
In Vitro Metabolism of Six C-Glycosidic Flavonoids from Passiflora incarnata L.

Abstract
1. Introduction
2. Results
2.1. Stability Testing
2.2. Cell Viability
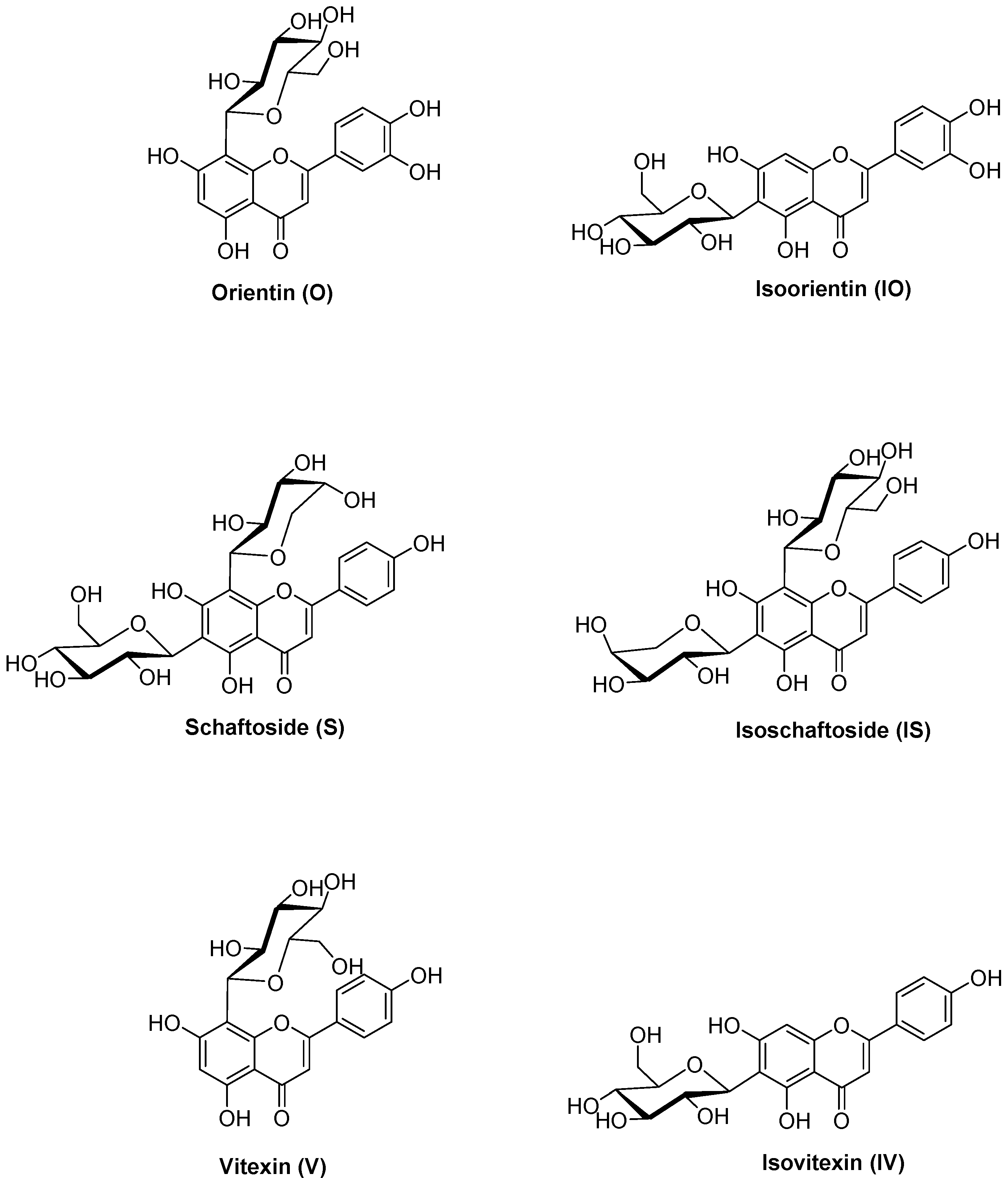
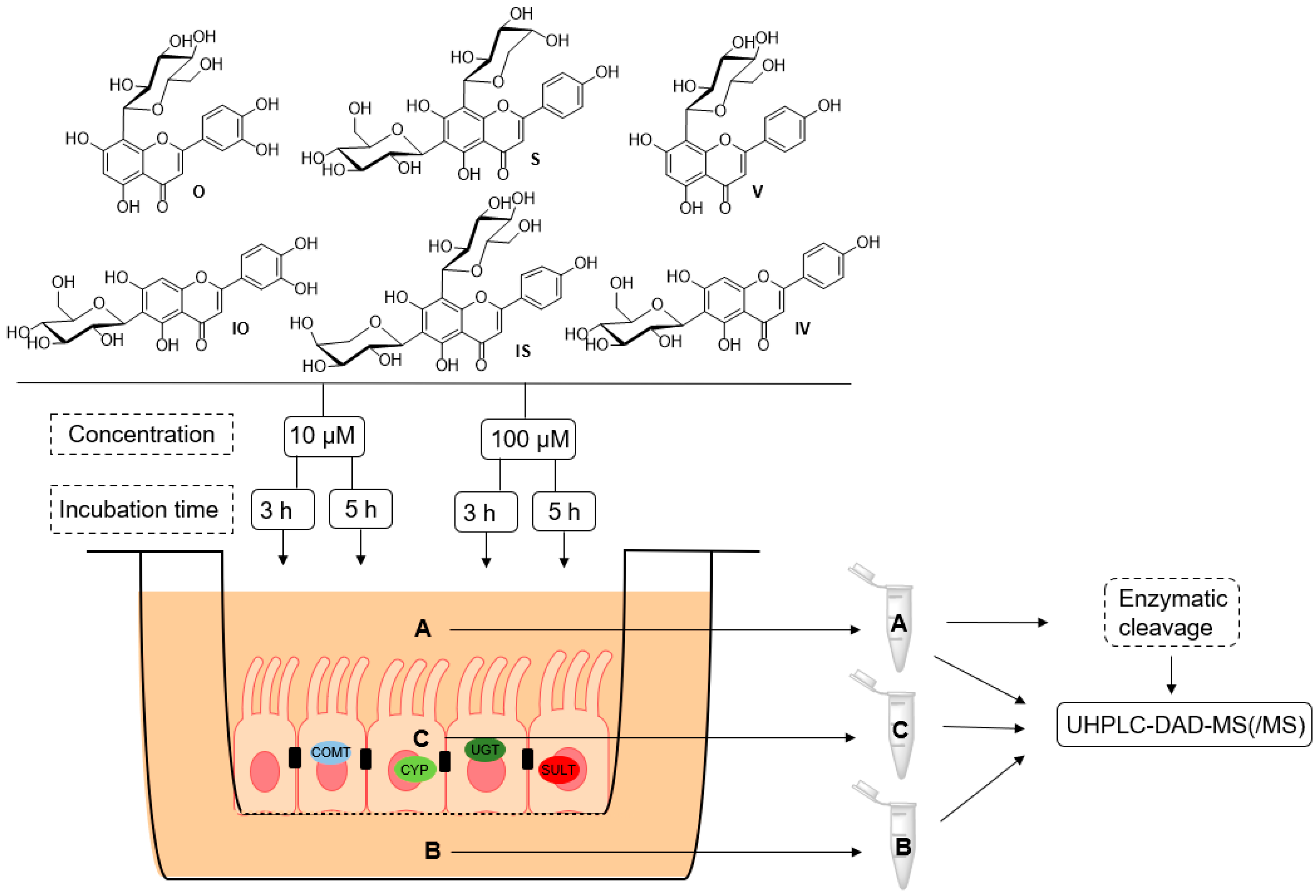
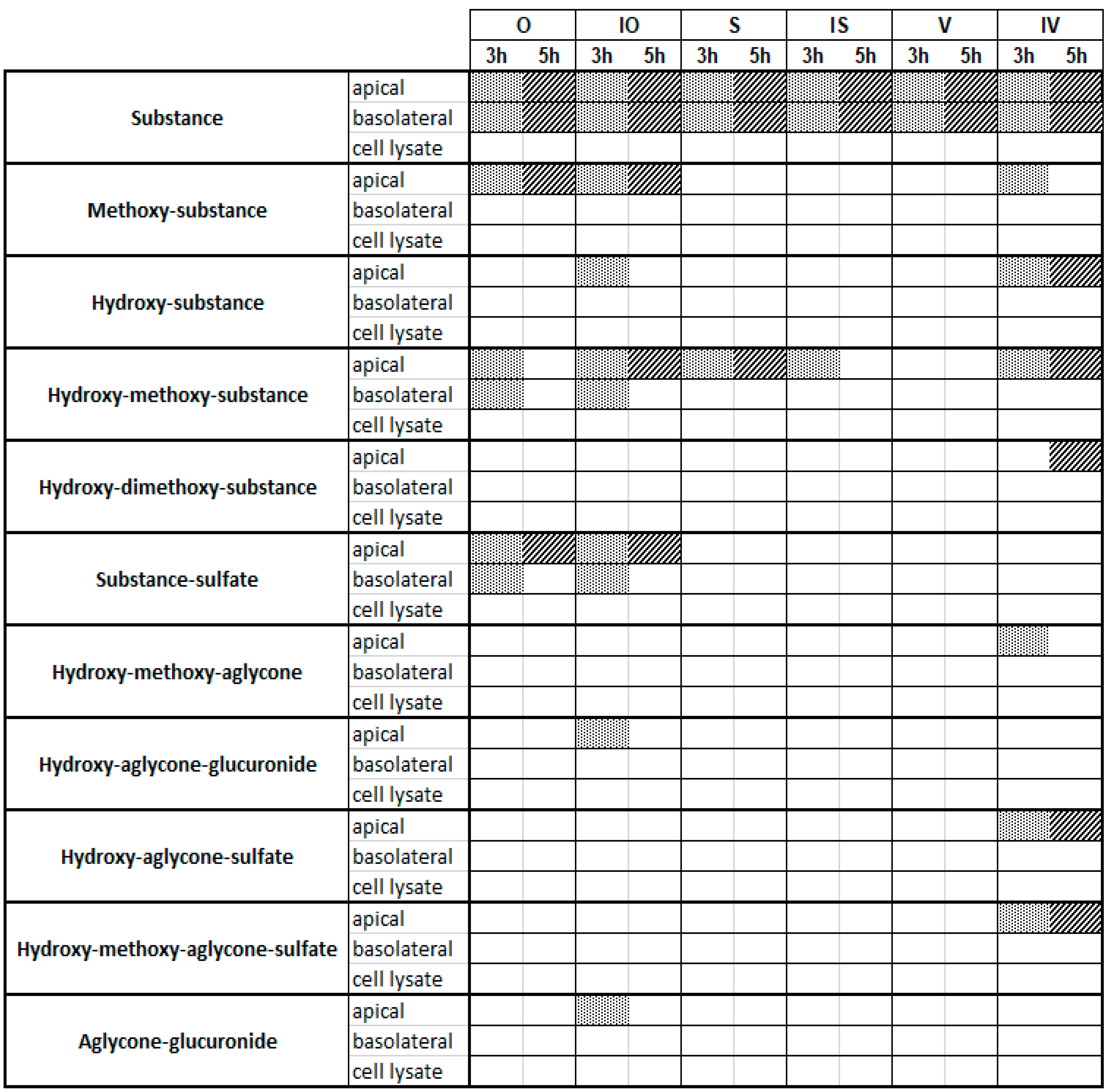
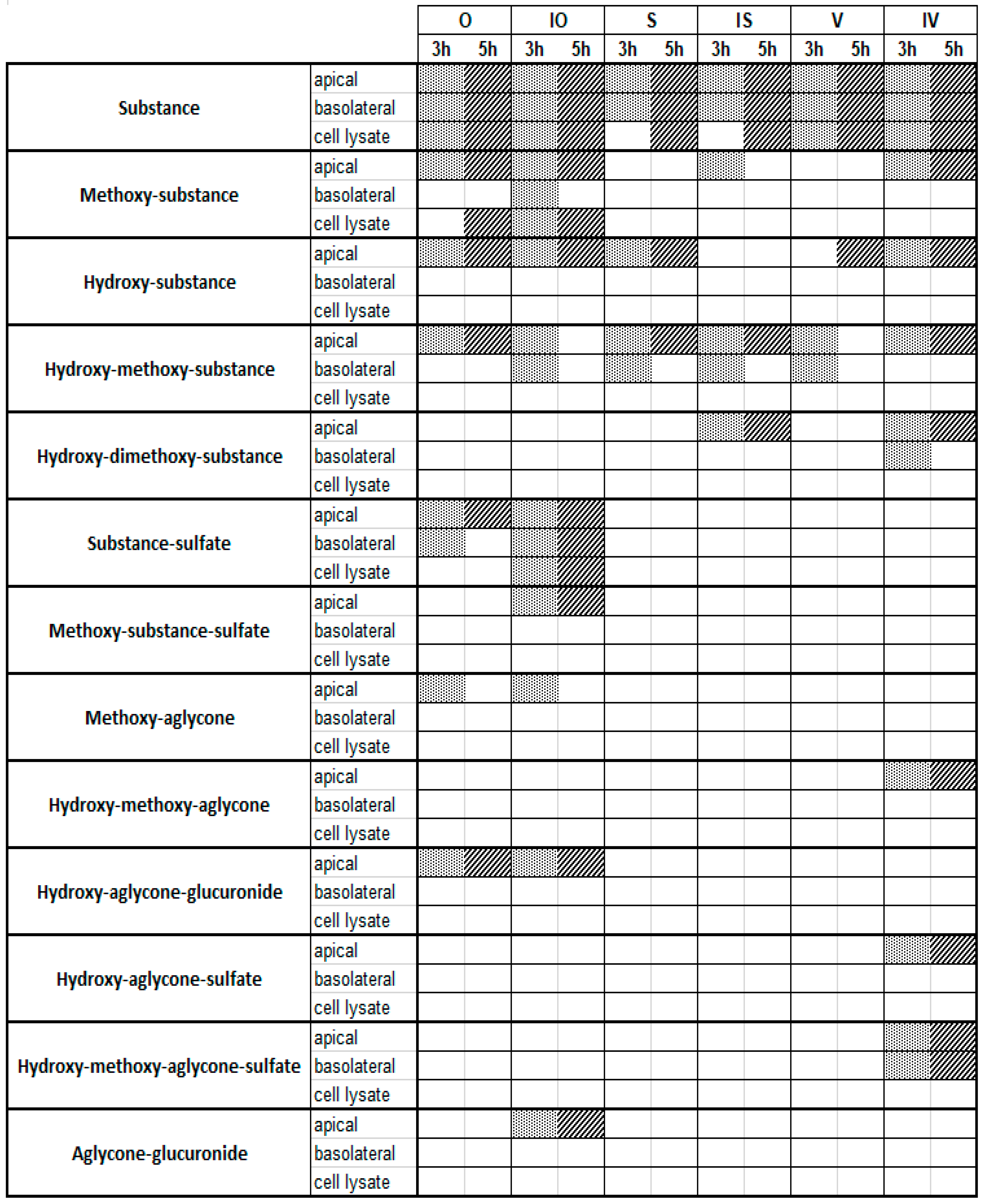
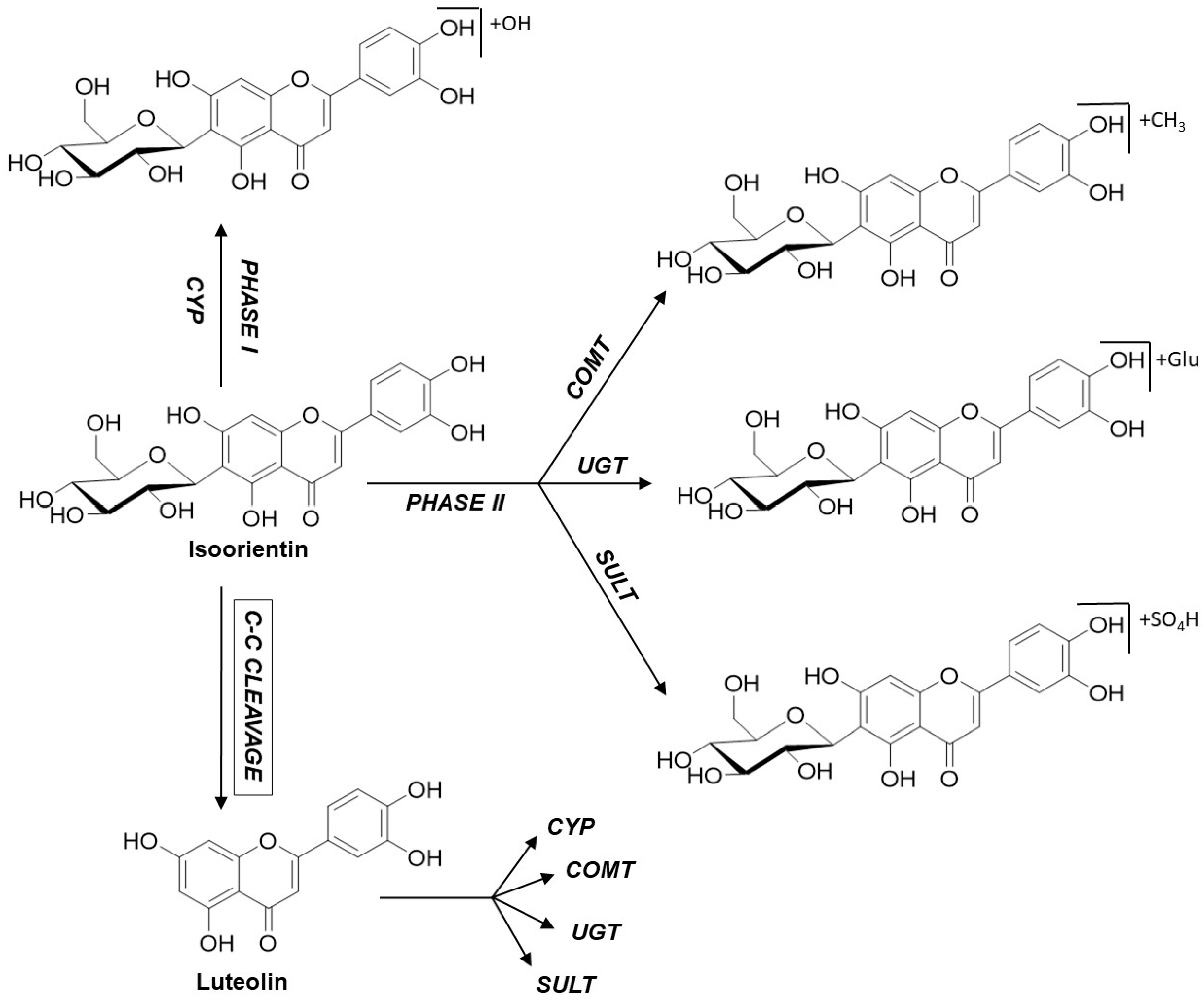
2.3. Metabolite Profiles of C-Glycosidic Flavonoids
3. Discussion
4. Material and Methods
4.1. Reagents and Chemicals
4.2. Stability Testing
4.3. HPLC-DAD
4.4. MTT Assay under Metabolic Conditions
4.5. Cell Culture
4.6. Cell Differentiation
4.7. Caco-2 Metabolism Experiment
4.8. Cell Lysis
4.9. Enzymatic Sample Preparation
4.10. UHPLC-DAD-MS
4.11. Aglycone Determination
