
Behavioral Phenotyping in a Murine Model of GBA1-Associated Parkinson Disease



Abstract
:1. Introduction
2. Results
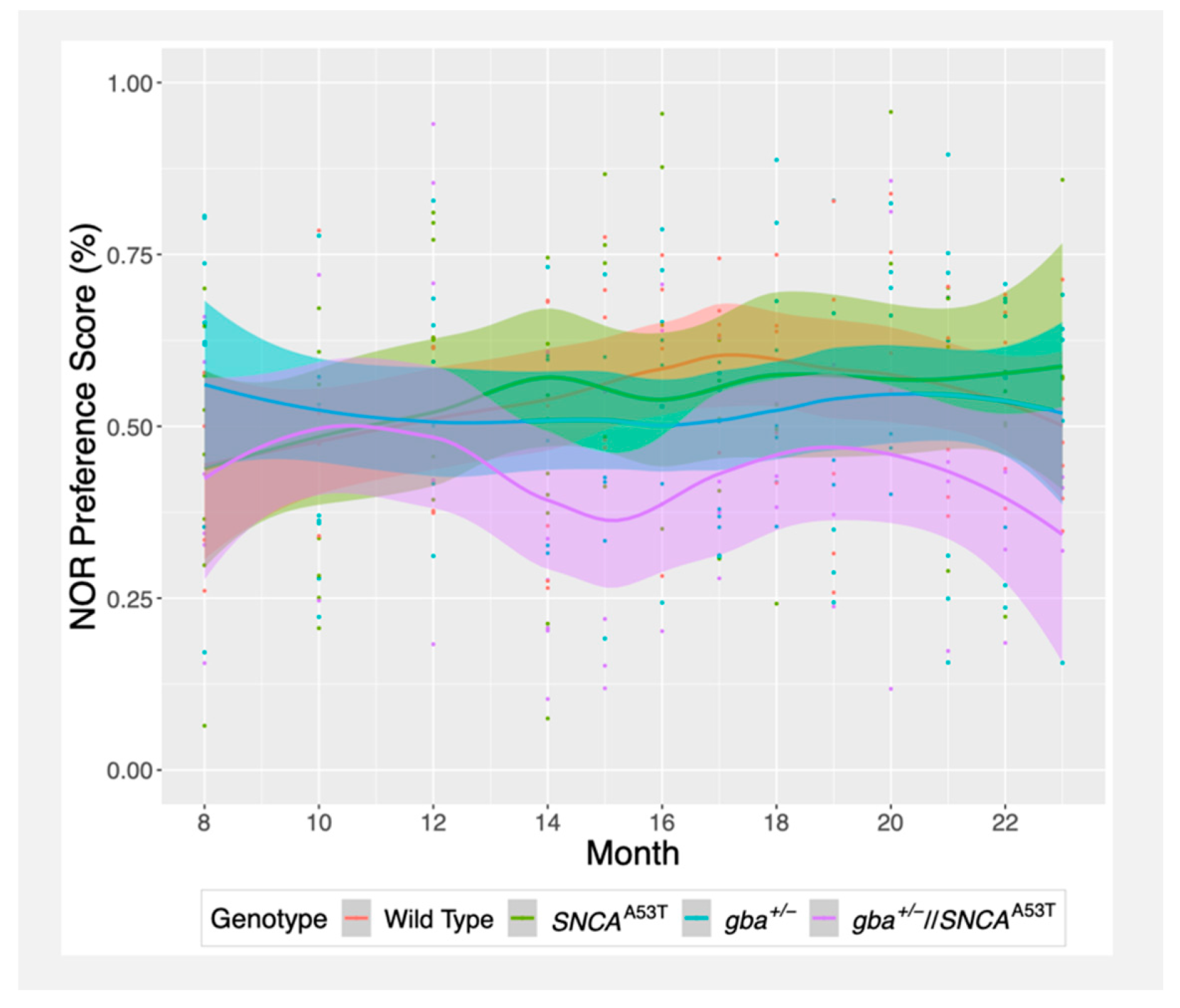
2.1. Novel Object Recognition (NOR)
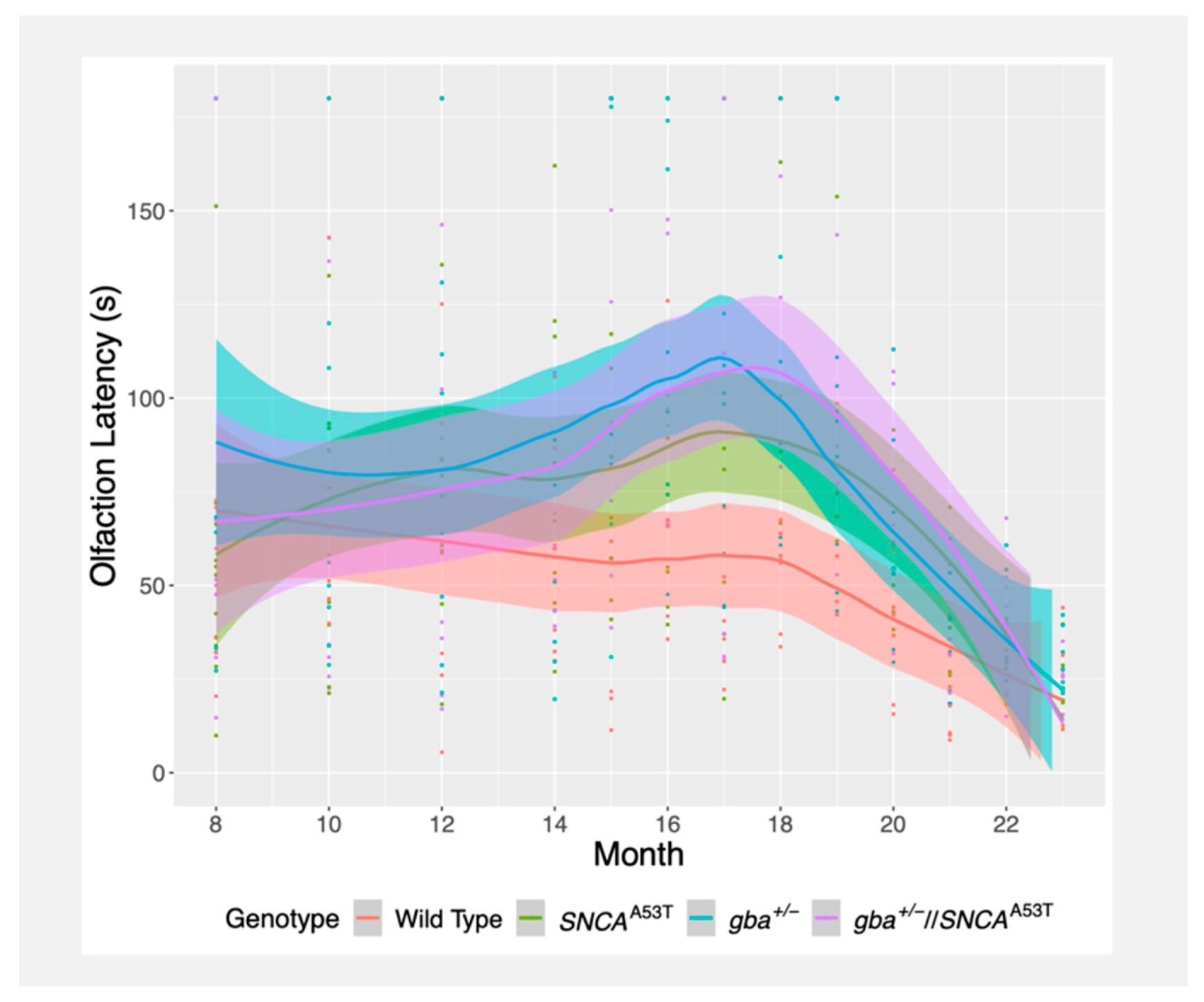
2.2. Buried Pellet Test
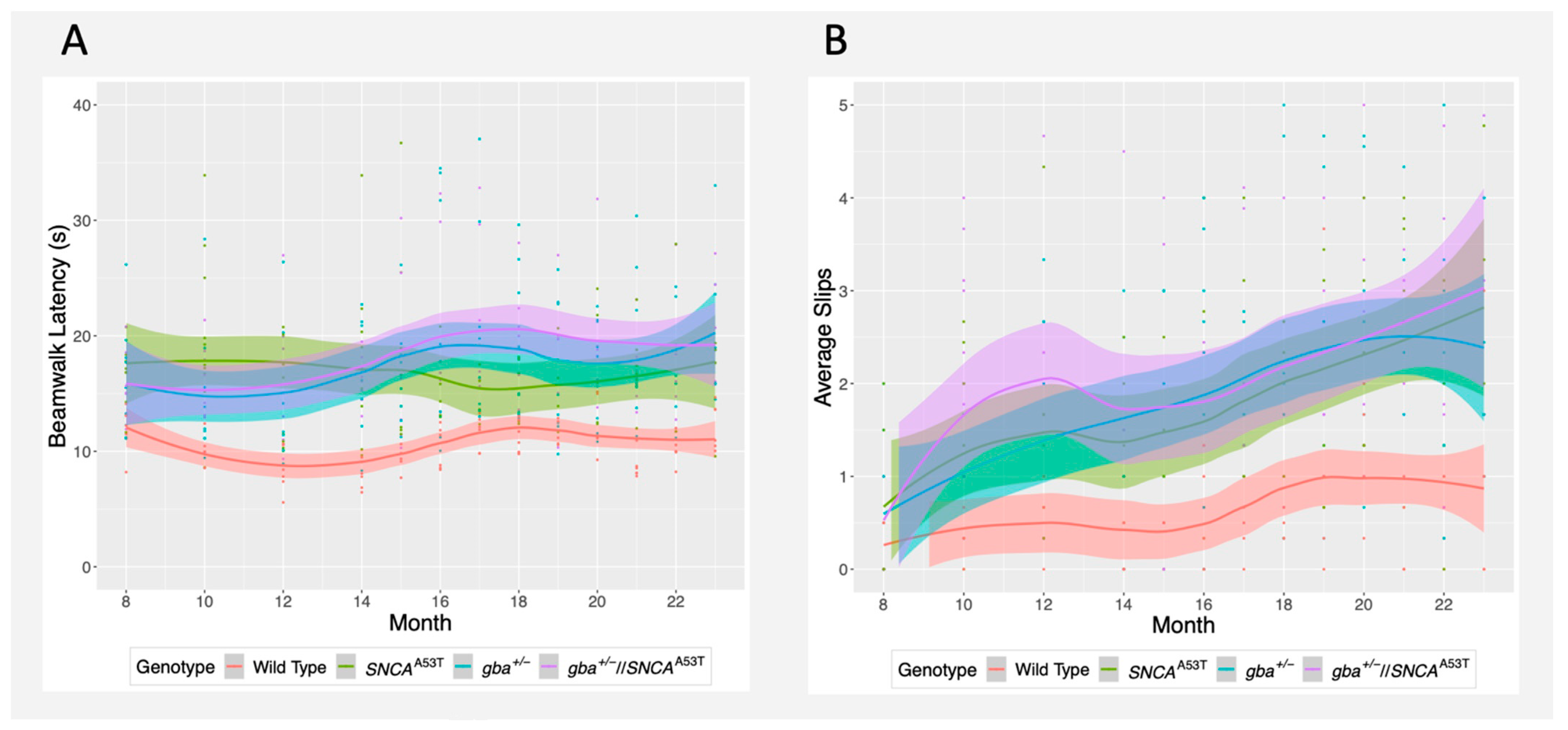
2.3. Traverse Beam Walk
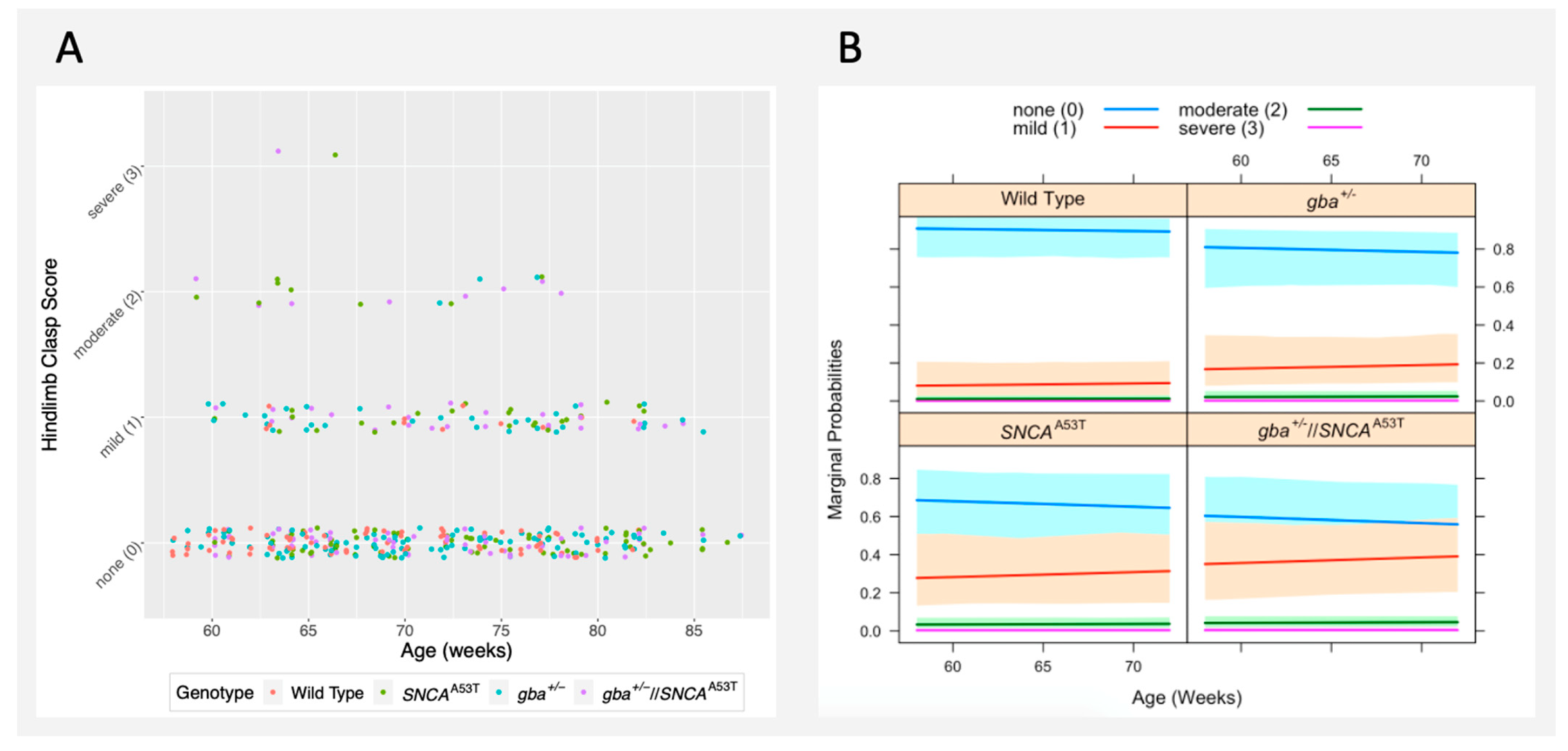
2.4. Hindlimb Clasp
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Testing Schedule
4.3. Novel Object Recognition (NOR)
4.4. Buried Pellet Test
4.5. Traverse Beam Walk
4.6. Hindlimb Clasp Score (HLC Score)
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dorsey, E.R.; Bloem, B.R. The Parkinson Pandemic-A Call to Action. JAMA Neurol. 2018, 75, 9–10. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Schaeffer, W.J. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010, 120, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Power, J.H.; Barnes, O.L.; Chegini, F. Lewy Bodies and the Mechanisms of Neuronal Cell Death in Parkinson’s Disease and Dementia with Lewy Bodies. Brain Pathol. 2017, 27, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Nalysnyk, L.; Rotella, P.; Simeone, J.C.; Hamed, A.; Weinreb, N. Gaucher disease epidemiology and natural history: A comprehensive review of the literature. Hematology 2017, 22, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Aerts, J.M.F.G.; Kuo, C.L.; Lelieveld, L.T.; Boer, D.E.C.; van der Lienden, M.J.C.; Overkleeft, H.S.; Artola, M. Glycosphingolipids and lysosomal storage disorders as illustrated by gaucher disease. Curr. Opin. Chem. Biol. 2019, 53, 204–215. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [Green Version]
