
The Different Faces of the TDP-43 Low-Complexity Domain: The Formation of Liquid Droplets and Amyloid Fibrils


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
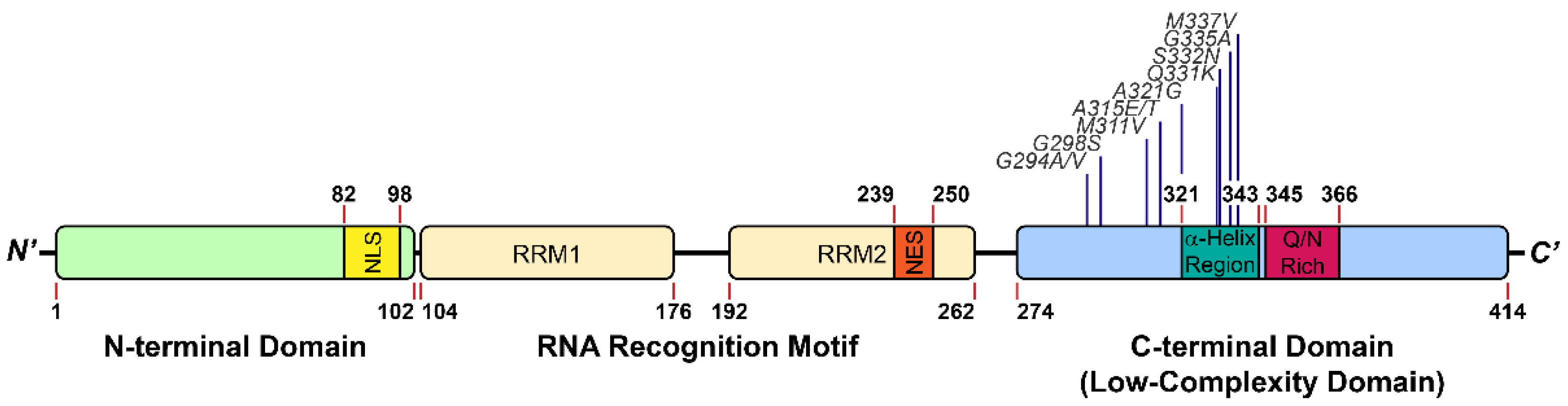
2. The Introduction of TDP-43 LCD
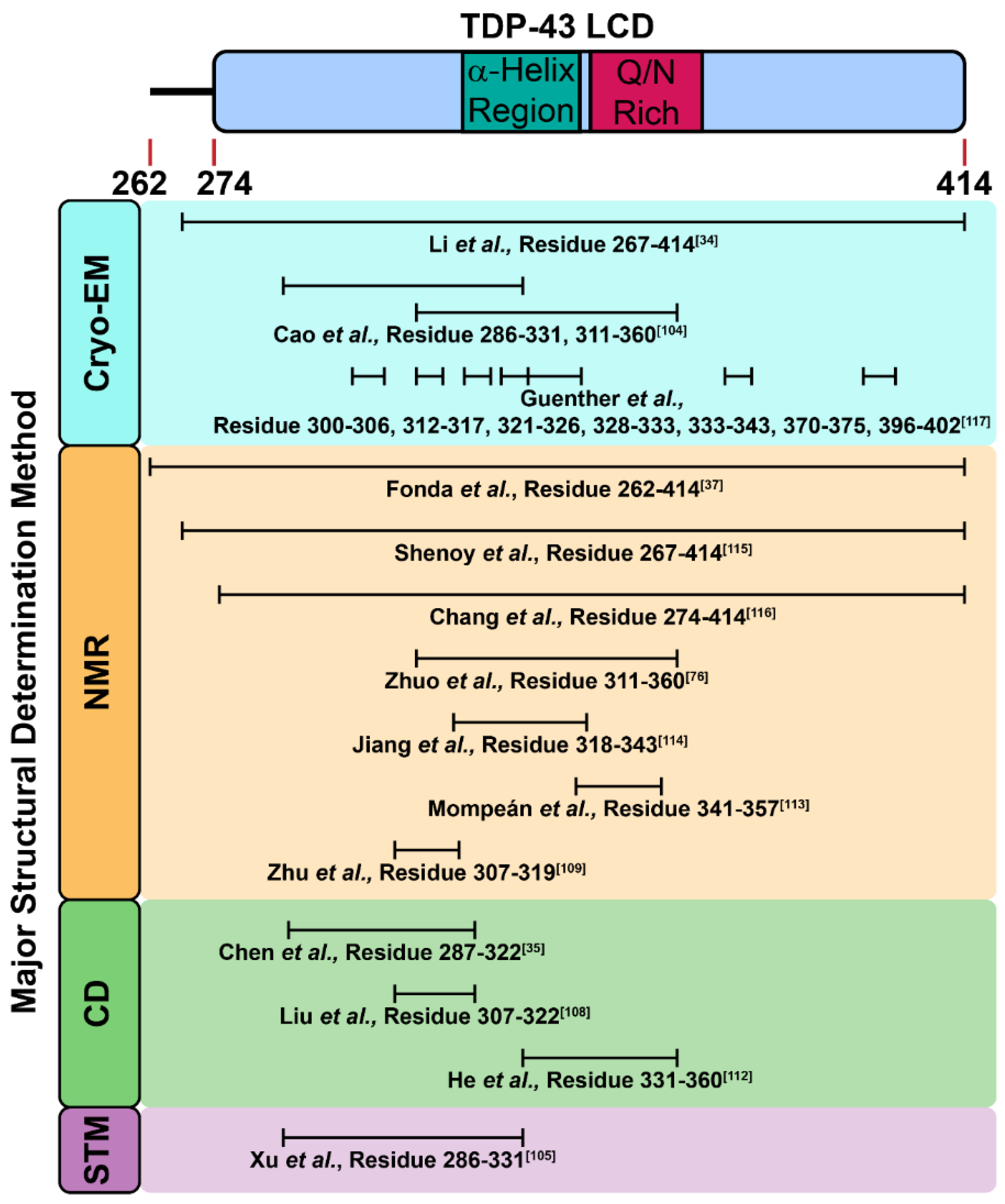
2.1. The Structure of TDP-43 LCD
2.2. The Possible Function of TDP-43 LCD
3. The Introduction of TDP-43 LCD
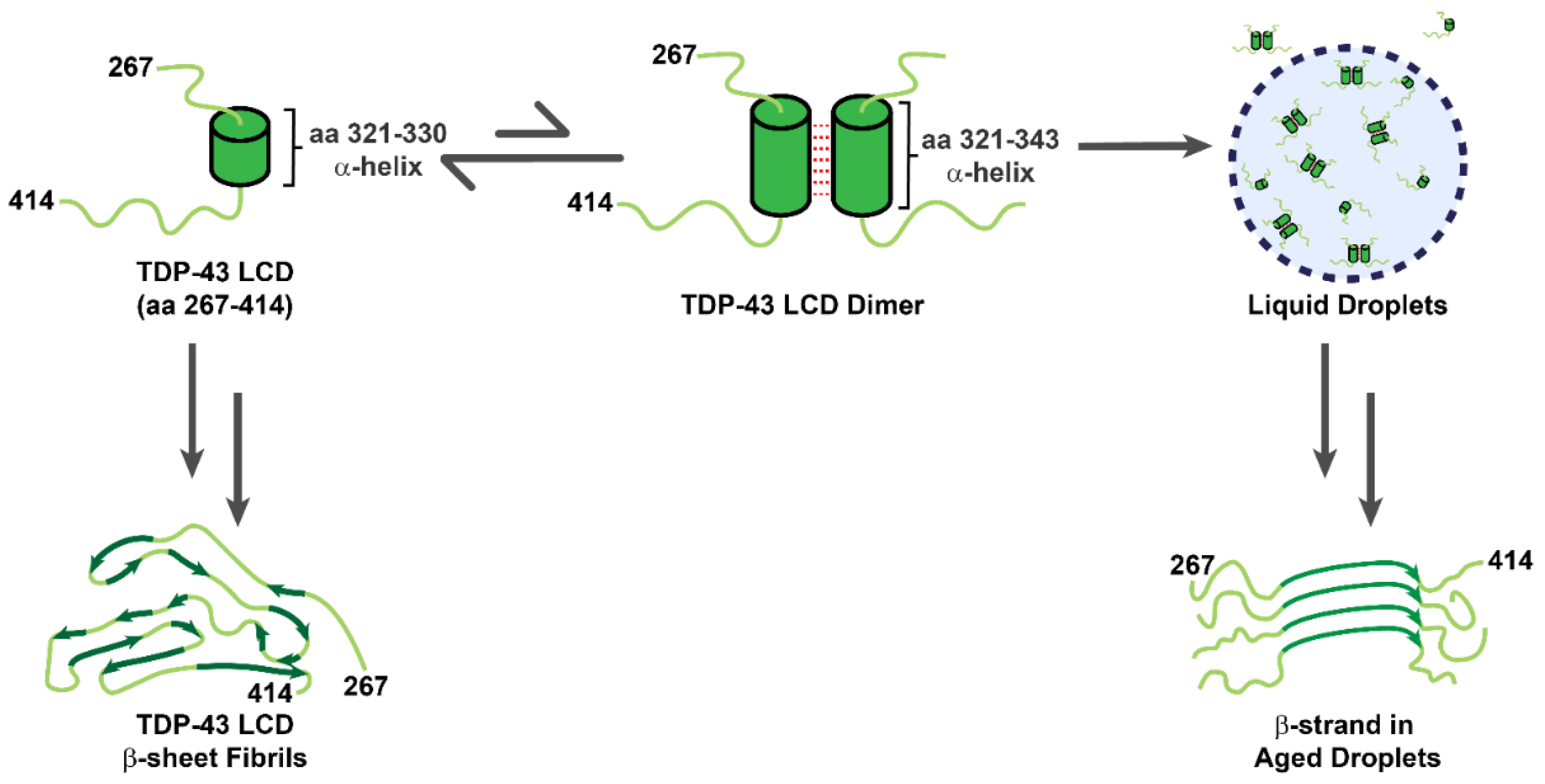
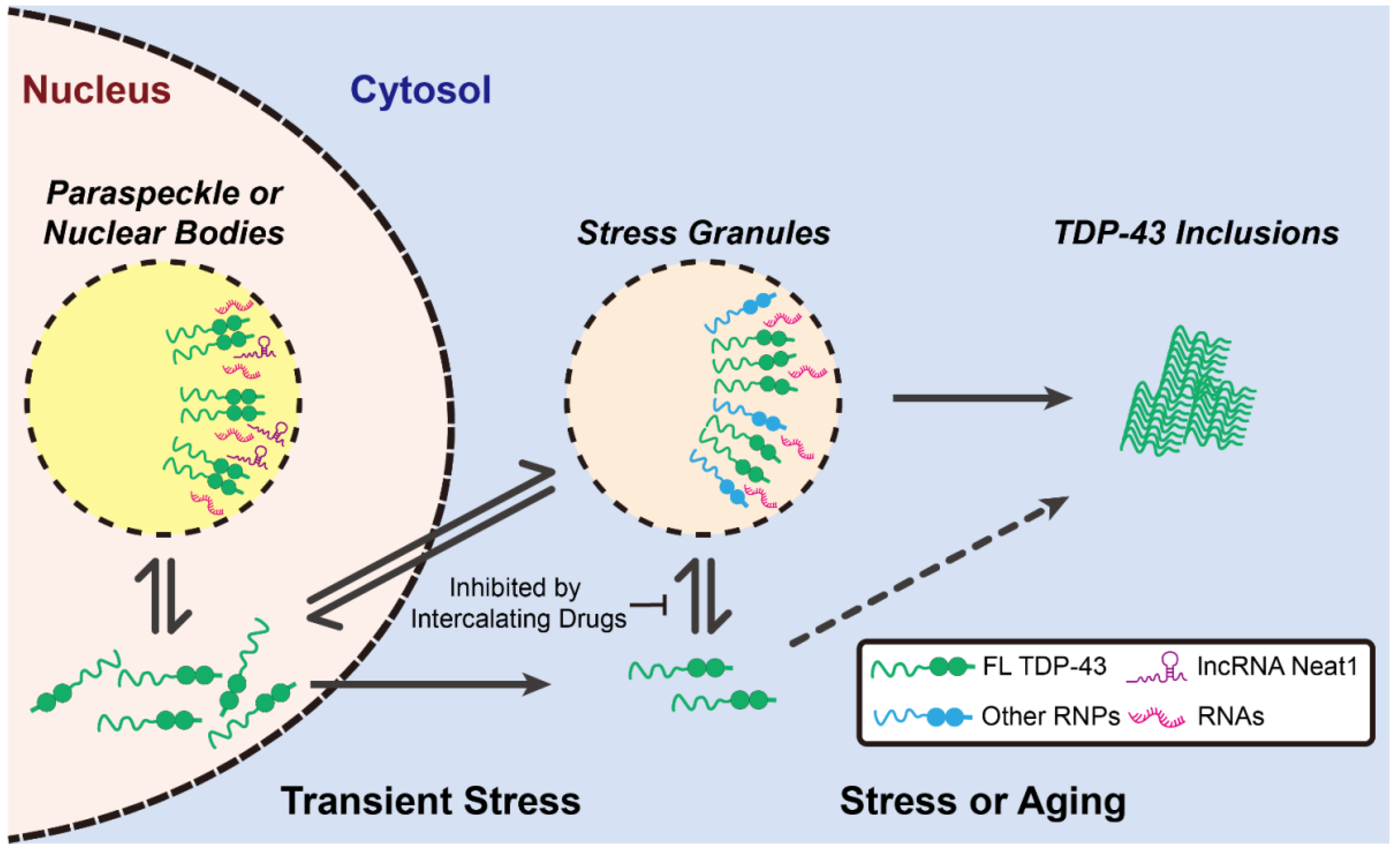
3.1. TDP-43 LCDs Form Liquid Droplets
3.2. TDP-43 LCDs Form Amyloid Fibrils
3.3. The Possible Toxicity of TDP-43 LCD
4. Concluding Remark
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sephton, C.F.; Cenik, B.; Cenik, B.K.; Herz, J.; Yu, G. TDP-43 in central nervous system development and function: Clues to TDP-43-associated neurodegeneration. Biol. Chem. 2012, 393, 589–594. [Google Scholar] [CrossRef]
- Donde, A.; Sun, M.; Ling, J.P.; Braunstein, K.E.; Pang, B.; Wen, X.; Cheng, X.; Chen, L.; Wong, P.C. Splicing repression is a major function of TDP-43 in motor neurons. Acta Neuropathol. 2019, 138, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Župunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.H.; Ke, Y.D.; Ittner, L.M.; Halliday, G.M. ALS/FTLD: Experimental models and reality. Acta Neuropathol. 2017, 133, 177–196. [Google Scholar] [CrossRef] [PubMed]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Berning, B.A.; Walker, A.K. The pathobiology of TDP-43 C-terminal fragments in ALS and FTLD. Front. Neurosci. 2019, 13, 335. [Google Scholar] [CrossRef]
- Afroz, T.; Hock, E.-M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.B.; Laferriere, F.; Maniecka, Z.; Plückthun, A.; Mittl, P.; et al. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 2017, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Lukavsky, P.J.; Daujotyte, D.; Tollervey, J.R.; Ule, J.; Stuani, C.; Buratti, E.; Baralle, F.E.; Damberger, F.F.; Allain, F.H.T. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 2013, 20, 1443–1449. [Google Scholar] [CrossRef]
- Kuo, P.-H.; Chiang, C.-H.; Wang, Y.-T.; Doudeva, L.G.; Yuan, H.S. The crystal structure of TDP-43 RRM1-DNA complex reveals the specific recognition for UG- and TG-rich nucleic acids. Nucleic Acids Res. 2014, 42, 4712–4722. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Ayala, Y.M.; Zago, P.; D’Ambrogio, A.; Xu, Y.-F.; Petrucelli, L.; Buratti, E.; Baralle, F.E. Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 2008, 121, 3778–3785. [Google Scholar] [CrossRef]
- Winton, M.J.; Igaz, L.M.; Wong, M.M.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M.Y. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 2008, 283, 13302–13309. [Google Scholar] [CrossRef]
- Jiang, L.-L.; Xue, W.; Hong, J.-Y.; Zhang, J.-T.; Li, M.-J.; Yu, S.-N.; He, J.-H.; Hu, H.-Y. The N-terminal dimerization is required for TDP-43 splicing activity. Sci. Rep. 2017, 7, 6196. [Google Scholar] [CrossRef]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.C.; Schuck, T.; Wheeler, J.M.; Robinson, L.C.; Trojanowski, J.Q.; Lee, V.M.Y.; Schellenberg, G.D. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010, 119, 409–419. [Google Scholar] [CrossRef]
- Ayala, Y.M.; De Conti, L.; Avendaño-Vázquez, S.E.; Dhir, A.; Romano, M.; D’Ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E.; et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011, 30, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.S.; McCaffery, J.M.; Lindquist, S.; Gitler, A.D. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 6439–6444. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.-S.; Cheng, W.-C.; Hou, S.-C.; Yan, Y.-T.; Jiang, S.-T.; Shen, C.K.J. TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis 2010, 48, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Mier, P.; Paladin, L.; Tamana, S.; Petrosian, S.; Hajdu-Soltész, B.; Urbanek, A.; Gruca, A.; Plewczynski, D.; Grynberg, M.; Bernadó, P.; et al. Disentangling the complexity of low complexity proteins. Brief. Bioinform. 2020, 21, 458–472. [Google Scholar] [CrossRef]