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CYP | cytochrome P450 |
| COMT | catechol-O-methyltransferase |
| SULT | sulfotransferase |
| UGT | uridine 5′-diphospho glucuronosyltransferase |
| O | orientin |
| IO | isoorientin |
| S | schaftoside |
| IS | isoschaftoside |
| V | vitexin |
| IV | isovitexin |
| DMSO | dimethyl sulfoxide |
| MTT | 3-(4,5-dimethylthiazolyl)-2,5-diphenyl tetrazolium bromide |
| SDS | sodium dodecyl sulfate |
| HBSS | Hanks’ balanced salt solution |
| DMEM | Dulbecco’s modified Eagle medium |
| PBS | phosphate-buffered saline |
| FBS | fetal bovine serum |
| EDTA | ethylenediaminetetraacetic acid |
| NEAA | nonessential amino acids |
| TEER | transepithelial electrical resistance |
| (U)HPLC | (ultra) high-performance liquid chromatography |
| DAD | diode array detector |
| MS | mass spectrometry |
| EIC | extracted ion chromatogram |
References
- Blaschek, W.; Hänsel, R.; Keller, K.; Reichling, J.; Rimpler, H.; Schneider, G. Hagers Handbuch der Pharmazeutischen Praxis. Folgeband 2: Drogen A-K, 5th ed.; Springer: Berlin/Heidelberg, Germany, 1998; Volume s.1. [Google Scholar]
- Krenn, L. Die Passionsblume (Passiflora incarnata L.)-ein bewährtes pflanzliches Sedativum. Wien. Med. Wochenschr. 2002, 152, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Lutomski, J.; Segiet, E.; Szpunar, K.; Grisse, K. Die Bedeutung der Passionsblume in der Heilkunde. Pharm. Unserer Zeit 1981, 10, 45–49. [Google Scholar] [CrossRef]
