
Fatty Acid Amide Hydrolase (FAAH) Inhibition Modulates Amyloid-Beta-Induced Microglia Polarization

 , , , ,
, , , ,  ,
,  , and
, and
Abstract
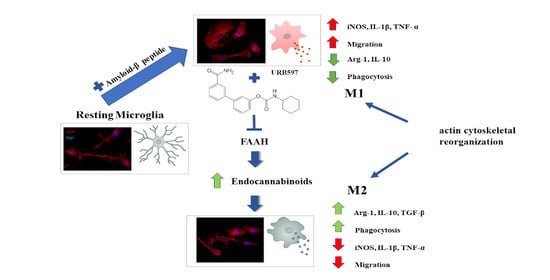
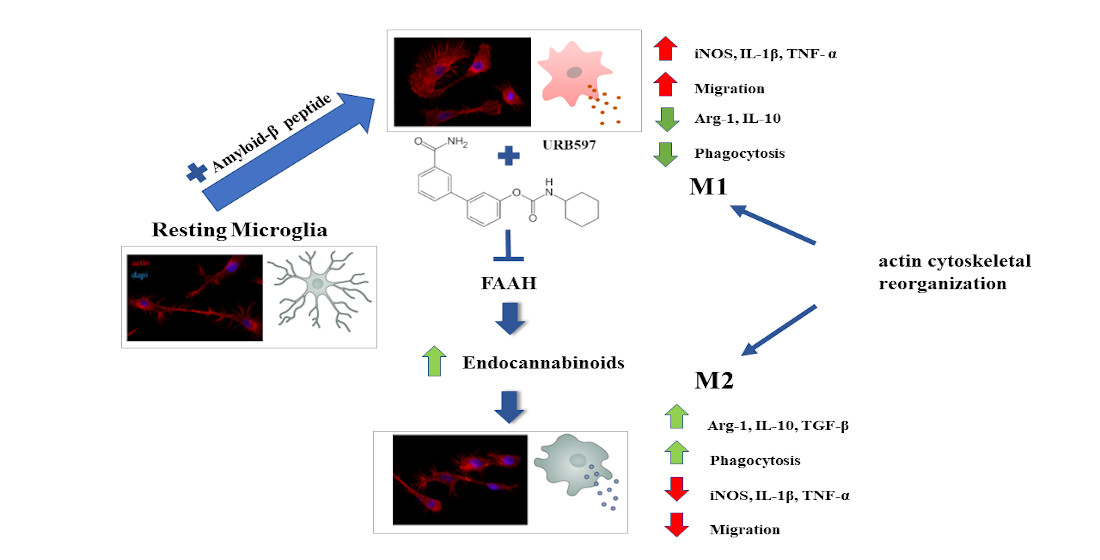
1. Introduction
2. Results
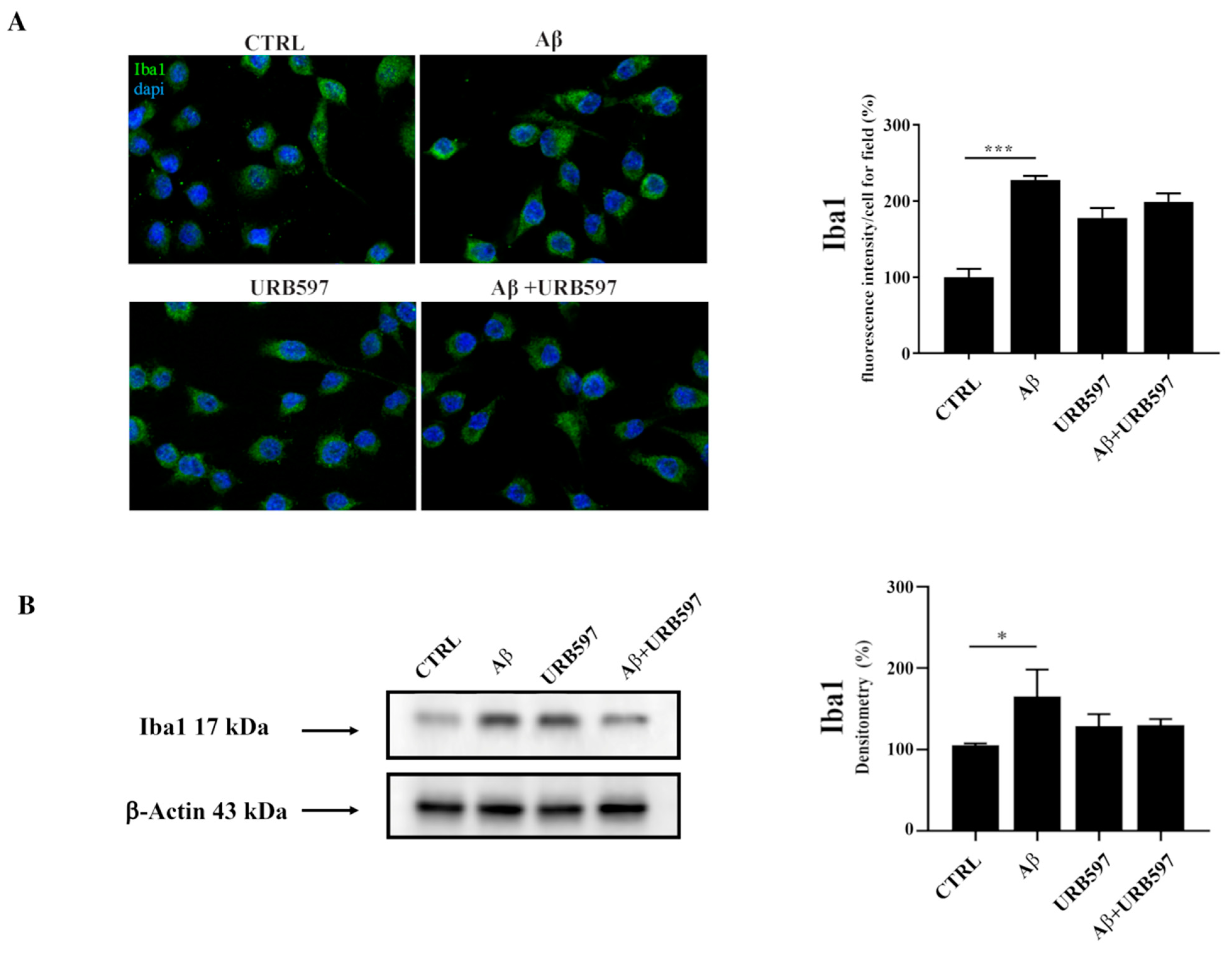
2.1. Aβ25–35 Induces Upregulation of the Iba1 Microglia Marker
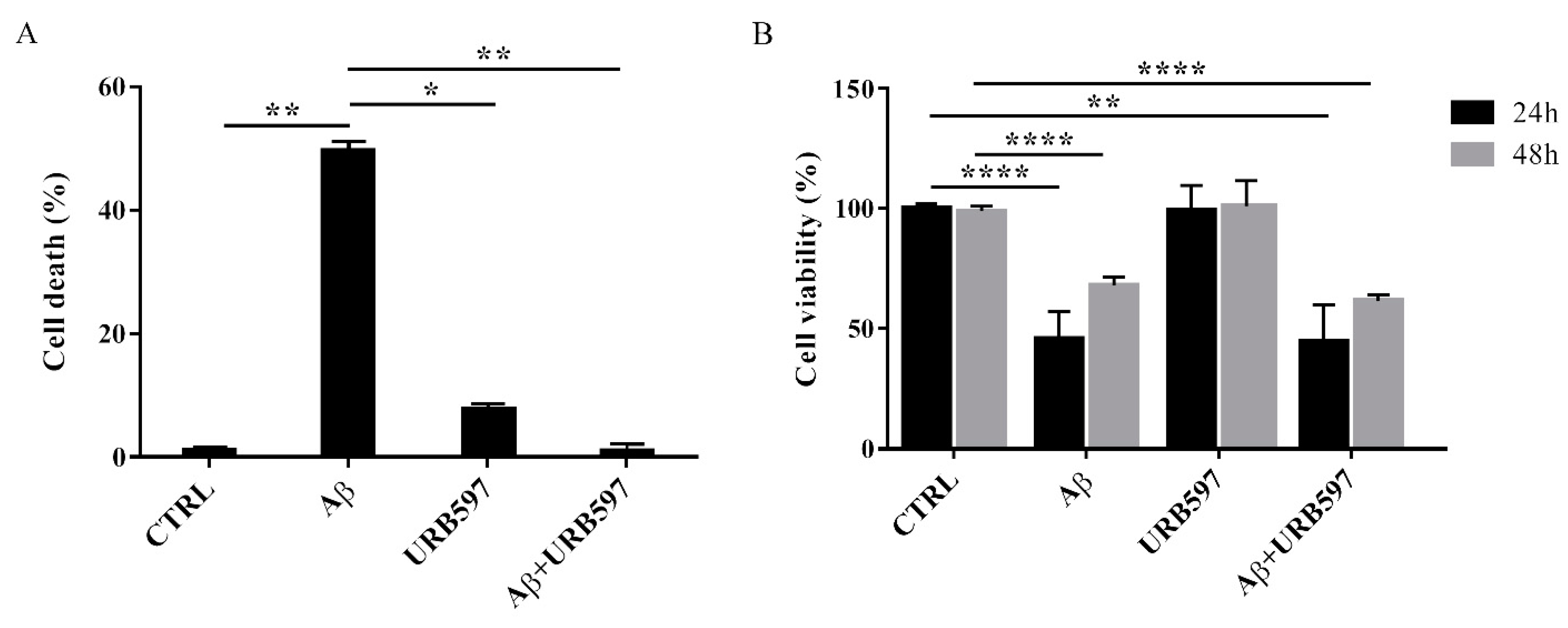
2.2. URB597 on BV-2 Cell Viability
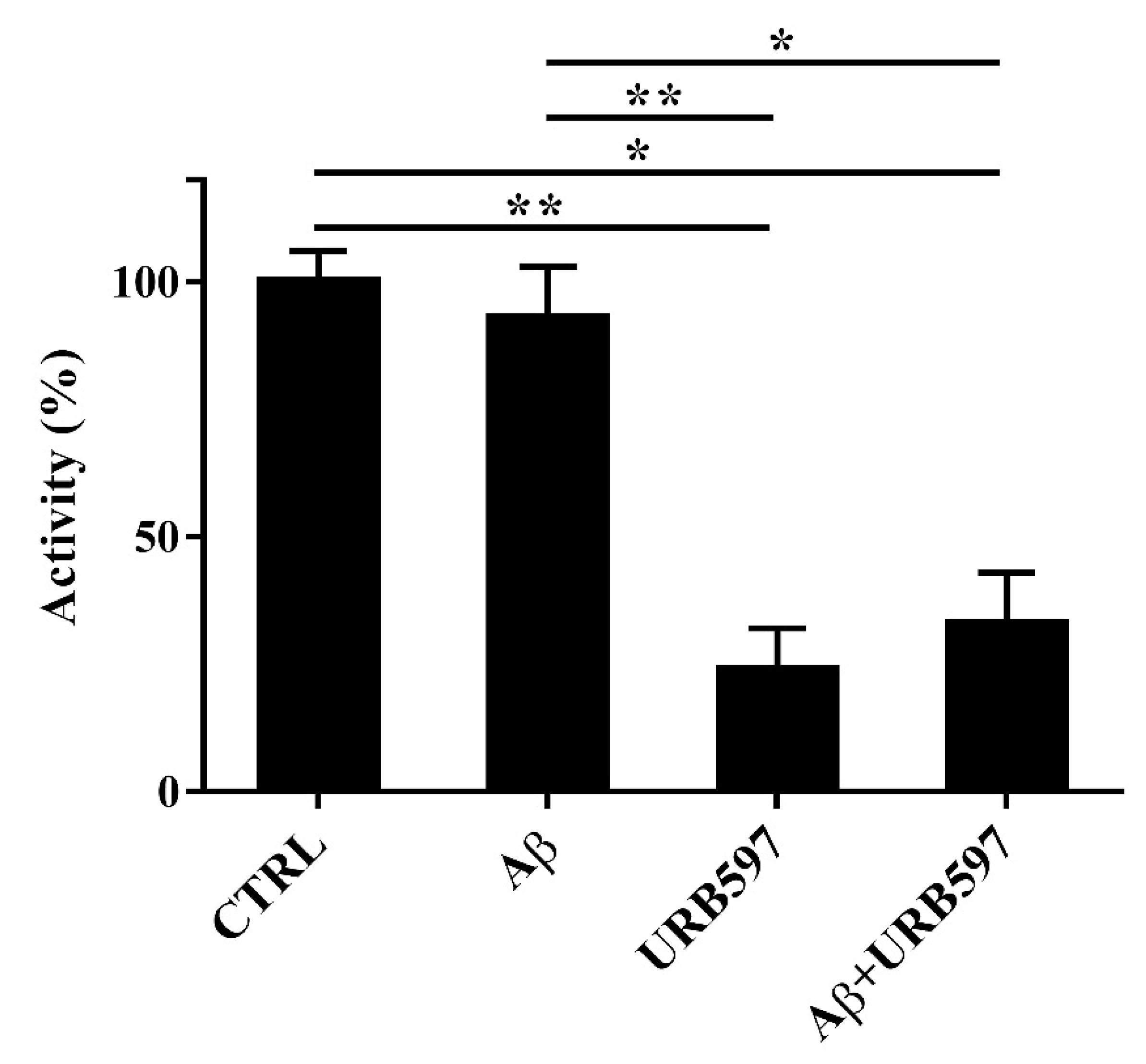
2.3. Aβ25–35 and URB597 Effects on FAAH Enzyme Activity
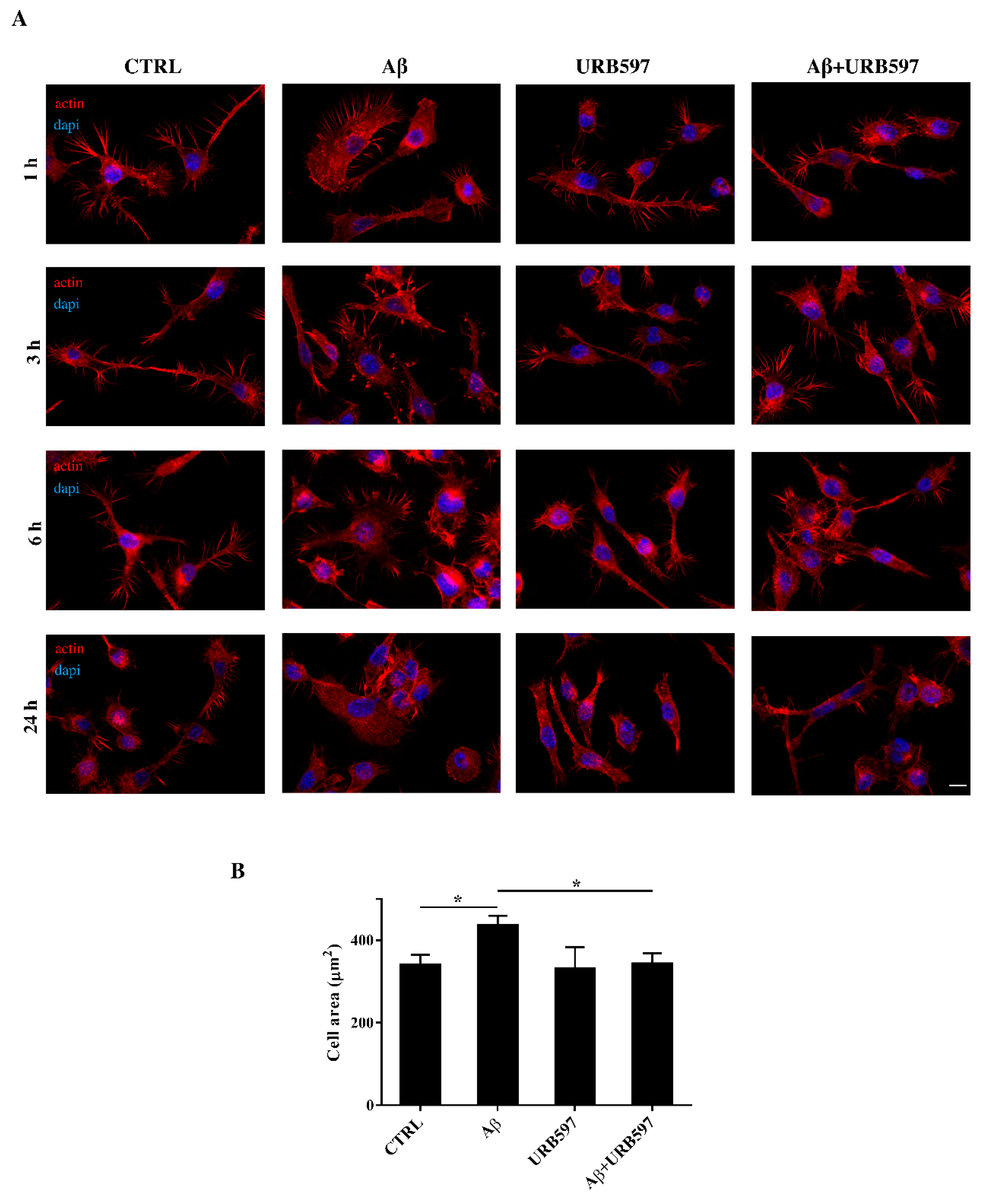
2.4. URB597 Reverts Morphological Changes Induced by Aβ25–35
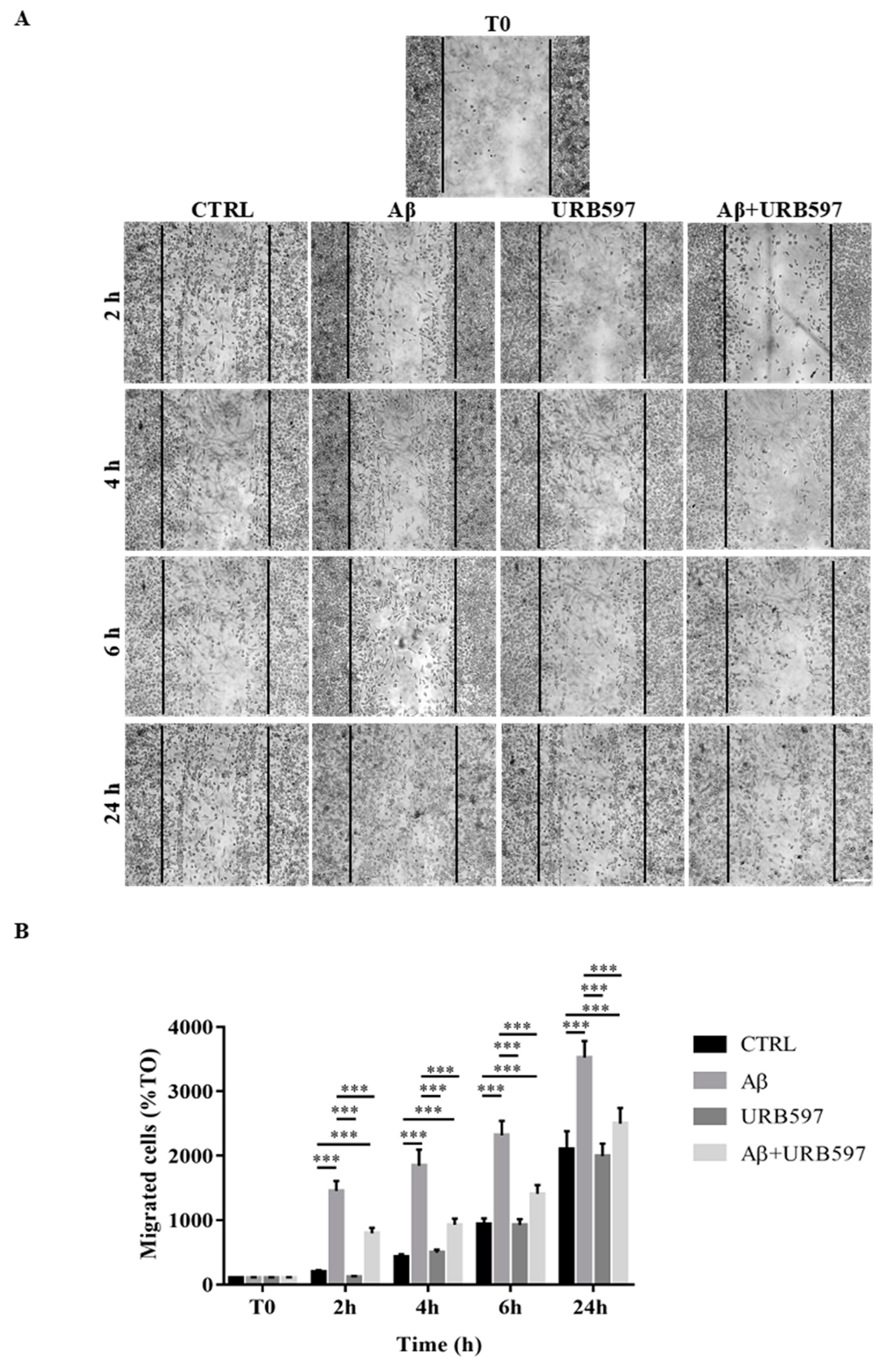
2.5. Effect of URB597 on Cellular Migration
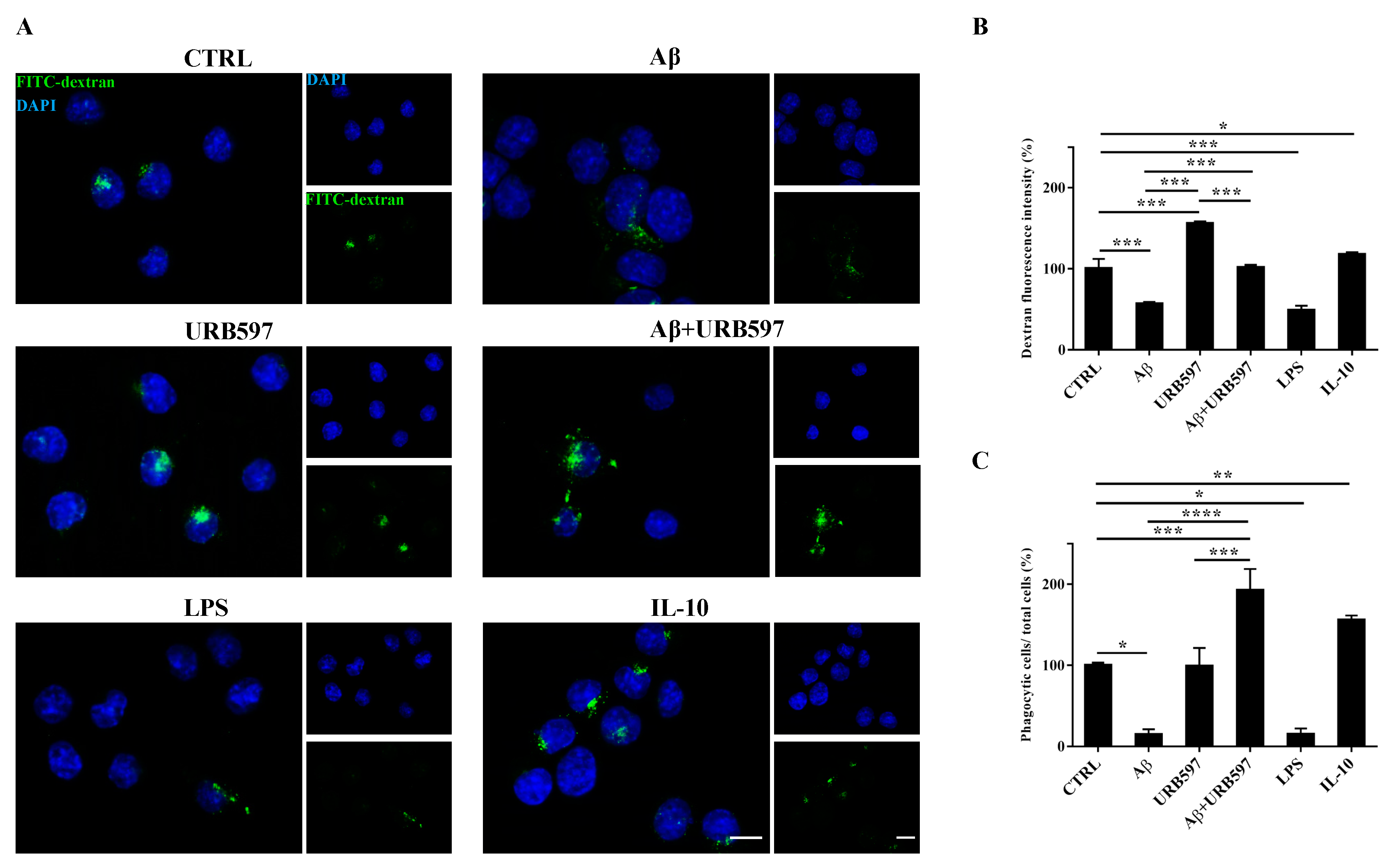
2.6. URB597 Increases Microglia Phagocytic Capacity
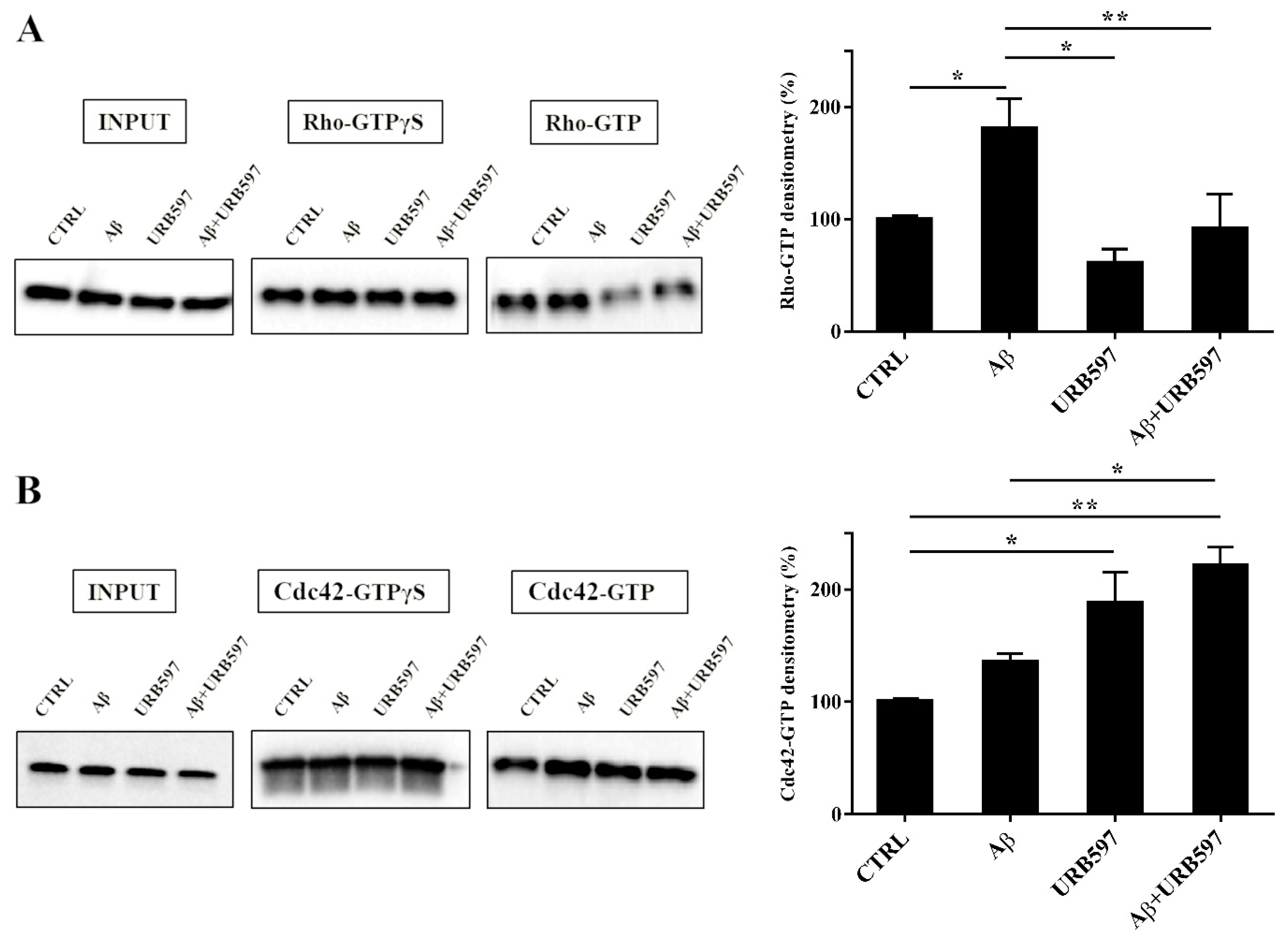
2.7. URB597 Affects Rho GTPases Activity
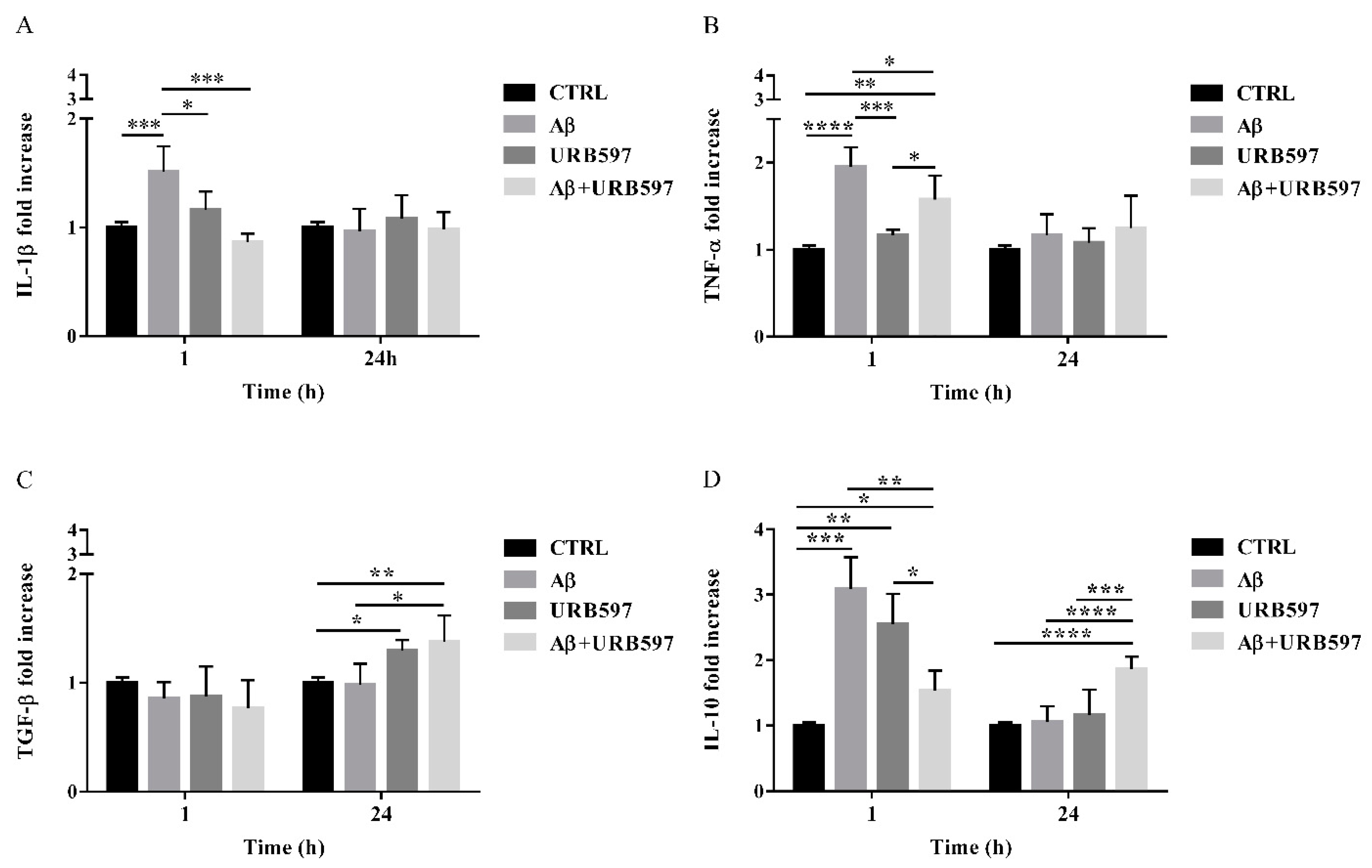
2.8. URB597 Reduces mRNA IL-1β and TNF-α Expression, Increases TGF-β and IL-10 Expression
2.9. URB597 Modulates Both iNOS and Arg-1 Expression
3. Discussion
4. Materials and Methods
4.1. Materials and Chemicals
4.2. Preparation of Aβ25–35 and URB597 Stock Solution
4.3. Fatty Acid Amide Hydrolase Assay
4.4. Cell Cultures and Treatment
4.5. Cell Viability Assays
4.6. Immunocytochemical Analyses
4.7. Migration Assay
4.8. Fluorescein Isothiocyanate (FITC)-Dextran Uptake Assay
4.9. Western Blotting
4.10. Pull-Down Assay for Activated Rho GTPases
4.11. Real-Time Quantitative PCR Analysis
4.12. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mayeux, R.; Sano, M. Treatment of Alzheimer’s disease. N. Engl. J. Med. 1999, 341, 1670–1679. [Google Scholar] [CrossRef]
- Walsh, D.M.; Selkoe, D.J. A beta oligomers—A decade of discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef]
- Yankner, B.A. New clues to Alzheimer’s disease: Unraveling the roles of amyloid and tau. Nat. Med. 1996, 2, 850–852. [Google Scholar] [CrossRef] [PubMed]
- Eikelenboom, P.; Zhan, S.S.; van Gool, W.A.; Allsop, D. Inflammatory mechanisms in Alzheimer’s disease. Trends Pharmacol. Sci. 1994, 15, 447–450. [Google Scholar] [CrossRef]
- McGeer, P.L.; Rogers, J.; McGeer, E.G. Neuroimmune mechanisms in Alzheimer disease pathogenesis. Alzheimer Dis. Assoc. Disord. 1994, 8, 149–158. [Google Scholar] [CrossRef]
- Shippy, D.C.; Ulland, T.K. Microglial Immunometabolism in Alzheimer’s Disease. Front. Cell. Neurosci. 2020, 14, 563446. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Arai, T.; Kondo, H.; Tanno, E.; Haga, C.; Ikeda, K. Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Dis. Assoc. Disord. 2000, 14 (Suppl. 1), S47–S53. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Yamasaki, T.R.; LaFerla, F.M. Microglia as a potential bridge between the amyloid beta-peptide and tau. Ann. N. Y. Acad. Sci. 2004, 1035, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.; Strohmeyer, R.; Kovelowski, C.J.; Li, R. Microglia and inflammatory mechanisms in the clearance of amyloid beta peptide. Glia 2002, 40, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.; Cash, A.D.; Smith, M.A. Alzheimer Disease and Oxidative Stress. J. Biomed. Biotechnol. 2002, 2, 120–123. [Google Scholar] [CrossRef]
