol. 2019, 431, 1671–1688. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E. TDP-43 post-translational modifications in health and disease. Expert Opin. Ther. Targets 2018, 22, 279–293. [Google Scholar] [CrossRef]
- Francois-Moutal, L.; Perez-Miller, S.; Scott, D.D.; Miranda, V.G.; Mollasalehi, N.; Khanna, M. Structural Insights Into TDP-43 and Effects of Post-translational Modifications. Front. Mol. Neurosci. 2019, 12, 301. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Xu, Y.F.; Cook, C.; Gendron, T.F.; Roettges, P.; Link, C.D.; Lin, W.L.; Tong, J.; Castanedes-Casey, M.; Ash, P.; et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 7607–7612. [Google Scholar] [CrossRef]
- Berning, B.A.; Walker, A.K. The Pathobiology of TDP-43 C-Terminal Fragments in ALS and FTLD. Front. Neurosci. 2019, 13, 335. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.H.; Kril, J.J.; Fatima, M.; McGeachie, A.; McCann, H.; Shepherd, C.; Forrest, S.L.; Affleck, A.; Kwok, J.B.; Hodges, J.R.; et al. TDP-43 proteinopathies: Pathological identification of brain regions differentiating clinical phenotypes. Brain 2015, 138, 3110–3122. [Google Scholar] [CrossRef] [PubMed]
- Steinacker, P.; Hendrich, C.; Sperfeld, A.D.; Jesse, S.; von Arnim, C.A.; Lehnert, S.; Pabst, A.; Uttner, I.; Tumani, H.; Lee, V.M.; et al. TDP-43 in cerebrospinal fluid of patients with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch. Neurol. 2008, 65, 1481–1487. [Google Scholar] [CrossRef]
- Geser, F.; Lee, V.M.; Trojanowski, J.Q. Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: A spectrum of TDP-43 proteinopathies. Neuropathology 2010, 30, 103–112. [Google Scholar] [CrossRef]
- Kasai, T.; Tokuda, T.; Ishigami, N.; Sasayama, H.; Foulds, P.; Mitchell, D.J.; Mann, D.M.; Allsop, D.; Nakagawa, M. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta. Neuropathol. 2009, 117, 55–62. [Google Scholar] [CrossRef]
- Rohn, T.T. Caspase-cleaved TAR DNA-binding protein-43 is a major pathological finding in Alzheimer’s disease. Brain Res. 2008, 1228, 189–198. [Google Scholar] [CrossRef]
- Nakashima-Yasuda, H.; Uryu, K.; Robinson, J.; Xie, S.X.; Hurtig, H.; Duda, J.E.; Arnold, S.E.; Siderowf, A.; Grossman, M.; Leverenz, J.B.; et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta. Neuropathol. 2007, 114, 221–229. [Google Scholar] [CrossRef]
- Schwab, C.; Arai, T.; Hasegawa, M.; Yu, S.; McGeer, P.L. Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1159–1165. [Google Scholar] [CrossRef]
- McKee, A.C.; Stein, T.D.; Kiernan, P.T.; Alvarez, V.E. The neuropathology of chronic traumatic encephalopathy. Brain Pathol. 2015, 25, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.K.; Daniels, C.M.; Goldman, J.E.; Trojanowski, J.Q.; Lee, V.M.; Messing, A. Astrocytic TDP-43 pathology in Alexander disease. J. Neurosci. 2014, 34, 6448–6458. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.A.; Mori, E.; Hamasaki, F.; Lieberman, A.P. TDP-43 proteinopathy occurs independently of autophagic substrate accumulation and underlies nuclear defects in Niemann-Pick C disease. Neuropathol. Appl. Neurobiol. 2021. [Google Scholar] [CrossRef]
- Pottier, C.; Ren, Y.; Perkerson, R.B., 3rd; Baker, M.; Jenkins, G.D.; van Blitterswijk, M.; DeJesus-Hernandez, M.; van Rooij, J.G.J.; Murray, M.E.; Christopher, E.; et al. Genome-wide analyses as part of the international FTLD-TDP whole-genome sequencing consortium reveals novel disease risk factors and increases support for immune dysfunction in FTLD. Acta Neuropathol. 2019, 137, 879–899. [Google Scholar] [CrossRef]
- Broce, I.; Karch, C.M.; Wen, N.; Fan, C.C.; Wang, Y.; Tan, C.H.; Kouri, N.; Ross, O.A.; Hoglinger, G.U.; Muller, U.; et al. Immune-related genetic enrichment in frontotemporal dementia: An analysis of genome-wide association studies. PLoS Med. 2018, 15, e1002487. [Google Scholar] [CrossRef]
- Wils, H.; Kleinberger, G.; Janssens, J.; Pereson, S.; Joris, G.; Cuijt, I.; Smits, V.; Ceuterick-de Groote, C.; Van Broeckhoven, C.; Kumar-Singh, S. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 3858–3863. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Jawaid, A.; Henstridge, C.M.; Valeri, A.; Merlini, M.; Robinson, J.L.; Lee, E.B.; Rose, J.; Appel, S.; Lee, V.M.; et al. TDP-43 Depletion in Microglia Promotes Amyloid Clearance but Also Induces Synapse Loss. Neuron 2017, 95, 297–308. [Google Scholar] [CrossRef]