- Kumari, B.; Kumar, R.; Kumar, M. Low complexity and disordered regions of proteins have different structural and amino acid preferences. Mol. BioSyst. 2015, 11, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Coletta, A.; Pinney, J.W.; Solís, D.Y.W.; Marsh, J.; Pettifer, S.R.; Attwood, T.K. Low-complexity regions within protein sequences have position-dependent roles. BMC Syst. Biol. 2010, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.; Dugast-Darzacq, C.; Liu, Z.; Dong, P.; Dailey, G.M.; Cattoglio, C.; Heckert, A.; Banala, S.; Lavis, L.; Darzacq, X.; et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 2018, 361, eaar2555. [Google Scholar] [CrossRef]
- Ntountoumi, C.; Vlastaridis, P.; Mossialos, D.; Stathopoulos, C.; Iliopoulos, I.; Promponas, V.; Oliver, S.G.; Amoutzias, G.D. Low complexity regions in the proteins of prokaryotes perform important functional roles and are highly conserved. Nucleic Acids Res. 2019, 47, 9998–10009. [Google Scholar] [CrossRef] [PubMed]
- Cascarina, S.M.; Elder, M.R.; Ross, E.D. Atypical structural tendencies among low-complexity domains in the Protein Data Bank proteome. PLOS Comput. Biol. 2020, 16, e1007487. [Google Scholar] [CrossRef]
- Franzmann, T.M.; Alberti, S. Prion-like low-complexity sequences: Key regulators of protein solubility and phase behavior. J. Biol. Chem. 2019, 294, 7128–7136. [Google Scholar] [CrossRef]
- Murthy, A.C.; Dignon, G.L.; Kan, Y.; Zerze, G.H.; Parekh, S.H.; Mittal, J.; Fawzi, N.L. Molecular interactions underlying liquid−liquid phase separation of the FUS low-complexity domain. Nat. Struct. Mol. Biol. 2019, 26, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.W.; Thomasen, F.E.; Milkovic, N.M.; Cuneo, M.J.; Grace, C.R.; Nourse, A.; Lindorff-Larsen, K.; Mittag, T. Interplay of folded domains and the disordered low-complexity domain in mediating hnRNPA1 phase separation. Nucleic Acids Res. 2021, 49, 2931–2945. [Google Scholar] [CrossRef]
- Murray, D.T.; Kato, M.; Lin, Y.; Thurber, K.R.; Hung, I.; McKnight, S.L.; Tycko, R. Structure of FUS protein fibrils and its relevance to self-assembly and phase separation of low-complexity domains. Cell 2017, 171, 615–627.e16. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Ghosh, U.; Thurber, K.R.; Kato, M.; Tycko, R. Molecular structure and interactions within amyloid-like fibrils formed by a low-complexity protein sequence from FUS. Nat. Commun. 2020, 11, 5735. [Google Scholar] [CrossRef]
- Lu, J.; Cao, Q.; Hughes, M.P.; Sawaya, M.R.; Boyer, D.R.; Cascio, D.; Eisenberg, D.S. CryoEM structure of the low-complexity domain of hnRNPA2 and its conversion to pathogenic amyloid. Nat. Commun. 2020, 11, 4090. [Google Scholar] [CrossRef]
- Murray, D.T.; Tycko, R. Side chain hydrogen-bonding interactions within amyloid-like fibrils formed by the low-complexity domain of FUS: Evidence from solid state nuclear magnetic resonance spectroscopy. Biochemistry 2020, 59, 364–378. [Google Scholar] [CrossRef]
- Li, Q.; Babinchak, W.M.; Surewicz, W.K. Cryo-EM structure of amyloid fibrils formed by the entire low complexity domain of TDP-43. Nat. Commun. 2021, 12, 1620. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.K.H.; Lin, R.Y.Y.; Hsieh, E.Z.J.; Tu, P.-H.; Chen, R.P.Y.; Liao, T.-Y.; Chen, W.; Wang, C.-H.; Huang, J.J.T. Induction of amyloid fibrils by the C-terminal fragments of TDP-43 in amyotrophic lateral sclerosis. J. Am. Chem. Soc. 2010, 132, 1186–1187. [Google Scholar] [CrossRef] [PubMed]
- Conicella, A.E.; Dignon, G.L.; Zerze, G.H.; Schmidt, H.B.; D’Ordine, A.M.; Kim, Y.C.; Rohatgi, R.; Ayala, Y.M.; Mittal, J.; Fawzi, N.L. TDP-43 α-helical structure tunes liquid–liquid phase separation and function. Proc. Natl. Acad. Sci. USA 2020, 117, 5883–5894. [Google Scholar] [CrossRef]
- Fonda, B.D.; Jami, K.M.; Boulos, N.R.; Murray, D.T. Identification of the rigid core for aged liquid droplets of an RNA-binding protein low complexity domain. J. Am. Chem. Soc. 2021, 143, 6657–6668. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Brindisi, A.; Giombi, M.; Tisminetzky, S.; Ayala, Y.M.; Baralle, F.E. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: An important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 2005, 280, 37572–37584. [Google Scholar] [CrossRef]