- Friedman, D.; French, J.A.; Maccarrone, M. Safety, efficacy, and mechanisms of action of cannabinoids in neurological disorders. Lancet Neurol. 2019, 18, 504–512. [Google Scholar] [CrossRef]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Malorni, W.; Bari, M.; Straface, E.; Battista, N.; Matarrese, P.; Finazzi-Agro, A.; Del Principe, D.; Maccarrone, M. Morphological evidence that 2-arachidonoylglycerol is a true agonist of human platelets. Thromb. Haemost. 2004, 92, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Fezza, F.; Bari, M.; Florio, R.; Talamonti, E.; Feole, M.; Maccarrone, M. Endocannabinoids, related compounds and their metabolic routes. Molecules 2014, 19, 17078–17106. [Google Scholar] [CrossRef]
- Jackson, S.J.; Diemel, L.T.; Pryce, G.; Baker, D. Cannabinoids and neuroprotection in CNS inflammatory disease. J. Neurol. Sci. 2005, 233, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Benito, C.; Nunez, E.; Tolon, R.M.; Carrier, E.J.; Rabano, A.; Hillard, C.J.; Romero, J. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J. Neurosci. 2003, 23, 11136–11141. [Google Scholar] [CrossRef]
- Ramirez, B.G.; Blazquez, C.; Gomez del Pulgar, T.; Guzman, M.; de Ceballos, M.L. Prevention of Alzheimer’s disease pathology by cannabinoids: Neuroprotection mediated by blockade of microglial activation. J. Neurosci. 2005, 25, 1904–1913. [Google Scholar] [CrossRef]
- Maccarrone, M.; Totaro, A.; Leuti, A.; Giacovazzo, G.; Scipioni, L.; Mango, D.; Coccurello, R.; Nistico, R.; Oddi, S. Early alteration of distribution and activity of hippocampal type-1 cannabinoid receptor in Alzheimer’s disease-like mice overexpressing the human mutant amyloid precursor protein. Pharmacol. Res. 2018, 130, 366–373. [Google Scholar] [CrossRef]
- Aso, E.; Juves, S.; Maldonado, R.; Ferrer, I. CB2 cannabinoid receptor agonist ameliorates Alzheimer-like phenotype in AbetaPP/PS1 mice. J. Alzheimers Dis. 2013, 35, 847–858. [Google Scholar] [CrossRef]
- Martin-Moreno, A.M.; Brera, B.; Spuch, C.; Carro, E.; Garcia-Garcia, L.; Delgado, M.; Pozo, M.A.; Innamorato, N.G.; Cuadrado, A.; de Ceballos, M.L. Prolonged oral cannabinoid administration prevents neuroinflammation, lowers beta-amyloid levels and improves cognitive performance in Tg APP 2576 mice. J. Neuroinflamm. 2012, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Walter, L.; Stella, N. Cannabinoids and neuroinflammation. Br. J. Pharmacol. 2004, 141, 775–785. [Google Scholar] [CrossRef]