- Sudria-Lopez, E.; Koppers, M.; de Wit, M.; van der Meer, C.; Westeneng, H.J.; Zundel, C.A.; Youssef, S.A.; Harkema, L.; de Bruin, A.; Veldink, J.H.; et al. Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta. Neuropathol. 2016, 132, 145–147. [Google Scholar] [CrossRef]
- Burberry, A.; Suzuki, N.; Wang, J.Y.; Moccia, R.; Mordes, D.A.; Stewart, M.H.; Suzuki-Uematsu, S.; Ghosh, S.; Singh, A.; Merkle, F.T.; et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med. 2016, 8, 347ra393. [Google Scholar] [CrossRef]
- Atanasio, A.; Decman, V.; White, D.; Ramos, M.; Ikiz, B.; Lee, H.C.; Siao, C.J.; Brydges, S.; LaRosa, E.; Bai, Y.; et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci. Rep. 2016, 6, 23204. [Google Scholar] [CrossRef]
- Thurner, L.; Preuss, K.D.; Fadle, N.; Regitz, E.; Klemm, P.; Zaks, M.; Kemele, M.; Hasenfus, A.; Csernok, E.; Gross, W.L.; et al. Progranulin antibodies in autoimmune diseases. J. Autoimmun. 2013, 42, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Thurner, L.; Fadle, N.; Regitz, E.; Kemele, M.; Klemm, P.; Zaks, M.; Stoger, E.; Bette, B.; Carbon, G.; Zimmer, V.; et al. The molecular basis for development of proinflammatory autoantibodies to progranulin. J. Autoimmun. 2015, 61, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, L.; Zhang, S.Y.; Casanova, J.L.; Sancho-Shimizu, V. Human TBK1: A Gatekeeper of Neuroinflammation. Trends Mol. Med. 2016, 22, 511–527. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef]
- Picher-Martel, V.; Dutta, K.; Phaneuf, D.; Sobue, G.; Julien, J.P. Ubiquilin-2 drives NF-kappaB activity and cytosolic TDP-43 aggregation in neuronal cells. Mol. Brain 2015, 8, 71. [Google Scholar] [CrossRef]
- Hjerpe, R.; Bett, J.S.; Keuss, M.J.; Solovyova, A.; McWilliams, T.G.; Johnson, C.; Sahu, I.; Varghese, J.; Wood, N.; Wightman, M.; et al. UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell 2016, 166, 935–949. [Google Scholar] [CrossRef]
- Dao, T.P.; Kolaitis, R.M.; Kim, H.J.; O’Donovan, K.; Martyniak, B.; Colicino, E.; Hehnly, H.; Taylor, J.P.; Castaneda, C.A. Ubiquitin Modulates Liquid-Liquid Phase Separation of UBQLN2 via Disruption of Multivalent Interactions. Mol. Cell 2018, 69, 965–978. [Google Scholar] [CrossRef]
- Watanabe, R.; Higashi, S.; Nonaka, T.; Kawakami, I.; Oshima, K.; Niizato, K.; Akiyama, H.; Yoshida, M.; Hasegawa, M.; Arai, T. Intracellular dynamics of Ataxin-2 in the human brains with normal and frontotemporal lobar degeneration with TDP-43 inclusions. Acta. Neuropathol. Commun. 2020, 8, 176. [Google Scholar] [CrossRef]
- Scoles, D.R.; Dansithong, W.; Pflieger, L.T.; Paul, S.; Gandelman, M.; Figueroa, K.P.; Rigo, F.; Bennett, C.F.; Pulst, S.M. ALS-associated genes in SCA2 mouse spinal cord transcriptomes. Hum. Mol. Genet. 2020, 29, 1658–1672. [Google Scholar] [CrossRef]
- Sieber, M.W.; Jaenisch, N.; Brehm, M.; Guenther, M.; Linnartz-Gerlach, B.; Neumann, H.; Witte, O.W.; Frahm, C. Attenuated inflammatory response in triggering receptor expressed on myeloid cells 2 (TREM2) knock-out mice following stroke. PLoS ONE 2013, 8, e52982. [Google Scholar] [CrossRef] [PubMed]
- Kawabori, M.; Kacimi, R.; Kauppinen, T.; Calosing, C.; Kim, J.Y.; Hsieh, C.L.; Nakamura, M.C.; Yenari, M.A. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J. Neurosci. 2015, 35, 3384–3396. [Google Scholar] [CrossRef] [PubMed]
- Saber, M.; Kokiko-Cochran, O.; Puntambekar, S.S.; Lathia, J.D.; Lamb, B.T. Triggering Receptor Expressed on Myeloid Cells 2 Deficiency Alters Acute Macrophage Distribution and Improves Recovery after Traumatic Brain Injury. J. Neurotrauma 2017, 34, 423–435. [Google Scholar] [CrossRef]
- Yu, L.; De Jager, P.L.; Yang, J.; Trojanowski, J.Q.; Bennett, D.A.; Schneider, J.A. The TMEM106B locus and TDP-43 pathology in older persons without FTLD. Neurology 2015, 84, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Rhinn, H.; Abeliovich, A. Differential Aging Analysis in Human Cerebral Cortex Identifies Variants in TMEM106B and GRN that Regulate Aging Phenotypes. Cell Syst. 2017, 4, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Osawa, T.; Mizuno, Y.; Fujita, Y.; Takatama, M.; Nakazato, Y.; Okamoto, K. Optineurin in neurodegenerative diseases. Neuropathology 2011, 31, 569–574. [Google Scholar] [CrossRef]
- Slowicka, K.; van Loo, G. Optineurin Functions for Optimal Immunity. Front. Immunol. 2018, 9, 769. [Google Scholar] [CrossRef]