- Ayala, Y.M.; Pantano, S.; D’Ambrogio, A.; Buratti, E.; Brindisi, A.; Marchetti, C.; Romano, M.; Baralle, F.E. Human, Drosophila, and C. elegans TDP43: Nucleic acid binding properties and splicing regulatory function. J. Mol. Biol. 2005, 348, 575–588. [Google Scholar] [CrossRef]
- D’Ambrogio, A.; Buratti, E.; Stuani, C.; Guarnaccia, C.; Romano, M.; Ayala, Y.M.; Baralle, F.E. Functional mapping of the interaction between TDP-43 and hnRNP A2 in vivo. Nucleic Acids Res. 2009, 37, 4116–4126. [Google Scholar] [CrossRef]
- Deng, H.-X.; Chen, W.; Hong, S.-T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef]
- Renaud, L.; Picher-Martel, V.; Codron, P.; Julien, J.-P. Key role of UBQLN2 in pathogenesis of amyotrophic lateral sclerosis and frontotemporal dementia. Acta Neuropathol. Commun. 2019, 7, 103. [Google Scholar] [CrossRef] [PubMed]
- Cassel, J.A.; Reitz, A.B. Ubiquilin-2 (UBQLN2) binds with high affinity to the C-terminal region of TDP-43 and modulates TDP-43 levels in H4 cells: Characterization of inhibition by nucleic acids and 4-aminoquinolines. Biochim. Biophys. Acta BBA Proteins Proteom. 2013, 1834, 964–971. [Google Scholar] [CrossRef] [PubMed]
- Hyman, A.A.; Weber, C.A.; Jülicher, F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.K.; Vibhute, M.A.; Spruijt, E. Biomolecular chemistry in liquid phase separated compartments. Front. Mol. Biosci. 2019, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, T.; Nozawa, R.-S.; Jia, T.Z.; Saio, T.; Mori, E. Biological phase separation: Cell biology meets biophysics. Biophys. Rev. 2020, 12, 519–539. [Google Scholar] [CrossRef] [PubMed]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Cuevas-Velazquez, C.L.; Dinneny, J.R. Organization out of disorder: Liquid–liquid phase separation in plants. Curr. Opin. Plant Biol. 2018, 45, 68–74. [Google Scholar] [CrossRef]
- Zhang, H.; Ji, X.; Li, P.; Liu, C.; Lou, J.; Wang, Z.; Wen, W.; Xiao, Y.; Zhang, M.; Zhu, X. Liquid-liquid phase separation in biology: Mechanisms, physiological functions and human diseases. Sci. China Life Sci. 2020, 63, 953–985. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Tompa, P.; Pappu, R.V. Polymer physics of intracellular phase transitions. Nat. Phys. 2015, 11, 899–904. [Google Scholar] [CrossRef]
- Owen, I.; Shewmaker, F. The role of post-translational modifications in the phase transitions of intrinsically disordered proteins. Int. J. Mol. Sci. 2019, 20, 5501. [Google Scholar] [CrossRef]
- Rai, A.K.; Chen, J.-X.; Selbach, M.; Pelkmans, L. Kinase-controlled phase transition of membraneless organelles in mitosis. Nature 2018, 559, 211–216. [Google Scholar] [CrossRef]
- Reineke, L.C.; Tsai, W.-C.; Jain, A.; Kaelber, J.T.; Jung, S.Y.; Lloyd, R.E. Casein kinase 2 is linked to stress granule dynamics through phosphorylation of the stress granule nucleating protein G3BP1. Mol. Cell. Biol. 2017, 37, e00596-16. [Google Scholar] [CrossRef] [PubMed]
- Monahan, Z.; Ryan, V.H.; Janke, A.M.; Burke, K.A.; Rhoads, S.N.; Zerze, G.H.; O’Meally, R.; Dignon, G.L.; Conicella, A.E.; Zheng, W.; et al. Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 2017, 36, 2951–2967. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, S.; Gu, J.; Tong, Y.; Li, Y.; Gui, X.; Long, H.; Wang, C.; Zhao, C.; Lu, J.; et al. Hsp27 chaperones FUS phase separation under the modulation of stress-induced phosphorylation. Nat. Struct. Mol. Biol. 2020, 27, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Nott, T.J.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.P.; Pawson, T.; Forman-Kay, J.D.; et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell 2015, 57, 936–947. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Hwang, A.W.; Restrepo, C.R.; Yuan, C.-X.; Trojanowski, J.Q.; Lee, V.M.Y. An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun. 2015, 6, 5845. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Hess, D.; Eglinger, J.; Fritsch, A.W.; Kreysing, M.; Weinert, B.T.; Choudhary, C.; Matthias, P. Acetylation of intrinsically disordered regions regulates phase separation. Nat. Chem. Biol. 2019, 15, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.R.; Carroll, N.J.; Rubinstein, M.; Chilkoti, A.; López, G.P. Programming molecular self-assembly of intrinsically disordered proteins containing sequences of low complexity. Nat. Chem. 2017, 9, 509–515. [Google Scholar] [CrossRef]