- Jung, K.M.; Astarita, G.; Yasar, S.; Vasilevko, V.; Cribbs, D.H.; Head, E.; Cotman, C.W.; Piomelli, D. An amyloid beta42-dependent deficit in anandamide mobilization is associated with cognitive dysfunction in Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1522–1532. [Google Scholar] [CrossRef]
- Chen, H.C.; Spiers, J.G.; Sernia, C.; Lavidis, N.A. Inhibition of Fatty Acid Amide Hydrolase by PF-3845 Alleviates the Nitrergic and Proinflammatory Response in Rat Hippocampus Following Acute Stress. Int. J. Neuropsychopharmacol. 2018, 21, 786–795. [Google Scholar] [CrossRef]
- Piro, J.R.; Benjamin, D.I.; Duerr, J.M.; Pi, Y.; Gonzales, C.; Wood, K.M.; Schwartz, J.W.; Nomura, D.K.; Samad, T.A. A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of Alzheimer’s disease. Cell Rep. 2012, 1, 617–623. [Google Scholar] [CrossRef]
- Vazquez, C.; Tolon, R.M.; Grande, M.T.; Caraza, M.; Moreno, M.; Koester, E.C.; Villaescusa, B.; Ruiz-Valdepenas, L.; Fernandez-Sanchez, F.J.; Cravatt, B.F.; et al. Endocannabinoid regulation of amyloid-induced neuroinflammation. Neurobiol. Aging 2015, 36, 3008–3019. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Yagyu, K.; Sackett, S.; Zhang, Y. Anti-Inflammatory Effects by Pharmacological Inhibition or Knockdown of Fatty Acid Amide Hydrolase in BV2 Microglial Cells. Cells 2019, 8, 491. [Google Scholar] [CrossRef] [PubMed]
- Harry, G.J. Microglia during development and aging. Pharmacol. Ther. 2013, 139, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Mori, I.; Imai, Y.; Kohsaka, S.; Kimura, Y. Upregulated expression of Iba1 molecules in the central nervous system of mice in response to neurovirulent influenza A virus infection. Microbiol. Immunol. 2000, 44, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Frozza, R.L.; Horn, A.P.; Hoppe, J.B.; Simao, F.; Gerhardt, D.; Comiran, R.A.; Salbego, C.G. A comparative study of beta-amyloid peptides Abeta1-42 and Abeta25-35 toxicity in organotypic hippocampal slice cultures. Neurochem. Res. 2009, 34, 295–303. [Google Scholar] [CrossRef]
- Lucin, K.M.; Wyss-Coray, T. Immune activation in brain aging and neurodegeneration: Too much or too little? Neuron 2009, 64, 110–122. [Google Scholar] [CrossRef]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef]
- Francos-Quijorna, I.; Amo-Aparicio, J.; Martinez-Muriana, A.; Lopez-Vales, R. IL-4 drives microglia and macrophages toward a phenotype conducive for tissue repair and functional recovery after spinal cord injury. Glia 2016, 64, 2079–2092. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006, 16, 522–529. [Google Scholar] [CrossRef]