- Meena, N.P.; Zhu, G.; Mittelstadt, P.R.; Giardino Torchia, M.L.; Pourcelot, M.; Arnoult, D.; Ashwell, J.D.; Munitic, I. The TBK1-binding domain of optineurin promotes type I interferon responses. FEBS Lett. 2016, 590, 1498–1508. [Google Scholar] [CrossRef]
- Chew, T.S.; O’Shea, N.R.; Sewell, G.W.; Oehlers, S.H.; Mulvey, C.M.; Crosier, P.S.; Godovac-Zimmermann, J.; Bloom, S.L.; Smith, A.M.; Segal, A.W. Optineurin deficiency in mice contributes to impaired cytokine secretion and neutrophil recruitment in bacteria-driven colitis. Dis. Model. Mech. 2015, 8, 817–829. [Google Scholar] [CrossRef]
- Gal, J.; Strom, A.L.; Kwinter, D.M.; Kilty, R.; Zhang, J.; Shi, P.; Fu, W.; Wooten, M.W.; Zhu, H. Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J. Neurochem. 2009, 111, 1062–1073. [Google Scholar] [CrossRef]
- Sanz, L.; Sanchez, P.; Lallena, M.J.; Diaz-Meco, M.T.; Moscat, J. The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. EMBO J. 1999, 18, 3044–3053. [Google Scholar] [CrossRef]
- Kratz, M.; Coats, B.R.; Hisert, K.B.; Hagman, D.; Mutskov, V.; Peris, E.; Schoenfelt, K.Q.; Kuzma, J.N.; Larson, I.; Billing, P.S.; et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014, 20, 614–625. [Google Scholar] [CrossRef]
- Gitcho, M.A.; Strider, J.; Carter, D.; Taylor-Reinwald, L.; Forman, M.S.; Goate, A.M.; Cairns, N.J. VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP-43 and induce cell death. J. Biol. Chem. 2009, 284, 12384–12398. [Google Scholar] [CrossRef]
- Dec, E.; Rana, P.; Katheria, V.; Dec, R.; Khare, M.; Nalbandian, A.; Leu, S.Y.; Radom-Aizik, S.; Llewellyn, K.; BenMohamed, L.; et al. Cytokine profiling in patients with VCP-associated disease. Clin. Transl. Sci. 2014, 7, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Nalbandian, A.; Khan, A.A.; Srivastava, R.; Llewellyn, K.J.; Tan, B.; Shukr, N.; Fazli, Y.; Kimonis, V.E.; BenMohamed, L. Activation of the NLRP3 Inflammasome Is Associated with Valosin-Containing Protein Myopathy. Inflammation 2017, 40, 21–41. [Google Scholar] [CrossRef]
- Dobson-Stone, C.; Hallupp, M.; Shahheydari, H.; Ragagnin, A.M.G.; Chatterton, Z.; Carew-Jones, F.; Shepherd, C.E.; Stefen, H.; Paric, E.; Fath, T.; et al. CYLD is a causative gene for frontotemporal dementia-amyotrophic lateral sclerosis. Brain 2020, 143, 783–799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lee, A.J.; Wu, X.; Sun, S.C. Regulation of antiviral innate immunity by deubiquitinase CYLD. Cell. Mol. Immunol. 2011, 8, 502–504. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kovalenko, A.; Chable-Bessia, C.; Cantarella, G.; Israel, A.; Wallach, D.; Courtois, G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 2003, 424, 801–805. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.N.; Wu, Y.; Odeh, H.M.; Gendron, T.F.; Jansen-West, K.; Del Rosso, G.; Yue, M.; Jiang, P.; Gomes, E.; Tong, J.; et al. C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci. Transl. Med. 2020, 12, 559. [Google Scholar] [CrossRef] [PubMed]
- Fredi, M.; Cavazzana, I.; Biasiotto, G.; Filosto, M.; Padovani, A.; Monti, E.; Tincani, A.; Franceschini, F.; Zanella, I. C9orf72 Intermediate Alleles in Patients with Amyotrophic Lateral Sclerosis, Systemic Lupus Erythematosus, and Rheumatoid Arthritis. Neuromol. Med. 2019, 21, 150–159. [Google Scholar] [CrossRef]
- Zhang, Y.; Burberry, A.; Wang, J.Y.; Sandoe, J.; Ghosh, S.; Udeshi, N.D.; Svinkina, T.; Mordes, D.A.; Mok, J.; Charlton, M.; et al. The C9orf72-interacting protein Smcr8 is a negative regulator of autoimmunity and lysosomal exocytosis. Genes. Dev. 2018, 32, 929–943. [Google Scholar] [CrossRef]
- O’Rourke, J.G.; Bogdanik, L.; Yanez, A.; Lall, D.; Wolf, A.J.; Muhammad, A.K.; Ho, R.; Carmona, S.; Vit, J.P.; Zarrow, J.; et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 2016, 351, 1324–1329. [Google Scholar] [CrossRef] [PubMed]
- Lall, D.; Baloh, R.H. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J. Clin. Investig. 2017, 127, 3250–3258. [Google Scholar] [CrossRef]
- Rostalski, H.; Leskela, S.; Huber, N.; Katisko, K.; Cajanus, A.; Solje, E.; Marttinen, M.; Natunen, T.; Remes, A.M.; Hiltunen, M.; et al. Astrocytes and Microglia as Potential Contributors to the Pathogenesis of C9orf72 Repeat Expansion-Associated FTLD and ALS. Front. Neurosci. 2019, 13, 486. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Baker, M.; Pickering-Brown, S.; Hsiung, G.Y.; Lindholm, C.; Dwosh, E.; Gass, J.; Cannon, A.; Rademakers, R.; Hutton, M.; et al. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain 2006, 129, 3081–3090. [Google Scholar] [CrossRef]