- Cui, M.; Wang, X.; An, B.; Zhang, C.; Gui, X.; Li, K.; Li, Y.; Ge, P.; Zhang, J.; Liu, C.; et al. Exploiting mammalian low-complexity domains for liquid-liquid phase separation–driven underwater adhesive coatings. Sci. Adv. 2019, 5, eaax3155. [Google Scholar] [CrossRef]
- Harrison, A.F.; Shorter, J. RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 2017, 474, 1417–1438. [Google Scholar] [CrossRef]
- Modic, M.; Grosch, M.; Rot, G.; Schirge, S.; Lepko, T.; Yamazaki, T.; Lee, F.C.Y.; Rusha, E.; Shaposhnikov, D.; Palo, M.; et al. Cross-regulation between TDP-43 and paraspeckles promotes pluripotency-differentiation transition. Mol. Cell 2019, 74, 951–965.e13. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, Y.; Nakagawa, S.; Hirose, T.; Okano, H.J.; Takao, M.; Shibata, S.; Suyama, S.; Kuwako, K.-i.; Imai, T.; Murayama, S.; et al. The long non-coding RNA nuclear-enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Mol. Brain 2013, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Khalfallah, Y.; Kuta, R.; Grasmuck, C.; Prat, A.; Durham, H.D.; Vande Velde, C. TDP-43 regulation of stress granule dynamics in neurodegenerative disease-relevant cell types. Sci. Rep. 2018, 8, 7551. [Google Scholar] [CrossRef]
- Droppelmann, C.A.; Campos-Melo, D.; Moszczynski, A.J.; Amzil, H.; Strong, M.J. TDP-43 aggregation inside micronuclei reveals a potential mechanism for protein inclusion formation in ALS. Sci. Rep. 2019, 9, 19928. [Google Scholar] [CrossRef] [PubMed]
- Gasset-Rosa, F.; Lu, S.; Yu, H.; Chen, C.; Melamed, Z.E.; Guo, L.; Shorter, J.; Da Cruz, S.; Cleveland, D.W. Cytoplasmic TDP-43 de-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron 2019, 102, 339–357.e7. [Google Scholar] [CrossRef] [PubMed]
- Gopal, P.P.; Nirschl, J.J.; Klinman, E.; Holzbaur, E.L.F. Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 RNP granules in neurons. Proc. Natl. Acad. Sci. USA 2017, 114, E2466–E2475. [Google Scholar] [CrossRef]
- Sun, Y.; Chakrabartty, A. Phase to phase with TDP-43. Biochemistry 2017, 56, 809–823. [Google Scholar] [CrossRef]
- Watanabe, S.; Inami, H.; Oiwa, K.; Murata, Y.; Sakai, S.; Komine, O.; Sobue, A.; Iguchi, Y.; Katsuno, M.; Yamanaka, K. Aggresome formation and liquid–liquid phase separation independently induce cytoplasmic aggregation of TAR DNA-binding protein 43. Cell Death Dis. 2020, 11, 909. [Google Scholar] [CrossRef]
- Babinchak, W.M.; Haider, R.; Dumm, B.K.; Sarkar, P.; Surewicz, K.; Choi, J.-K.; Surewicz, W.K. The role of liquid-liquid phase separation in aggregation of the TDP-43 low-complexity domain. J. Biol. Chem. 2019, 294, 6306–6317. [Google Scholar] [CrossRef]
- Schmidt, H.B.; Rohatgi, R. In vivo formation of vacuolated multi-phase compartments lacking membranes. Cell Rep. 2016, 16, 1228–1236. [Google Scholar] [CrossRef]
- Li, H.-R.; Chiang, W.-C.; Chou, P.-C.; Wang, W.-J.; Huang, J.-r. TAR DNA-binding protein 43 (TDP-43) liquid-liquid phase separation is mediated by just a few aromatic residues. J. Biol. Chem. 2018, 293, 6090–6098. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-R.; Chen, T.-C.; Hsiao, C.-L.; Shi, L.; Chou, C.-Y.; Huang, J.-R. The physical forces mediating self-association and phase-separation in the C-terminal domain of TDP-43. Biochim. Biophys. Acta BBA Proteins Proteom. 2018, 1866, 214–223. [Google Scholar] [CrossRef]