- Stankiewicz, T.R.; Linseman, D.A. Rho family GTPases: Key players in neuronal development, neuronal survival, and neurodegeneration. Front. Cell. Neurosci. 2014, 8, 314. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Das, R.; Chinnathambi, S. Actin-mediated Microglial Chemotaxis via G-Protein Coupled Purinergic Receptor in Alzheimer’s Disease. Neuroscience 2020, 448, 325–336. [Google Scholar] [CrossRef]
- Sferra, A.; Nicita, F.; Bertini, E. Microtubule Dysfunction: A Common Feature of Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 7354. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Cabral, G.A.; Ferreira, G.A.; Jamerson, M.J. Endocannabinoids and the Immune System in Health and Disease. Handb. Exp. Pharmacol. 2015, 231, 185–211. [Google Scholar] [CrossRef]
- Talarico, G.; Trebbastoni, A.; Bruno, G.; de Lena, C. Modulation of the Cannabinoid System: A New Perspective for the Treatment of the Alzheimer’s Disease. Curr. Neuropharmacol. 2019, 17, 176–183. [Google Scholar] [CrossRef]
- Laezza, C.; Pagano, C.; Navarra, G.; Pastorino, O.; Proto, M.C.; Fiore, D.; Piscopo, C.; Gazzerro, P.; Bifulco, M. The Endocannabinoid System: A Target for Cancer Treatment. Int. J. Mol. Sci. 2020, 21, 747. [Google Scholar] [CrossRef]
- Stella, N. Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia 2010, 58, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Benard, G.; Massa, F.; Puente, N.; Lourenco, J.; Bellocchio, L.; Soria-Gomez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB(1) receptors regulate neuronal energy metabolism. Nat. Neurosci. 2012, 15, 558–564. [Google Scholar] [CrossRef]
- Zaccagnino, P.; Corcelli, A.; Baronio, M.; Lorusso, M. Anandamide inhibits oxidative phosphorylation in isolated liver mitochondria. FEBS Lett. 2011, 585, 429–434. [Google Scholar] [CrossRef]
- Pagani, L.; Eckert, A. Amyloid-Beta interaction with mitochondria. Int. J. Alzheimers Dis. 2011, 2011, 925050. [Google Scholar] [CrossRef]
- Sharma, C.; Kim, S.; Nam, Y.; Jung, U.J.; Kim, S.R. Mitochondrial Dysfunction as a Driver of Cognitive Impairment in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4850. [Google Scholar] [CrossRef]
- Mikheeva, I.B.; Shubina, L.; Matveeva, N.; Pavlik, L.L.; Kitchigina, V.F. Fatty acid amide hydrolase inhibitor URB597 may protect against kainic acid-induced damage to hippocampal neurons: Dependence on the degree of injury. Epilepsy Res. 2017, 137, 84–94. [Google Scholar] [CrossRef]
- Piomelli, D.; Tarzia, G.; Duranti, A.; Tontini, A.; Mor, M.; Compton, T.R.; Dasse, O.; Monaghan, E.P.; Parrott, J.A.; Putman, D. Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597). CNS Drug Rev. 2006, 12, 21–38. [Google Scholar] [CrossRef]
- Ehrhart, J.; Obregon, D.; Mori, T.; Hou, H.; Sun, N.; Bai, Y.; Klein, T.; Fernandez, F.; Tan, J.; Shytle, R.D. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J. Neuroinflamm. 2005, 2, 29. [Google Scholar] [CrossRef]