- Baker, M.; Mackenzie, I.R.; Pickering-Brown, S.M.; Gass, J.; Rademakers, R.; Lindholm, C.; Snowden, J.; Adamson, J.; Sadovnick, A.D.; Rollinson, S.; et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006, 442, 916–919. [Google Scholar] [CrossRef]
- Mukherjee, O.; Pastor, P.; Cairns, N.J.; Chakraverty, S.; Kauwe, J.S.; Shears, S.; Behrens, M.I.; Budde, J.; Hinrichs, A.L.; Norton, J.; et al. HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann. Neurol. 2006, 60, 314–322. [Google Scholar] [CrossRef]
- Pickford, F.; Marcus, J.; Camargo, L.M.; Xiao, Q.; Graham, D.; Mo, J.R.; Burkhardt, M.; Kulkarni, V.; Crispino, J.; Hering, H.; et al. Progranulin is a chemoattractant for microglia and stimulates their endocytic activity. Am. J. Pathol. 2011, 178, 284–295. [Google Scholar] [CrossRef]
- Townley, R.A.; Boeve, B.F.; Benarroch, E.E. Progranulin: Functions and neurologic correlations. Neurology 2018, 90, 118–125. [Google Scholar] [CrossRef]
- Cruts, M.; Gijselinck, I.; van der Zee, J.; Engelborghs, S.; Wils, H.; Pirici, D.; Rademakers, R.; Vandenberghe, R.; Dermaut, B.; Martin, J.J.; et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006, 442, 920–924. [Google Scholar] [CrossRef]
- Milanesi, E.; Bonvicini, C.; Alberici, A.; Pilotto, A.; Cattane, N.; Premi, E.; Gazzina, S.; Archetti, S.; Gasparotti, R.; Cancelli, V.; et al. Molecular signature of disease onset in granulin mutation carriers: A gene expression analysis study. Neurobiol. Aging 2013, 34, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Matsuwaki, T.; Yamanouchi, K.; Nishihara, M. Increased lysosomal biogenesis in activated microglia and exacerbated neuronal damage after traumatic brain injury in progranulin-deficient mice. Neuroscience 2013, 250, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Martens, L.H.; Zhang, J.; Barmada, S.J.; Zhou, P.; Kamiya, S.; Sun, B.; Min, S.W.; Gan, L.; Finkbeiner, S.; Huang, E.J.; et al. Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J. Clin. Investig. 2012, 122, 3955–3959. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Banerjee, R.; Thomas, B.; Zhou, P.; Qian, L.; Jia, T.; Ma, X.; Ma, Y.; Iadecola, C.; Beal, M.F.; et al. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J. Exp. Med. 2010, 207, 117–128. [Google Scholar] [CrossRef]
- Huang, M.; Modeste, E.; Dammer, E.; Merino, P.; Taylor, G.; Duong, D.M.; Deng, Q.; Holler, C.J.; Gearing, M.; Dickson, D.; et al. Network analysis of the progranulin-deficient mouse brain proteome reveals pathogenic mechanisms shared in human frontotemporal dementia caused by GRN mutations. Acta. Neuropathol. Commun. 2020, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016, 165, 921–935. [Google Scholar] [CrossRef] [PubMed]
- Pottier, C.; Bieniek, K.F.; Finch, N.; van de Vorst, M.; Baker, M.; Perkersen, R.; Brown, P.; Ravenscroft, T.; van Blitterswijk, M.; Nicholson, A.M.; et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta. Neuropathol. 2015, 130, 77–92. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Muller, K.; Marroquin, N.; Nordin, F.; Hubers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- van der Zee, J.; Gijselinck, I.; Van Mossevelde, S.; Perrone, F.; Dillen, L.; Heeman, B.; Baumer, V.; Engelborghs, S.; De Bleecker, J.; Baets, J.; et al. TBK1 Mutation Spectrum in an Extended European Patient Cohort with Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Hum. Mutat. 2017, 38, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar] [CrossRef] [PubMed]
- Kounatidis, I.; Chtarbanova, S.; Cao, Y.; Hayne, M.; Jayanth, D.; Ganetzky, B.; Ligoxygakis, P. NF-kappaB Immunity in the Brain Determines Fly Lifespan in Healthy Aging and Age-Related Neurodegeneration. Cell Rep. 2017, 19, 836–848. [Google Scholar] [CrossRef]
- Wang, Z.R.; Li, Y.X.; Lei, H.Y.; Yang, D.Q.; Wang, L.Q.; Luo, M.Y. Regulating effect of activated NF-kappaB on edema induced by traumatic brain injury of rats. Asian Pac. J. Trop. Med. 2016, 9, 274–277. [Google Scholar] [CrossRef]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-B interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019, 21, 101059. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 1–9. [Google Scholar] [CrossRef]