- Bhopatkar, A.A.; Uversky, V.N.; Rangachari, V. Granulins modulate liquid–liquid phase separation and aggregation of the prion-like C-terminal domain of the neurodegeneration-associated protein TDP-43. J. Biol. Chem. 2020, 295, 2506–2519. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, X.-F.; Wang, J.; Zhang, J.; Jiang, L.-L.; Hu, H.-Y.; Lu, J.-X. Solid-state NMR reveals the structural transformation of the TDP-43 amyloidogenic region upon fibrillation. J. Am. Chem. Soc. 2020, 142, 3412–3421. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-C.; Bose, J.K.; Majumder, P.; Lee, K.-H.; Huang, J.-T.J.; Huang, J.K.; Shen, C.-K.J. Metabolism and mis-metabolism of the neuropathological signature protein TDP-43. J. Cell Sci. 2014, 127, 3024–3038. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Conicella, A.E.; Schmidt, H.B.; Martin, E.W.; Rhoads, S.N.; Reeb, A.N.; Nourse, A.; Ramirez Montero, D.; Ryan, V.H.; Rohatgi, R.; et al. A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation, and RNA splicing. EMBO J. 2018, 37, e97452. [Google Scholar] [CrossRef]
- Chang, C.-K.; Wu, T.-H.; Wu, C.-Y.; Chiang, M.-H.; Toh, E.K.-W.; Hsu, Y.-C.; Lin, K.-F.; Liao, Y.-H.; Huang, T.-H.; Huang, J.J.-T. The N-terminus of TDP-43 promotes its oligomerization and enhances DNA binding affinity. Biochem. Biophys. Res. Commun. 2012, 425, 219–224. [Google Scholar] [CrossRef]
- Gordon, D.; Dafinca, R.; Scaber, J.; Alegre-Abarrategui, J.; Farrimond, L.; Scott, C.; Biggs, D.; Kent, L.; Oliver, P.L.; Davies, B.; et al. Single-copy expression of an amyotrophic lateral sclerosis-linked TDP-43 mutation (M337V) in BAC transgenic mice leads to altered stress granule dynamics and progressive motor dysfunction. Neurobiol. Dis. 2019, 121, 148–162. [Google Scholar] [CrossRef]
- Colombrita, C.; Zennaro, E.; Fallini, C.; Weber, M.; Sommacal, A.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 2009, 111, 1051–1061. [Google Scholar] [CrossRef]
- McDonald, K.K.; Aulas, A.; Destroismaisons, L.; Pickles, S.; Beleac, E.; Camu, W.; Rouleau, G.A.; Vande Velde, C. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 2011, 20, 1400–1410. [Google Scholar] [CrossRef]
- Fang, M.Y.; Markmiller, S.; Vu, A.Q.; Javaherian, A.; Dowdle, W.E.; Jolivet, P.; Bushway, P.J.; Castello, N.A.; Baral, A.; Chan, M.Y.; et al. Small-molecule modulation of TDP-43 recruitment to stress granules prevents persistent TDP-43 accumulation in ALS/FTD. Neuron 2019, 103, 802–819.e11. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.R.; Gleixner, A.M.; Mauna, J.C.; Gomes, E.; DeChellis-Marks, M.R.; Needham, P.G.; Copley, K.E.; Hurtle, B.; Portz, B.; Pyles, N.J.; et al. RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron 2019, 102, 321–338.e8. [Google Scholar] [CrossRef]
- Wang, C.; Duan, Y.; Duan, G.; Wang, Q.; Zhang, K.; Deng, X.; Qian, B.; Gu, J.; Ma, Z.; Zhang, S.; et al. Stress induces dynamic, cytotoxicity-antagonizing TDP-43 nuclear bodies via paraspeckle LncRNA NEAT1-mediated liquid-liquid phase separation. Mol. Cell 2020, 79, 443–458.e7. [Google Scholar] [CrossRef]
- Yu, H.; Lu, S.; Gasior, K.; Singh, D.; Vazquez-Sanchez, S.; Tapia, O.; Toprani, D.; Beccari, M.S.; Yates, J.R.; Da Cruz, S.; et al. HSP70 chaperones RNA-free TDP-43 into anisotropic intranuclear liquid spherical shells. Science 2021, 371, eabb4309. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.; Luo, F.; Li, Y.; Zhou, H.; Qin, Z.; Liu, Z.; Gu, J.; Xie, M.; Zhao, K.; Dai, B.; et al. Structural basis for reversible amyloids of hnRNPA1 elucidates their role in stress granule assembly. Nat. Commun. 2019, 10, 2006. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Ke, P.C.; Zhou, R.; Serpell, L.C.; Riek, R.; Knowles, T.P.J.; Lashuel, H.A.; Gazit, E.; Hamley, I.W.; Davis, T.P.; Fändrich, M.; et al. Half a century of amyloids: Past, present and future. Chem. Soc. Rev. 2020, 49, 5473–5509. [Google Scholar] [CrossRef] [PubMed]
- Adamcik, J.; Mezzenga, R. Amyloid polymorphism in the protein folding and aggregation energy landscape. Angew. Chem. Int. Ed. 2018, 57, 8370–8382. [Google Scholar] [CrossRef]
- Lee, S.J.C.; Nam, E.; Lee, H.J.; Savelieff, M.G.; Lim, M.H. Towards an understanding of amyloid-β oligomers: Characterization, toxicity mechanisms, and inhibitors. Chem. Soc. Rev. 2017, 46, 310–323. [Google Scholar] [CrossRef]