- Guida, F.; Luongo, L.; Boccella, S.; Giordano, M.E.; Romano, R.; Bellini, G.; Manzo, I.; Furiano, A.; Rizzo, A.; Imperatore, R.; et al. Palmitoylethanolamide induces microglia changes associated with increased migration and phagocytic activity: Involvement of the CB2 receptor. Sci. Rep. 2017, 7, 375. [Google Scholar] [CrossRef] [PubMed]
- Romero-Sandoval, E.A.; Horvath, R.; Landry, R.P.; DeLeo, J.A. Cannabinoid receptor type 2 activation induces a microglial anti-inflammatory phenotype and reduces migration via MKP induction and ERK dephosphorylation. Mol. Pain 2009, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Benito, C.; Tolon, R.M.; Castillo, A.I.; Ruiz-Valdepenas, L.; Martinez-Orgado, J.A.; Fernandez-Sanchez, F.J.; Vazquez, C.; Cravatt, B.F.; Romero, J. beta-Amyloid exacerbates inflammation in astrocytes lacking fatty acid amide hydrolase through a mechanism involving PPAR-alpha, PPAR-gamma and TRPV1, but not CB(1) or CB(2) receptors. Br. J. Pharmacol. 2012, 166, 1474–1489. [Google Scholar] [CrossRef]
- Bosier, B.; Muccioli, G.G.; Lambert, D.M. The FAAH inhibitor URB597 efficiently reduces tyrosine hydroxylase expression through CB(1)- and FAAH-independent mechanisms. Br. J. Pharmacol. 2013, 169, 794–807. [Google Scholar] [CrossRef]
- Zhang, D.; Saraf, A.; Kolasa, T.; Bhatia, P.; Zheng, G.Z.; Patel, M.; Lannoye, G.S.; Richardson, P.; Stewart, A.; Rogers, J.C.; et al. Fatty acid amide hydrolase inhibitors display broad selectivity and inhibit multiple carboxylesterases as off-targets. Neuropharmacology 2007, 52, 1095–1105. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Rajendran, L.; Paolicelli, R.C. Microglia-Mediated Synapse Loss in Alzheimer’s Disease. J. Neurosci. 2018, 38, 2911–2919. [Google Scholar] [CrossRef]
- Lau, S.F.; Fu, A.K.Y.; Ip, N.Y. Cytokine signaling convergence regulates the microglial state transition in Alzheimer’s disease. Cell. Mol. Life Sci. 2021, 78, 4703–4712. [Google Scholar] [CrossRef]
- Victor, T.R.; Tsirka, S.E. Microglial contributions to aberrant neurogenesis and pathophysiology of epilepsy. Neuroimmunol. Neuroinflamm. 2020, 7, 234–247. [Google Scholar] [CrossRef]
- Hao, S.; Dey, A.; Yu, X.; Stranahan, A.M. Dietary obesity reversibly induces synaptic stripping by microglia and impairs hippocampal plasticity. Brain Behav. Immun. 2016, 51, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Balmik, A.A.; Chinnathambi, S. Phagocytosis of full-length Tau oligomers by Actin-remodeling of activated microglia. J. Neuroinflamm. 2020, 17, 10. [Google Scholar] [CrossRef]
- Uhlemann, R.; Gertz, K.; Boehmerle, W.; Schwarz, T.; Nolte, C.; Freyer, D.; Kettenmann, H.; Endres, M.; Kronenberg, G. Actin dynamics shape microglia effector functions. Brain Struct. Funct. 2016, 221, 2717–2734. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Khoshbouei, H.; Bechmann, I. Dystrophic microglia in late-onset Alzheimer’s disease. Glia 2020, 68, 845–854. [Google Scholar] [CrossRef]