- Correia, A.S.; Patel, P.; Dutta, K.; Julien, J.P. Inflammation Induces TDP-43 Mislocalization and Aggregation. PLoS ONE 2015, 10, e0140248. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Bell, S.; Wang, J.; Wen, S.; Baloh, R.H.; Appel, S.H. TDP-43 activates microglia through NF-kappaB and NLRP3 inflammasome. Exp. Neurol. 2015, 273, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef]
- Dutta, K.; Thammisetty, S.S.; Boutej, H.; Bareil, C.; Julien, J.P. Mitigation of ALS Pathology by Neuron-Specific Inhibition of Nuclear Factor Kappa B Signaling. J. Neurosci. 2020, 40, 5137–5154. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Cynader, M.S.; Jia, W. TDP-43 Inhibits NF-kappaB Activity by Blocking p65 Nuclear Translocation. PLoS ONE 2015, 10, e0142296. [Google Scholar] [CrossRef]
- Swarup, V.; Phaneuf, D.; Dupre, N.; Petri, S.; Strong, M.; Kriz, J.; Julien, J.P. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB-mediated pathogenic pathways. J. Exp. Med. 2011, 208, 2429–2447. [Google Scholar] [CrossRef]
- Pozzi, S.; Thammisetty, S.S.; Codron, P.; Rahimian, R.; Plourde, K.V.; Soucy, G.; Bareil, C.; Phaneuf, D.; Kriz, J.; Gravel, C.; et al. Virus-mediated delivery of antibody targeting TAR DNA-binding protein-43 mitigates associated neuropathology. J. Clin. Investig. 2019, 129, 1581–1595. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Huntley, M.L.; Perry, G.; Wang, X. Pathomechanisms of TDP-43 in neurodegeneration. J. Neurochem. 2018, 146, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.D.; van Hummel, A.; Stevens, C.H.; Gladbach, A.; Ippati, S.; Bi, M.; Lee, W.S.; Kruger, S.; van der Hoven, J.; Volkerling, A.; et al. Short-term suppression of A315T mutant human TDP-43 expression improves functional deficits in a novel inducible transgenic mouse model of FTLD-TDP and ALS. Acta Neuropathol. 2015, 130, 661–678. [Google Scholar] [CrossRef] [PubMed]
- Ohta, Y.; Tremblay, C.; Schneider, J.A.; Bennett, D.A.; Calon, F.; Julien, J.P. Interaction of transactive response DNA binding protein 43 with nuclear factor kappaB in mild cognitive impairment with episodic memory deficits. Acta Neuropathol. Commun. 2014, 2, 37. [Google Scholar] [CrossRef] [PubMed]
- Kallstig, E.; McCabe, B.D.; Schneider, B.L. The Links between ALS and NF-kappaB. Int. J. Mol. Sci. 2021, 22, 3875. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H.; Bohr, V.A. Signaling by cGAS-STING in Neurodegeneration, Neuroinflammation, and Aging. Trends Neurosci. 2021, 44, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649. [Google Scholar] [CrossRef]
- Magrane, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhao, W. NLRP3 Inflammasome-A Key Player in Antiviral Responses. Front. Immunol. 2020, 11, 211. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef]
- Song, L.; Pei, L.; Yao, S.; Wu, Y.; Shang, Y. NLRP3 Inflammasome in Neurological Diseases, from Functions to Therapies. Front. Cell. Neurosci. 2017, 11, 63. [Google Scholar] [CrossRef]
- Kadhim, H.; Deltenre, P.; Martin, J.J.; Sebire, G. In-situ expression of Interleukin-18 and associated mediators in the human brain of sALS patients: Hypothesis for a role for immune-inflammatory mechanisms. Med. Hypotheses 2016, 86, 14–17. [Google Scholar] [CrossRef]
- Zhuang, J.; Wen, X.; Zhang, Y.Q.; Shan, Q.; Zhang, Z.F.; Zheng, G.H.; Fan, S.H.; Li, M.Q.; Wu, D.M.; Hu, B.; et al. TDP-43 upregulation mediated by the NLRP3 inflammasome induces cognitive impairment in 2 2’,4,4’-tetrabromodiphenyl ether (BDE-47)-treated mice. Brain Behav. Immun. 2017, 65, 99–110. [Google Scholar] [CrossRef]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Hebron, M.L.; Lonskaya, I.; Sharpe, K.; Weerasinghe, P.P.; Algarzae, N.K.; Shekoyan, A.R.; Moussa, C.E. Parkin ubiquitinates Tar-DNA binding protein-43 (TDP-43) and promotes its cytosolic accumulation via interaction with histone deacetylase 6 (HDAC6). J. Biol. Chem. 2013, 288, 4103–4115. [Google Scholar] [CrossRef]
- Wenqiang, C.; Lonskaya, I.; Hebron, M.L.; Ibrahim, Z.; Olszewski, R.T.; Neale, J.H.; Moussa, C.E. Parkin-mediated reduction of nuclear and soluble TDP-43 reverses behavioral decline in symptomatic mice. Hum. Mol. Genet. 2014, 23, 4960–4969. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Duan, Y.; Qin, C.; Li, J.C.; Duan, G.; Deng, X.; Ni, J.; Cao, X.; Xiang, K.; Tian, K.; et al. Distinct multilevel misregulations of Parkin and PINK1 revealed in cell and animal models of TDP-43 proteinopathy. Cell Death Dis. 2018, 9, 953. [Google Scholar] [CrossRef] [PubMed]