- Nalls, M.A.; Duran, R.; Lopez, G.; Kurzawa-Akanbi, M.; McKeith, I.G.; Chinnery, P.F.; Morris, C.M.; Theuns, J.; Crosiers, D.; Cras, P.; et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. 2013, 70, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balestrino, R.; Tunesi, S.; Tesei, S.; Lopiano, L.; Zecchinelli, A.L.; Goldwurm, S. Penetrance of Glucocerebrosidase (GBA) Mutations in Parkinson’s Disease: A Kin Cohort Study. Mov. Disord. 2020, 35, 2111–2114. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.L.; Gruschus, J.M.; Velayati, A.; Westbroek, W.; Goldin, E.; Moaven, N.; Sidransky, E.; Lee, J.C. Alpha-synuclein interacts with Glucocerebrosidase providing a molecular link between Parkinson and Gaucher diseases. J. Biol Chem. 2011, 286, 28080–28088. [Google Scholar] [CrossRef] [Green Version]
- Massaro, G.; Mattar, C.N.Z.; Wong, A.M.S.; Sirka, E.; Buckley, S.M.K.; Herbert, B.R.; Karlsson, S.; Perocheau, D.P.; Burke, D.; Heales, S.; et al. Fetal gene therapy for neurodegenerative disease of infants. Nat. Med. 2018, 24, 1317–1323. [Google Scholar] [CrossRef]
- Sardi, S.P.; Viel, C.; Clarke, J.; Treleaven, C.M.; Richards, A.M.; Park, H.; Olszewski, M.A.; Dodge, J.C.; Marshall, J.; Makino, E.; et al. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc. Natl. Acad. Sci. USA 2017, 114, 2699–2704. [Google Scholar] [CrossRef] [Green Version]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef] [Green Version]
- Farfel-Becker, T.; Do, J.; Tayebi, N.; Sidransky, E. Can GBA1-Associated Parkinson Disease Be Modeled in the Mouse? Trends Neurosci. 2019, 42, 631–643. [Google Scholar] [CrossRef]