- Streit, W.J. Microglial senescence: Does the brain’s immune system have an expiration date? Trends Neurosci. 2006, 29, 506–510. [Google Scholar] [CrossRef]
- Burridge, K.; Wennerberg, K. Rho and Rac take center stage. Cell 2004, 116, 167–179. [Google Scholar] [CrossRef]
- Moon, M.Y.; Kim, H.J.; Li, Y.; Kim, J.G.; Jeon, Y.J.; Won, H.Y.; Kim, J.S.; Kwon, H.Y.; Choi, I.G.; Ro, E.; et al. Involvement of small GTPase RhoA in the regulation of superoxide production in BV2 cells in response to fibrillar Abeta peptides. Cell Signal. 2013, 25, 1861–1869. [Google Scholar] [CrossRef]
- Johnson, D.I. Cdc42: An essential Rho-type GTPase controlling eukaryotic cell polarity. Microbiol. Mol. Biol. Rev. 1999, 63, 54–105. [Google Scholar] [CrossRef] [PubMed]
- Govek, E.E.; Wu, Z.; Acehan, D.; Molina, H.; Rivera, K.; Zhu, X.; Fang, Y.; Tessier-Lavigne, M.; Hatten, M.E. Cdc42 Regulates Neuronal Polarity during Cerebellar Axon Formation and Glial-Guided Migration. iScience 2018, 1, 35–48. [Google Scholar] [CrossRef]
- Aguilar, B.J.; Zhu, Y.; Lu, Q. Rho GTPases as therapeutic targets in Alzheimer’s disease. Alzheimers Res. Ther. 2017, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Petratos, S.; Li, Q.X.; George, A.J.; Hou, X.; Kerr, M.L.; Unabia, S.E.; Hatzinisiriou, I.; Maksel, D.; Aguilar, M.I.; Small, D.H. The beta-amyloid protein of Alzheimer’s disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain 2008, 131, 90–108. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Alonso, J.; de Salas-Quiroga, A.; Paraiso-Luna, J.; Garcia-Rincon, D.; Garcez, P.P.; Parsons, M.; Andradas, C.; Sanchez, C.; Guillemot, F.; Guzman, M.; et al. Loss of Cannabinoid CB1 Receptors Induces Cortical Migration Malformations and Increases Seizure Susceptibility. Cereb. Cortex 2017, 27, 5303–5317. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kaplan, H.M.; Pazarci, P. Effects of chronic Delta(9)-tetrahydrocannabinol treatment on Rho/Rho-kinase signalization pathway in mouse brain. Saudi Pharm. J. 2017, 25, 1078–1081. [Google Scholar] [CrossRef]
- Mendoza-Naranjo, A.; Gonzalez-Billault, C.; Maccioni, R.B. Abeta1-42 stimulates actin polymerization in hippocampal neurons through Rac1 and Cdc42 Rho GTPases. J. Cell Sci. 2007, 120, 279–288. [Google Scholar] [CrossRef]
- Kurihara, R.; Tohyama, Y.; Matsusaka, S.; Naruse, H.; Kinoshita, E.; Tsujioka, T.; Katsumata, Y.; Yamamura, H. Effects of peripheral cannabinoid receptor ligands on motility and polarization in neutrophil-like HL60 cells and human neutrophils. J. Biol. Chem. 2006, 281, 12908–12918. [Google Scholar] [CrossRef] [PubMed]