- De Simone, A.; Esposito, L.; Pedone, C.; Vitagliano, L. Insights into stability and toxicity of amyloid-like oligomers by replica exchange molecular dynamics analyses. Biophys. J. 2008, 95, 1965–1973. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.; Basdevant, N.; Prévost, C.; Ha-Duong, T. Structure of ring-shaped Aβ42 oligomers determined by conformational selection. Sci. Rep. 2016, 6, 21429. [Google Scholar] [CrossRef]
- Lee, M.; Kim, J.I.; Na, S.; Eom, K. Metal ions affect the formation and stability of amyloid β aggregates at multiple length scales. Phys. Chem. Chem. Phys. 2018, 20, 8951–8961. [Google Scholar] [CrossRef] [PubMed]
- Alam, P.; Bousset, L.; Melki, R.; Otzen, D.E. α-synuclein oligomers and fibrils: A spectrum of species, a spectrum of toxicities. J. Neurochem. 2019, 150, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Ramamoorthy, A.; Sahoo, B.R.; Zheng, J.; Faller, P.; Straub, J.E.; Dominguez, L.; Shea, J.-E.; Dokholyan, N.V.; De Simone, A.; et al. Amyloid oligomers: A joint experimental/computational perspective on Alzheimer’s disease, Parkinson’s disease, type II diabetes, and amyotrophic lateral sclerosis. Chem. Rev. 2021, 121, 2545–2647. [Google Scholar] [CrossRef] [PubMed]
- Fontana, I.C.; Zimmer, A.R.; Rocha, A.S.; Gosmann, G.; Souza, D.O.; Lourenco, M.V.; Ferreira, S.T.; Zimmer, E.R. Amyloid-β oligomers in cellular models of Alzheimer’s disease. J. Neurochem. 2020, 155, 348–369. [Google Scholar] [CrossRef]
- Surguchev, A.; Surguchov, A. Conformational diseases: Looking into the eyes. Brain Res. Bull. 2010, 81, 12–24. [Google Scholar] [CrossRef]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef]
- Bigio, E.H.; Wu, J.Y.; Deng, H.-X.; Bit-Ivan, E.N.; Mao, Q.; Ganti, R.; Peterson, M.; Siddique, N.; Geula, C.; Siddique, T.; et al. Inclusions in frontotemporal lobar degeneration with TDP-43 proteinopathy (FTLD-TDP) and amyotrophic lateral sclerosis (ALS), but not FTLD with FUS proteinopathy (FTLD-FUS), have properties of amyloid. Acta Neuropathol. 2013, 125, 463–465. [Google Scholar] [CrossRef]
- Robinson, J.L.; Geser, F.; Stieber, A.; Umoh, M.; Kwong, L.K.; Van Deerlin, V.M.; Lee, V.M.Y.; Trojanowski, J.Q. TDP-43 skeins show properties of amyloid in a subset of ALS cases. Acta Neuropathol. 2013, 125, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Chen, Y.; Zhou, X.; Kar, A.; Ray, P.; Chen, X.; Rao, E.J.; Yang, M.; Ye, H.; Zhu, L.; et al. An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat. Struct. Mol. Biol. 2011, 18, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, T.; Hasegawa, M. Prion-like properties of assembled TDP-43. Curr. Opin. Neurobiol. 2020, 61, 23–28. [Google Scholar] [CrossRef]
- Budini, M.; Buratti, E.; Stuani, C.; Guarnaccia, C.; Romano, V.; De Conti, L.; Baralle, F.E. Cellular model of TAR DNA-binding protein 43 (TDP-43) aggregation based on its C-terminal Gln/Asn-rich region. J. Biol. Chem. 2012, 287, 7512–7525. [Google Scholar] [CrossRef]
- Cao, Q.; Boyer, D.R.; Sawaya, M.R.; Ge, P.; Eisenberg, D.S. Cryo-EM structures of four polymorphic TDP-43 amyloid cores. Nat. Struct. Mol. Biol. 2019, 26, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhu, L.; Liu, J.; Yang, Y.; Wu, J.Y.; Wang, C. Characterization of β-domains in C-terminal fragments of TDP-43 by scanning tunneling microscopy. J. Struct. Biol. 2013, 181, 11–16. [Google Scholar] [CrossRef]
- Sun, C.-S.; Wang, C.Y.-H.; Chen, B.P.-W.; He, R.-Y.; Liu, G.C.-H.; Wang, C.-H.; Chen, W.; Chern, Y.; Huang, J.J.-T. The influence of pathological mutations and proline substitutions in TDP-43 glycine-rich peptides on its amyloid properties and cellular toxicity. PLoS ONE 2014, 9, e103644. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Khan, A.; Huang, J.J.-T.; Ulmschneider, M.B. Mechanisms of membrane pore formation by amyloidogenic peptides in amyotrophic lateral sclerosis. Chem. Eur. J. 2016, 22, 9958–9961. [Google Scholar] [CrossRef]