- Deora, V.; Lee, J.D.; Albornoz, E.A.; McAlary, L.; Jagaraj, C.J.; Robertson, A.A.B.; Atkin, J.D.; Cooper, M.A.; Schroder, K.; Yerbury, J.J.; et al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia 2020, 68, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Trageser, K.J.; Smith, C.; Herman, F.J.; Ono, K.; Pasinetti, G.M. Mechanisms of Immune Activation by c9orf72-Expansions in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2019, 13, 1298. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Vandoorne, T.; Steyaert, J.; Staats, K.A.; Van Den Bosch, L. The multifaceted role of kinases in amyotrophic lateral sclerosis: Genetic, pathological and therapeutic implications. Brain 2020, 143, 1651–1673. [Google Scholar] [CrossRef] [PubMed]
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 5. [Google Scholar] [CrossRef]
- Asih, P.R.; Prikas, E.; Stefanoska, K.; Tan, A.R.P.; Ahel, H.I.; Ittner, A. Functions of p38 MAP Kinases in the Central Nervous System. Front. Mol. Neurosci. 2020, 13, 570586. [Google Scholar] [CrossRef]
- Zhan, L.; Xie, Q.; Tibbetts, R.S. Opposing roles of p38 and JNK in a Drosophila model of TDP-43 proteinopathy reveal oxidative stress and innate immunity as pathogenic components of neurodegeneration. Hum. Mol. Genet. 2015, 24, 757–772. [Google Scholar] [CrossRef]
- Van Deerlin, V.M.; Leverenz, J.B.; Bekris, L.M.; Bird, T.D.; Yuan, W.; Elman, L.B.; Clay, D.; Wood, E.M.; Chen-Plotkin, A.S.; Martinez-Lage, M.; et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: A genetic and histopathological analysis. Lancet Neurol. 2008, 7, 409–416. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, Z.F.; Qin, F.W.; Tang, W.; Liu, D.H.; Wu, P.Y.; Jiao, F. The mechanism of TDP-43 gene expression on inflammatory factors and the JNK and p38 MAPK signalling pathways in ischaemic hypoxic stress dependence. Int. Wound J. 2019, 16, 724–729. [Google Scholar] [CrossRef]
- Woodruff, T.M.; Costantini, K.J.; Crane, J.W.; Atkin, J.D.; Monk, P.N.; Taylor, S.M.; Noakes, P.G. The complement factor C5a contributes to pathology in a rat model of amyotrophic lateral sclerosis. J. Immunol. 2008, 181, 8727–8734. [Google Scholar] [CrossRef]
- Sta, M.; Sylva-Steenland, R.M.; Casula, M.; de Jong, J.M.; Troost, D.; Aronica, E.; Baas, F. Innate and adaptive immunity in amyotrophic lateral sclerosis: Evidence of complement activation. Neurobiol. Dis. 2011, 42, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.D.; Levin, S.C.; Willis, E.F.; Li, R.; Woodruff, T.M.; Noakes, P.G. Complement components are upregulated and correlate with disease progression in the TDP-43(Q331K) mouse model of amyotrophic lateral sclerosis. J. Neuroinflamm. 2018, 15, 171. [Google Scholar] [CrossRef] [PubMed]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The complement system: An unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 2012, 35, 369–389. [Google Scholar] [CrossRef] [PubMed]
- Heyburn, L.; Hebron, M.L.; Smith, J.; Winston, C.; Bechara, J.; Li, Z.; Lonskaya, I.; Burns, M.P.; Harris, B.T.; Moussa, C.E. Tyrosine kinase inhibition reverses TDP-43 effects on synaptic protein expression, astrocytic function and amino acid dis-homeostasis. J. Neurochem. 2016, 139, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Sephton, C.F.; Yu, G. The function of RNA-binding proteins at the synapse: Implications for neurodegeneration. Cell. Mol. Life Sci. 2015, 72, 3621–3635. [Google Scholar] [CrossRef] [PubMed]
- Naviaux, J.C.; Wang, L.; Li, K.; Bright, A.T.; Alaynick, W.A.; Williams, K.R.; Powell, S.B.; Naviaux, R.K. Antipurinergic therapy corrects the autism-like features in the Fragile X (Fmr1 knockout) mouse model. Mol. Autism 2015, 6, 1. [Google Scholar] [CrossRef]
- Zhang, J.; Velmeshev, D.; Hashimoto, K.; Huang, Y.H.; Hofmann, J.W.; Shi, X.; Chen, J.; Leidal, A.M.; Dishart, J.G.; Cahill, M.K.; et al. Neurotoxic microglia promote TDP-43 proteinopathy in progranulin deficiency. Nature 2020, 588, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Miller, Z.A.; Rankin, K.P.; Graff-Radford, N.R.; Takada, L.T.; Sturm, V.E.; Cleveland, C.M.; Criswell, L.A.; Jaeger, P.A.; Stan, T.; Heggeli, K.A.; et al. TDP-43 frontotemporal lobar degeneration and autoimmune disease. J. Neurol. Neurosurg. Psychiatry 2013, 84, 956–962. [Google Scholar] [CrossRef]