- Carlisle, S.J.; Marciano-Cabral, F.; Staab, A.; Ludwick, C.; Cabral, G.A. Differential expression of the CB2 cannabinoid receptor by rodent macrophages and macrophage-like cells in relation to cell activation. Int. Immunopharmacol. 2002, 2, 69–82. [Google Scholar] [CrossRef]
- Mecha, M.; Feliu, A.; Carrillo-Salinas, F.J.; Rueda-Zubiaurre, A.; Ortega-Gutierrez, S.; de Sola, R.G.; Guaza, C. Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav. Immun. 2015, 49, 233–245. [Google Scholar] [CrossRef]
- Luo, X.Q.; Li, A.; Yang, X.; Xiao, X.; Hu, R.; Wang, T.W.; Dou, X.Y.; Yang, D.J.; Dong, Z. Paeoniflorin exerts neuroprotective effects by modulating the M1/M2 subset polarization of microglia/macrophages in the hippocampal CA1 region of vascular dementia rats via cannabinoid receptor 2. Chin. Med. 2018, 13, 14. [Google Scholar] [CrossRef]
- Tanaka, M.; Sackett, S.; Zhang, Y. Endocannabinoid Modulation of Microglial Phenotypes in Neuropathology. Front. Neurol. 2020, 11, 87. [Google Scholar] [CrossRef]
- Raza, A.; Crothers, J.W.; McGill, M.M.; Mawe, G.M.; Teuscher, C.; Krementsov, D.N. Anti-inflammatory roles of p38alpha MAPK in macrophages are context dependent and require IL-10. J. Leukoc. Biol. 2017, 102, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.L.; Li, X.Y.; Zhou, X.; Yuan, L.H.; Ma, W.W.; Xi, Y.D.; Zhao, X.; Wu, J.; Xiao, R. Beta amyloid peptide (25–35) leading to inflammation through Toll-like receptors and the anti-inflammatory effect of genistein in BV-2 cells. J. Mol. Neurosci. 2013, 51, 771–778. [Google Scholar] [CrossRef]
- Guillot-Sestier, M.V.; Doty, K.R.; Gate, D.; Rodriguez, J., Jr.; Leung, B.P.; Rezai-Zadeh, K.; Town, T. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron 2015, 85, 534–548. [Google Scholar] [CrossRef]
- Asselineau, D.; Benlhassan, K.; Arosio, B.; Mari, D.; Ferri, E.; Casati, M.; Gussago, C.; Tedone, E.; Annoni, G.; Mazzola, P.; et al. Interleukin-10 Production in Response to Amyloid-beta Differs between Slow and Fast Decliners in Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2015, 46, 837–842. [Google Scholar] [CrossRef]
- Tchantchou, F.; Tucker, L.B.; Fu, A.H.; Bluett, R.J.; McCabe, J.T.; Patel, S.; Zhang, Y. The fatty acid amide hydrolase inhibitor PF-3845 promotes neuronal survival, attenuates inflammation and improves functional recovery in mice with traumatic brain injury. Neuropharmacology 2014, 85, 427–439. [Google Scholar] [CrossRef]
- Rivera, P.; Fernandez-Arjona, M.D.M.; Silva-Pena, D.; Blanco, E.; Vargas, A.; Lopez-Avalos, M.D.; Grondona, J.M.; Serrano, A.; Pavon, F.J.; Rodriguez de Fonseca, F.; et al. Pharmacological blockade of fatty acid amide hydrolase (FAAH) by URB597 improves memory and changes the phenotype of hippocampal microglia despite ethanol exposure. Biochem. Pharmacol. 2018, 157, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Tham, C.S.; Whitaker, J.; Luo, L.; Webb, M. Inhibition of microglial fatty acid amide hydrolase modulates LPS stimulated release of inflammatory mediators. FEBS Lett. 2007, 581, 2899–2904. [Google Scholar] [CrossRef]