- Liu, G.C.-H.; Chen, B.P.-W.; Ye, N.T.-J.; Wang, C.-H.; Chen, W.; Lee, H.-M.; Chan, S.I.; Huang, J.J.-T. Delineating the membrane-disrupting and seeding properties of the TDP-43 amyloidogenic core. Chem. Commun. 2013, 49, 11212–11214. [Google Scholar] [CrossRef]
- Zhu, L.; Xu, M.; Yang, M.; Yang, Y.; Li, Y.; Deng, J.; Ruan, L.; Liu, J.; Du, S.; Liu, X.; et al. An ALS-mutant TDP-43 neurotoxic peptide adopts an anti-parallel β-structure and induces TDP-43 redistribution. Hum. Mol. Genet. 2014, 23, 6863–6877. [Google Scholar] [CrossRef]
- Saini, A.; Chauhan, V.S. Delineation of the core aggregation sequences of TDP-43 C-terminal fragment. ChemBioChem 2011, 12, 2495–2501. [Google Scholar] [CrossRef] [PubMed]
- Budini, M.; Romano, V.; Avendaño-Vázquez, S.E.; Bembich, S.; Buratti, E.; Baralle, F.E. Role of selected mutations in the Q/N rich region of TDP-43 in EGFP-12xQ/N-induced aggregate formation. Brain Res. 2012, 1462, 139–150. [Google Scholar] [CrossRef]
- He, R.-Y.; Huang, Y.-C.; Chiang, C.-W.; Tsai, Y.-J.; Ye, T.-J.; Gao, H.-D.; Wu, C.-Y.; Lee, H.-M.; Huang, J.J.-T. Characterization and real-time imaging of the FTLD-related protein aggregation induced by amyloidogenic peptides. Chem. Commun. 2015, 51, 8652–8655. [Google Scholar] [CrossRef] [PubMed]
- Mompeán, M.; Hervás, R.; Xu, Y.; Tran, T.H.; Guarnaccia, C.; Buratti, E.; Baralle, F.; Tong, L.; Carrión-Vázquez, M.; McDermott, A.E.; et al. Structural evidence of amyloid fibril formation in the putative aggregation domain of TDP-43. J. Phys. Chem. Lett. 2015, 6, 2608–2615. [Google Scholar] [CrossRef]
- Jiang, L.-L.; Che, M.-X.; Zhao, J.; Zhou, C.-J.; Xie, M.-Y.; Li, H.-Y.; He, J.-H.; Hu, H.-Y. Structural transformation of the amyloidogenic core region of TDP-43 protein initiates its aggregation and cytoplasmic inclusion. J. Biol. Chem. 2013, 288, 19614–19624. [Google Scholar] [CrossRef]
- Shenoy, J.; El Mammeri, N.; Dutour, A.; Berbon, M.; Saad, A.; Lends, A.; Morvan, E.; Grélard, A.; Lecomte, S.; Kauffmann, B.; et al. Structural dissection of amyloid aggregates of TDP-43 and its C-terminal fragments TDP-35 and TDP-16. FEBS J. 2020, 287, 2449–2467. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Deng, J.; Zhao, W.; Yang, J. Amyloid-like aggregation and fibril core determination of TDP-43 C-terminal domain. Biochem. Biophys. Res. Commun. 2020, 532, 655–661. [Google Scholar] [CrossRef]
- Guenther, E.L.; Cao, Q.; Trinh, H.; Lu, J.; Sawaya, M.R.; Cascio, D.; Boyer, D.R.; Rodriguez, J.A.; Hughes, M.P.; Eisenberg, D.S. Atomic structures of TDP-43 LCD segments and insights into reversible or pathogenic aggregation. Nat. Struct. Mol. Biol. 2018, 25, 463–471. [Google Scholar] [CrossRef]
- Goldschmidt, L.; Teng, P.K.; Riek, R.; Eisenberg, D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2010, 107, 3487–3492. [Google Scholar] [CrossRef]
- Salazar, D.A.; Butler, V.J.; Argouarch, A.R.; Hsu, T.-Y.; Mason, A.; Nakamura, A.; McCurdy, H.; Cox, D.; Ng, R.; Pan, G.; et al. The progranulin cleavage products, granulins, exacerbate TDP-43 toxicity and increase TDP-43 levels. J. Neurosci. 2015, 35, 9315–9328. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Xu, Y.-F.; Cook, C.; Gendron, T.F.; Roettges, P.; Link, C.D.; Lin, W.-L.; Tong, J.; Castanedes-Casey, M.; Ash, P.; et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 7607–7612. [Google Scholar] [CrossRef] [PubMed]
- Fallini, C.; Bassell, G.J.; Rossoll, W. The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum. Mol. Genet. 2012, 21, 3703–3718. [Google Scholar] [CrossRef]
- Chou, C.-C.; Alexeeva, O.M.; Yamada, S.; Pribadi, A.; Zhang, Y.; Mo, B.; Williams, K.R.; Zarnescu, D.C.; Rossoll, W. PABPN1 suppresses TDP-43 toxicity in ALS disease models. Hum. Mol. Genet. 2015, 24, 5154–5173. [Google Scholar] [CrossRef]
- Che, M.-X.; Jiang, Y.-J.; Xie, Y.-Y.; Jiang, L.-L.; Hu, H.-Y. Aggregation of the 35-kDa fragment of TDP-43 causes formation of cytoplasmic inclusions and alteration of RNA processing. FASEB J. 2011, 25, 2344–2353. [Google Scholar] [CrossRef]
- Igaz, L.M.; Kwong, L.K.; Chen-Plotkin, A.; Winton, M.J.; Unger, T.L.; Xu, Y.; Neumann, M.; Trojanowski, J.Q.; Lee, V.M.Y. Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J. Biol. Chem. 2009, 284, 8516–8524. [Google Scholar] [CrossRef]