- Meyer, R.C.; Giddens, M.M.; Coleman, B.M.; Hall, R.A. The protective role of prosaposin and its receptors in the nervous system. Brain Res. 2014, 1585, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Tayebi, N.; Parisiadou, L.; Berhe, B.; Gonzalez, A.N.; Serra-Vinardell, J.; Tamargo, R.J.; Maniwang, E.; Sorrentino, Z.; Fujiwara, H.; Grey, R.J.; et al. Glucocerebrosidase haploinsufficiency in A53T α-synuclein mice impacts disease onset and course. Mol. Genet. Metab. 2017, 122, 198–208. [Google Scholar] [CrossRef]
- Kim, D.; Hwang, H.; Choi, S.; Kwon, S.H.; Lee, S.; Park, J.H.; Kim, S.; Ko, H.S. D409H GBA1 mutation accelerates the progression of pathology in A53T α-synuclein transgenic mouse model. Acta Neuropathol. Commun. 2018, 6, 32. [Google Scholar] [CrossRef]
- Migdalska-Richards, A.; Wegrzynowicz, M.; Rusconi, R.; Deangeli, G.; Di Monte, D.A.; Spillantini, M.G.; Schapira, A.H.V. The L444P Gba1 mutation enhances alpha-synuclein induced loss of nigral dopaminergic neurons in mice. Brain 2017, 140, 2706–2721. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.H.; Sun, Y.; Ran, H.; Quinn, B.; Witte, D.; Grabowski, G.A. Accumulation and distribution of α-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol. Genet. Metab. 2011, 102, 436–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, E.; Jantrachotechatchawan, C.; Buhidma, Y.; Broadstock, M.; Yu, L.; Howlett, D.; Aarsland, D.; Ballard, C.; Francis, P.T. Age-related neurochemical and behavioural changes in D409V/WT GBA1 mouse: Relevance to lewy body dementia. Neurochem. Int. 2019, 129, 104502. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Giladi, N.; Rozovski, U.; Shifrin, C.; Rosner, S.; Gurevich, T.; Bar-Shira, A.; Orr-Urtreger, A. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 2008, 70, 2277–2283. [Google Scholar] [CrossRef]