- Miller, Z.A.; Sturm, V.E.; Camsari, G.B.; Karydas, A.; Yokoyama, J.S.; Grinberg, L.T.; Boxer, A.L.; Rosen, H.J.; Rankin, K.P.; Gorno-Tempini, M.L.; et al. Increased prevalence of autoimmune disease within C9 and FTD/MND cohorts: Completing the picture. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e301. [Google Scholar] [CrossRef]
- Katisko, K.; Solje, E.; Koivisto, A.M.; Kruger, J.; Kinnunen, T.; Hartikainen, P.; Helisalmi, S.; Korhonen, V.; Herukka, S.K.; Haapasalo, A.; et al. Prevalence of immunological diseases in a Finnish frontotemporal lobar degeneration cohort with the C9orf72 repeat expansion carriers and non-carriers. J. Neuroimmunol. 2018, 321, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Sullivan, P.M.; Hu, F. SMCR8 negatively regulates AKT and MTORC1 signaling to modulate lysosome biogenesis and tissue homeostasis. Autophagy 2019, 15, 871–885. [Google Scholar] [CrossRef] [PubMed]
- Jian, J.; Li, G.; Hettinghouse, A.; Liu, C. Progranulin: A key player in autoimmune diseases. Cytokine 2018, 101, 48–55. [Google Scholar] [CrossRef]
- Thurner, L.; Stoger, E.; Fadle, N.; Klemm, P.; Regitz, E.; Kemele, M.; Bette, B.; Held, G.; Dauer, M.; Lammert, F.; et al. Proinflammatory progranulin antibodies in inflammatory bowel diseases. Dig. Dis. Sci. 2014, 59, 1733–1742. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. Neuroinflamm. 2019, 16, 142. [Google Scholar] [CrossRef]
- Lee, E.B.; Lee, V.M.; Trojanowski, J.Q.; Neumann, M. TDP-43 immunoreactivity in anoxic, ischemic and neoplastic lesions of the central nervous system. Acta. Neuropathol. 2008, 115, 305–311. [Google Scholar] [CrossRef]
- Kanazawa, M.; Kakita, A.; Igarashi, H.; Takahashi, T.; Kawamura, K.; Takahashi, H.; Nakada, T.; Nishizawa, M.; Shimohata, T. Biochemical and histopathological alterations in TAR DNA-binding protein-43 after acute ischemic stroke in rats. J. Neurochem. 2011, 116, 957–965. [Google Scholar] [CrossRef]
- Thammisetty, S.S.; Pedragosa, J.; Weng, Y.C.; Calon, F.; Planas, A.; Kriz, J. Age-related deregulation of TDP-43 after stroke enhances NF-kappaB-mediated inflammation and neuronal damage. J. Neuroinflamm. 2018, 15, 312. [Google Scholar] [CrossRef]
- He, T.; Zuo, Y.; Ai-Zakwani, K.; Luo, J.; Zhu, H.; Yan, X.X.; Liu, F. Subarachnoid hemorrhage enhances the expression of TDP-43 in the brain of experimental rats and human subjects. Exp. Ther. Med. 2018, 16, 3363–3368. [Google Scholar] [CrossRef]
- Kahl, A.; Blanco, I.; Jackman, K.; Baskar, J.; Milaganur Mohan, H.; Rodney-Sandy, R.; Zhang, S.; Iadecola, C.; Hochrainer, K. Cerebral ischemia induces the aggregation of proteins linked to neurodegenerative diseases. Sci. Rep. 2018, 8, 2701. [Google Scholar] [CrossRef]
- Sun, M.; Yamashita, T.; Shang, J.; Liu, N.; Deguchi, K.; Liu, W.; Ikeda, Y.; Feng, J.; Abe, K. Acceleration of TDP43 and FUS/TLS protein expressions in the preconditioned hippocampus following repeated transient ischemia. J. Neurosci. Res. 2014, 92, 54–63. [Google Scholar] [CrossRef]
- Blennow, K.; Hardy, J.; Zetterberg, H. The neuropathology and neurobiology of traumatic brain injury. Neuron 2012, 76, 886–899. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Gavett, B.E.; Stern, R.A.; Nowinski, C.J.; Cantu, R.C.; Kowall, N.W.; Perl, D.P.; Hedley-Whyte, E.T.; Price, B.; Sullivan, C.; et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J. Neuropathol. Exp. Neurol. 2010, 69, 918–929. [Google Scholar] [CrossRef]
- McKee, A.C.; Robinson, M.E. Military-related traumatic brain injury and neurodegeneration. Alzheimers Dement. 2014, 10, S242–S253. [Google Scholar] [CrossRef]
- Kenney, K.; Iacono, D.; Edlow, B.L.; Katz, D.I.; Diaz-Arrastia, R.; Dams-O’Connor, K.; Daneshvar, D.H.; Stevens, A.; Moreau, A.L.; Tirrell, L.S.; et al. Dementia After Moderate-Severe Traumatic Brain Injury: Coexistence of Multiple Proteinopathies. J. Neuropathol. Exp. Neurol. 2018, 77, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.K.; Lee, Y.C.; Huang, C.Y.; Liliang, P.C.; Lu, K.; Chen, H.J.; Li, Y.C.; Tsai, K.J. Traumatic brain injury causes frontotemporal dementia and TDP-43 proteolysis. Neuroscience 2015, 300, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Lee, Y.C.; Li, P.C.; Liliang, P.C.; Lu, K.; Wang, K.W.; Chang, L.C.; Shiu, L.Y.; Chen, M.F.; Sun, Y.T.; et al. TDP-43 proteolysis is associated with astrocyte reactivity after traumatic brain injury in rodents. J. Neuroimmunol. 2017, 313, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Teng, Z.; Song, Y.; Hu, M.; Chen, C. Inhibition of monoacylglycerol lipase prevents chronic traumatic encephalopathy-like neuropathology in a mouse model of repetitive mild closed head injury. J. Cereb. Blood Flow Metab. 2015, 35, 706. [Google Scholar] [CrossRef]