- Yang, Z.; Lin, F.; Robertson, C.S.; Wang, K.K.W. Dual Vulnerability of TDP-43 to calpain and caspase-3 proteolysis after neurotoxic conditions and traumatic brain injury. J. Cereb. Blood Flow Metab. 2014, 34, 1444–1452. [Google Scholar] [CrossRef]
- Kitamura, A.; Nakayama, Y.; Shibasaki, A.; Taki, A.; Yuno, S.; Takeda, K.; Yahara, M.; Tanabe, N.; Kinjo, M. Interaction of RNA with a C-terminal fragment of the amyotrophic lateral sclerosis-associated TDP43 reduces cytotoxicity. Sci. Rep. 2016, 6, 19230. [Google Scholar] [CrossRef]
- Wang, X.; Ma, M.; Teng, J.; Che, X.; Zhang, W.; Feng, S.; Zhou, S.; Zhang, Y.; Wu, E.; Ding, X. Valproate attenuates 25-kDa C-terminal fragment of TDP-43-induced neuronal toxicity via suppressing endoplasmic reticulum stress and activating autophagy. Int. J. Biol. Sci. 2015, 11, 752–761. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M.Y. Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 2020, 16, 199–212. [Google Scholar] [CrossRef]
- Nonaka, T.; Masuda-Suzukake, M.; Arai, T.; Hasegawa, Y.; Akatsu, H.; Obi, T.; Yoshida, M.; Murayama, S.; Mann, D.M.A.; Akiyama, H.; et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013, 4, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Smethurst, P.; Newcombe, J.; Troakes, C.; Simone, R.; Chen, Y.-R.; Patani, R.; Sidle, K. In vitro prion-like behaviour of TDP-43 in ALS. Neurobiol. Dis. 2016, 96, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Feiler, M.S.; Strobel, B.; Freischmidt, A.; Helferich, A.M.; Kappel, J.; Brewer, B.M.; Li, D.; Thal, D.R.; Walther, P.; Ludolph, A.C.; et al. TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 2015, 211, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, Y.; Eid, L.; Parent, M.; Soucy, G.; Bareil, C.; Riku, Y.; Kawai, K.; Takagi, S.; Yoshida, M.; Katsuno, M.; et al. Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 2016, 139, 3187–3201. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Ma, M.; Teng, J.; Teng, R.K.F.; Zhou, S.; Yin, J.; Fonkem, E.; Huang, J.H.; Wu, E.; Wang, X. Exposure to ALS-FTD-CSF generates TDP-43 aggregates in glioblastoma cells through exosomes and TNTs-like structure. Oncotarget 2015, 6, 24178. [Google Scholar] [CrossRef]
- Fang, Y.-S.; Tsai, K.-J.; Chang, Y.-J.; Kao, P.; Woods, R.; Kuo, P.-H.; Wu, C.-C.; Liao, J.-Y.; Chou, S.-C.; Lin, V.; et al. Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat. Commun. 2014, 5, 4824. [Google Scholar] [CrossRef]
- Kao, P.F.; Chen, Y.-R.; Liu, X.-B.; DeCarli, C.; Seeley, W.W.; Jin, L.-W. Detection of TDP-43 oligomers in frontotemporal lobar degeneration–TDP. Ann. Neurol. 2015, 78, 211–221. [Google Scholar] [CrossRef]
- Jo, M.; Lee, S.; Jeon, Y.-M.; Kim, S.; Kwon, Y.; Kim, H.-J. The role of TDP-43 propagation in neurodegenerative diseases: Integrating insights from clinical and experimental studies. Exp. Mol. Med. 2020, 52, 1652–1662. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chien, H.-M.; Lee, C.-C.; Huang, J.J.-T. The Different Faces of the TDP-43 Low-Complexity Domain: The Formation of Liquid Droplets and Amyloid Fibrils. Int. J. Mol. Sci. 2021, 22, 8213. https://doi.org/10.3390/ijms22158213
Chien H-M, Lee C-C, Huang JJ-T. The Different Faces of the TDP-43 Low-Complexity Domain: The Formation of Liquid Droplets and Amyloid Fibrils. International Journal of Molecular Sciences. 2021; 22(15):8213. https://doi.org/10.3390/ijms22158213
Chicago/Turabian StyleChien, Hung-Ming, Chi-Chang Lee, and Joseph Jen-Tse Huang. 2021. "The Different Faces of the TDP-43 Low-Complexity Domain: The Formation of Liquid Droplets and Amyloid Fibrils" International Journal of Molecular Sciences 22, no. 15: 8213. https://doi.org/10.3390/ijms22158213
APA StyleChien, H.-M., Lee, C.-C., & Huang, J. J.-T. (2021). The Different Faces of the TDP-43 Low-Complexity Domain: The Formation of Liquid Droplets and Amyloid Fibrils. International Journal of Molecular Sciences, 22(15), 8213. https://doi.org/10.3390/ijms22158213