- Aharon-Peretz, J.; Rosenbaum, H.; Gershoni-Baruch, R. Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 2004, 351, 1972–1977. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.; Tong, J.; Fook-Chong, S.; Yih, Y.; Meng- Wong, C.; Pavanni, R.; Zhao, Y. Glucocerebrosidase Mutations and Risk of Parkinson Disease in Chinese Patients. Arch Neurol. 2007, 64, 1056–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, Y.E.; Chiang, M.S.R.; Locascio, J.J.; Liao, Z.; Liu, G.; Choudhury, K.; Kuras, Y.I.; Tuncali, I.; Videnovic, A.; Hunt, A.L.; et al. β-Glucocerebrosidase activity in GBA-linked Parkinson disease: The type of mutation matters. Neurology 2020, 95, e685–e696. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qin, L.; Pan, H.; Liu, Z.; Jiang, L.; He, Y.; Zeng, Q.; Zhou, X.; Zhou, X.; Zhou, Y.; et al. The role of genetics in Parkinson’s disease: A large cohort study in Chinese mainland population. Brain 2020, 143, 2220–2234. [Google Scholar] [CrossRef] [PubMed]
- Avenali, M.; Toffoli, M.; Mullin, S.; McNeil, A.; Hughes, D.A.; Mehta, A.; Blandini, F.; Schapira, A.H.V. Evolution of prodromal parkinsonian features in a cohort of GBA mutation-positive individuals: A 6-year longitudinal study. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Mullin, S.; Beavan, M.; Bestwick, J.; McNeill, A.; Proukakis, C.; Cox, T.; Hughes, D.; Mehta, A.; Zetterberg, H.; Schapira, A.H.V. Evolution and clustering of prodromal parkinsonian features in GBA1 carriers. Mov. Disord. 