- Wiesner, D.; Tar, L.; Linkus, B.; Chandrasekar, A.; Olde Heuvel, F.; Dupuis, L.; Tsao, W.; Wong, P.C.; Ludolph, A.; Roselli, F. Reversible induction of TDP-43 granules in cortical neurons after traumatic injury. Exp. Neurol. 2018, 299, 15–25. [Google Scholar] [CrossRef]
- Heyburn, L.; Sajja, V.; Long, J.B. The Role of TDP-43 in Military-Relevant TBI and Chronic Neurodegeneration. Front. Neurol. 2019, 10, 680. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Brettschneider, J.; Ludolph, A.C.; Lee, V.M.; Trojanowski, J.Q.; Del Tredici, K. Amyotrophic lateral sclerosis--a model of corticofugal axonal spread. Nat. Rev. Neurol. 2013, 9, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.N.; Gochenaur, L.; Singh, A.; Grant, R.; Patel, K.; Watkins, S.; Wu, J.Y.; Pandey, U.B. Traumatic injury induces stress granule formation and enhances motor dysfunctions in ALS/FTD models. Hum. Mol. Genet. 2018, 27, 1366–1381. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.N.; Morera, A.A.; Kour, S.; Cherry, J.D.; Ramesh, N.; Gleixner, A.; Schwartz, J.C.; Ebmeier, C.; Old, W.; Donnelly, C.J.; et al. Traumatic injury compromises nucleocytoplasmic transport and leads to TDP-43 pathology. eLife 2021, 10, e67587. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene | Protein | Relationship to TDP-43 | Associated Diseases | Published Evidence |
|---|---|---|---|---|
| TARDBP | TDP-43 | Gene encoding for TDP-43 protein | ALS, FTD rare variant |
|
| C9orf72 | Guanine nucleotide exchange C9orf72 | TDP-43 is pathological feature of C9orf72 expansion in ALS and FTD | ALS, FTD, AD |
|
| GRN | Progranulin | TDP-43 is pathological feature of GRN mutation in FTD | FTLD-GRN, CLN11 disease |
|
| TBK1 | Serine/threonine-protein kinase TBK1 | 3rd most common genetic cause of FTLD-TDP [46] | ALS, FTD rare variant | |
| UBQLN2 | Ubiquilin-2 | UBQLN2 dysregulation in neurons can drive NF-κβ activation and cytosolic TDP-43 aggregation [57] | FTD rare variant |
|
| ATXN2 | Ataxin-2 | Link between ATXN2 and TDP-43 proteinopathy established [60] | ALS, Parkinson’s disease (late onset), Spinocerebellar ataxia type 2 (SCA2) |
|
| TREM2 | Triggering receptor expressed on myeloid cells 2 | Reported novel interaction between TREM2 and TDP-43 | Susceptibility gene FTD |
|
| TMEM106B | Transmembrane protein 106B | Common variants in TMEM106B serve as a distinct risk factor for TDP-43 pathology in older individuals without FTLD [65] | Susceptibility gene FTD |
|
| OPTN | Optineurin | Optineurin inclusions detected in small subset ALS and FTD with TDP pathology [67] | Glaucoma, ALS with or without FTD rare variant |
|
| SQSTM1 | Sequestosome-1, p62 | Sequestration of SQTSM1 into TDP-43 aggregates, leads to inhibition of proteasome function and autophagy and promotes the accumulation of toxic, misfolded proteins [71] | Paget disease of bone 3, FTD/ALS rare variant |
|
| VCP | Transitional endoplasmic reticulum ATPase | Major component of ubiquitinated inclusions of FTLD with VCP mutation is TDP-43 [74] | FTD |
|
| CYLD | Ubiquitin carboxyl-terminal hydrolase CYLD | CYLD directly interacts with TBK1, OPTN, and p62 [77] | ALS, FTD |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bright, F.; Chan, G.; van Hummel, A.; Ittner, L.M.; Ke, Y.D. TDP-43 and Inflammation: Implications for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Int. J. Mol. Sci. 2021, 22, 7781. https://doi.org/10.3390/ijms22157781
Bright F, Chan G, van Hummel A, Ittner LM, Ke YD. TDP-43 and Inflammation: Implications for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. International Journal of Molecular Sciences. 2021; 22(15):7781. https://doi.org/10.3390/ijms22157781
Chicago/Turabian StyleBright, Fiona, Gabriella Chan, Annika van Hummel, Lars M. Ittner, and Yazi D. Ke. 2021. "TDP-43 and Inflammation: Implications for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia" International Journal of Molecular Sciences 22, no. 15: 7781. https://doi.org/10.3390/ijms22157781
APA StyleBright, F., Chan, G., van Hummel, A., Ittner, L. M., & Ke, Y. D. (2021). TDP-43 and Inflammation: Implications for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. International Journal of Molecular Sciences, 22(15), 7781. https://doi.org/10.3390/ijms22157781