- Su, S.H.; Wu, Y.F.; Lin, Q.; Hai, J. Cannabinoid receptor agonist WIN55,212-2 and fatty acid amide hydrolase inhibitor URB597 ameliorate neuroinflammatory responses in chronic cerebral hypoperfusion model by blocking NF-kappaB pathways. Naunyn Schmiedebergs Arch. Pharmacol. 2017, 390, 1189–1200. [Google Scholar] [CrossRef]
- Colombo, R.; Atherton, E.; Sheppard, R.C.; Woolley, V. 4-Chloromethylphenoxyacetyl polystyrene and polyamide supports for solid-phase peptide synthesis. Int. J. Pept. Protein Res. 1983, 21, 118–126. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENE | Forward Primer (5′–3′) | Reverse Primer (5′–3′) | Accession Numbers |
|---|---|---|---|
| mIL-1β | GAAATGCCACCTTTTGACAGTG | TGGATGCTCTCATCAGGACAG | NM_008361.4 |
| mTNF-α | CTGAACTTCGGGGTGATCGG | GGCTTGTCACTCGAATTTTGAGA | BC137720.1 |
| mTGF-β | CTCCCGTGGCTTCTAGTGC | GCCTTAGTTTGGACAGGATCTG | NM_011577.2 |
| mIL-10 | GCCCTTTGCTATGGTGTCCTTTC | TCCCTGGTTTCTCTTCCCAAGAC | NM_010548.2 |
| mR18s | AAATCAGTTATGGTTCCTTTGGTC | GCTCTAGAATTACCACAGTTATCCAA | M27358 |
| mARG1 | ATGTGCCCTCTGTCTTTTAGGG | GGTCTCTCACGTCATACTCTGT | NM_007482.3 |
| miNOS | GGCAGCCTGTGAGACCTTTG | GCATTGGAAGTGAAGCGTTTC | AF427516.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grieco, M.; De Caris, M.G.; Maggi, E.; Armeli, F.; Coccurello, R.; Bisogno, T.; D’Erme, M.; Maccarrone, M.; Mancini, P.; Businaro, R. Fatty Acid Amide Hydrolase (FAAH) Inhibition Modulates Amyloid-Beta-Induced Microglia Polarization. Int. J. Mol. Sci. 2021, 22, 7711. https://doi.org/10.3390/ijms22147711
Grieco M, De Caris MG, Maggi E, Armeli F, Coccurello R, Bisogno T, D’Erme M, Maccarrone M, Mancini P, Businaro R. Fatty Acid Amide Hydrolase (FAAH) Inhibition Modulates Amyloid-Beta-Induced Microglia Polarization. International Journal of Molecular Sciences. 2021; 22(14):7711. https://doi.org/10.3390/ijms22147711
Chicago/Turabian StyleGrieco, Maddalena, Maria Giovanna De Caris, Elisa Maggi, Federica Armeli, Roberto Coccurello, Tiziana Bisogno, Maria D’Erme, Mauro Maccarrone, Patrizia Mancini, and Rita Businaro. 2021. "Fatty Acid Amide Hydrolase (FAAH) Inhibition Modulates Amyloid-Beta-Induced Microglia Polarization" International Journal of Molecular Sciences 22, no. 14: 7711. https://doi.org/10.3390/ijms22147711
APA StyleGrieco, M., De Caris, M. G., Maggi, E., Armeli, F., Coccurello, R., Bisogno, T., D’Erme, M., Maccarrone, M., Mancini, P., & Businaro, R. (2021). Fatty Acid Amide Hydrolase (FAAH) Inhibition Modulates Amyloid-Beta-Induced Microglia Polarization. International Journal of Molecular Sciences, 22(14), 7711. https://doi.org/10.3390/ijms22147711