2019, 34, 1365–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerson, J.E.; Farmer, K.M.; Henson, N.; Castillo-Carranza, D.L.; Carretero Murillo, M.; Sengupta, U.; Barrett, A.; Kayed, R. Tau oligomers mediate α-synuclein toxicity and can be targeted by immunotherapy. Mol. Neurodegener. 2018, 13, 13. [Google Scholar] [CrossRef] [PubMed]
- Campos, F.L.; Carvalho, M.M.; Cristovão, A.C.; Je, G.; Baltazar, G.; Salgado, A.J.; Kim, Y.-S.; Sousa, N. Rodent models of Parkinson’s disease: Beyond the motor symptomatology. Front. Behav. Neurosci. 2013, 7, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rial, D.; Castro, A.A.; Machado, N.; Garção, P.; Gonçalves, F.Q.; Silva, H.B.; Tomé, A.R.; Köfalvi, A.; Corti, O.; Raisman-Vozari, R.; et al. Behavioral phenotyping of Parkin-deficient mice: Looking for early preclinical features of Parkinson’s disease. PLoS ONE 2014, 9, e114216, Erratum in: PLoS ONE 2015, 10, e0118526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antunes, M.; Biala, G. The novel object recognition memory: Neurobiology, test procedure, and its modifications. Cogn. Process. 2012, 13, 93–110. [Google Scholar] [CrossRef] [Green Version]
- Dawson, P.A.; Steane, S.E.; Markovich, D. Impaired memory and olfactory performance in NaSi-1 sulphate transporter deficient mice. Behav. Brain Res. 2005, 159, 15–20. [Google Scholar] [CrossRef]
- Yang, M.; Crawley, J.N. Simple behavioral assessment of mouse olfaction. Curr. Protoc. Neurosci. 2009, 8, 8–24. [Google Scholar] [CrossRef]
- Zou, J.; Wang, W.; Pan, Y.W.; Lu, S.; Xia, Z. Methods to measure olfactory behavior in mice. Curr. Protoc. Toxicol. 2015, 63, 11–18. [Google Scholar] [CrossRef]
- Luong, T.N.; Carlisle, H.J.; Southwell, A.; Patterson, P.H. Assessment of motor balance and coordination in mice using the balance beam. J. Vis. Exp. 2011, 49, 2376. [Google Scholar] [CrossRef] [Green Version]
- Guyenet, S.J.; Furrer, S.A.; Damian, V.M.; Baughan, T.D.; La Spada, A.R.; Garden, G.A. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J. Vis. Exp. 2010, 39, 1787. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test | Genotype | Baseline Value (8 Months) | Slope | p-Val vs. Wildtype (Baseline) | p-Val vs. Wildtype (Slope) | p-Val vs. gba+/− (Baseline) | p-Val vs. gba+/− (Slope) | p-Val vs. SNCAA53T (Baseline) | p-Val vs. SNCAA53T (Slope) |
|---|---|---|---|---|---|---|---|---|---|
| Novel Object Recognition | Wildtype | 0.503 ± 0.051 1 | 0.0049 ± 0.0052 1 | ~ | ~ | ~ | ~ | ~ | ~ |
| gba+/− | 0.520 ± 0.041 1 | 0.0006 ± 0.0044 1 | 0.790 | 0.532 | ~ | ~ | ~ | ~ | |
| SNCAA53T | 0.473 ± 0.043 1 | 0.0086 ± 0.0052 1 | 0.666 | 0.461 | ~ | ~ | ~ | ~ | |
| gba+/−//SNCAA53T | 0.464 ± 0.043 1 | −0.0043 ± 0.0051 1 | 0.580 | 0.225 | 0.884 | 0.493 | 0.985 | 0.055 | |
| Buried Pellet Test | Wildtype | 76.1 ± 11.3 | −3.07 ± 1.18 | ~ | ~ | ~ | ~ | ~ | ~ |
| gba+/− | 105.0 ± 9.2 | −3.36 ± 0.97 | 0.057 | 0.849 | ~ | ~ | ~ | ~ | |
| SNCAA53T | 80.6 ± 9.2 | −1.24 ± 1.06 | 0.758 | 0.249 | ~ | ~ | ~ | ~ | |
| gba+/−//SNCAA53T | 88.9 ± 10.0 | −1.54 ± 1.10 | 0.401 | 0.344 | 0.071 | 0.214 | 0.545 | 0.841 | |
| Beam Walk Latency | Wildtype | 10.0 ± 1.45 | 0.08 ± 0.13 | ~ | ~ | ~ | ~ | ~ | ~ |
| gba+/− | 15.4 ± 1.81 | 0.27 ± 0.11 | 0.028 | 0.549 | ~ | ~ | ~ | ~ | |
| SNCAA53T | 18.5 ± 1.45 | −0.03 ± 0.13 | 0.007 | 0.260 | ~ | ~ | ~ | ~ | |
| gba+/−//SNCAA53T | 16.3 ± 1.60 | 0.27 ± 0.13 | 0.014 | 0.314 | 0.693 | 0.969 | 0.299 | 0.105 | |
| Beam Walk Slips | Wildtype | 0.28 ± 0.46 | 0.05 ± 0.04 | ~ | ~ | ~ | ~ | ~ | ~ |
| gba+/− | 0.91 ± 0.37 | 0.17 ± 0.03 | 0.294 | 0.017 | ~ | ~ | ~ | ~ | |
| SNCAA53T | 1.31 ± 0.37 | 0.21 ± 0.04 | 0.089 | 0.005 | ~ | ~ | ~ | ~ | |
| gba+/−//SNCAA53T | 1.31 ± 0.40 | 0.19 ± 0.04 | 0.101 | 0.014 | 0.684 | 0.796 | 0.816 | 0.690 |
| Test | Genotype | Odds Ratio Relative to Wildtype | p-Val vs. Wildtype | p-Val vs. gba+/− | p-Val vs. SNCAA53T |
|---|---|---|---|---|---|
| Hindlimb Clasp | Wildtype | ~ | ~ | ~ | ~ |
| gba+/− | 2.29 1 (0.65–8.09) | 0.196 | ~ | ~ | |
| SNCAA53T | 4.39 1 (1.19–16.23) | 0.027 | ~ | ~ | |
| gba+/−//SNCAA53T | 6.17 1 (1.57–24.24) | 0.009 | 0.098 | 0.571 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Do, J.; Perez, G.; Berhe, B.; Tayebi, N.; Sidransky, E. Behavioral Phenotyping in a Murine Model of GBA1-Associated Parkinson Disease. Int. J. Mol. Sci. 2021, 22, 6826. https://doi.org/10.3390/ijms22136826
Do J, Perez G, Berhe B, Tayebi N, Sidransky E. Behavioral Phenotyping in a Murine Model of GBA1-Associated Parkinson Disease. International Journal of Molecular Sciences. 2021; 22(13):6826. https://doi.org/10.3390/ijms22136826
Chicago/Turabian StyleDo, Jenny, Gani Perez, Bahafta Berhe, Nahid Tayebi, and Ellen Sidransky. 2021. "Behavioral Phenotyping in a Murine Model of GBA1-Associated Parkinson Disease" International Journal of Molecular Sciences 22, no. 13: 6826. https://doi.org/10.3390/ijms22136826
APA StyleDo, J., Perez, G., Berhe, B., Tayebi, N., & Sidransky, E. (2021). Behavioral Phenotyping in a Murine Model of GBA1-Associated Parkinson Disease. International Journal of Molecular Sciences, 22(13), 6826. https://doi.org/10.3390/ijms22136826