- Marchart, E.; Krenn, L.; Kopp, B. Quantification of the flavonoid glycosides in Passiflora incarnata by capillary electrophoresis. Planta Med. 2003, 69, 452–456. [Google Scholar] [PubMed]
- Vissiennon, C.; Nieber, K.; Kelber, O.; Butterweck, V. Route of administration determines the anxiolytic activity of the flavonols kaempferol, quercetin and myricetin—are they prodrugs? J. Nutr. Biochem. 2012, 23, 733–740. [Google Scholar] [CrossRef]
- Leong, C.N.A.; Tako, M.; Hanashiro, I.; Tamaki, H. Antioxidant flavonoid glycosides from the leaves of Ficus pumila L. Food Chem. 2008, 109, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Wang, J.; Xiao, H.; Xiao, C.; Wang, Y.; Liu, X. Isoorientin induces apoptosis through mitochondrial dysfunction and inhibition of PI3K/Akt signaling pathway in HepG2 cancer cells. Toxicol. Appl. Pharmacol. 2012, 265, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. Pharmacokinetics of vitexin in rats after intravenous and oral administration. Afr. J. Pharm. Pharmacol. 2012, 6, 2368–2373. [Google Scholar] [CrossRef]
- Chen, Y.-G.; Li, P.; Li, P.; Yan, R.; Zhang, X.-Q.; Wang, Y.; Zhang, X.-T.; Ye, W.-C.; Zhang, Q.-W. α-Glucosidase inhibitory effect and simultaneous quantification of three major flavonoid glycosides in Microctis folium. Molecules 2013, 18, 4221–4232. [Google Scholar] [CrossRef]
- Hawas, U.W.; Soliman, G.M.; Abou El-Kassem, L.T.; Farrag, A.R.H.; Mahmoud, K.; León, F. A new flavonoid C-glycoside from Solanum elaeagnifolium with hepatoprotective and curative activities against paracetamol-induced liver injury in mice. Z. Naturforsch. C 2013, 68, 19–28. [Google Scholar]
- Polimanti, R.; Piacentini, S.; Manfellotto, D.; Fuciarelli, M. Human genetic variation of CYP450 superfamily. Analysis of functional diversity in worldwide populations. Pharmacogenomics 2012, 13, 1951–1960. [Google Scholar] [CrossRef]
- Wilkinson, G.R. Drug metabolism and variability among patients in drug response. N. Engl. J. Med. 2005, 352, 2211–2221. [Google Scholar] [CrossRef] [PubMed]
- Männistö, P.T.; Kaakkola, S. Catechol-O-methyltransferase (COMT). Biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol. Rev. 1999, 51, 593–628. [Google Scholar]
- Chen, Z.-J.; Dai, Y.-Q.; Kong, S.-S.; Song, F.-F.; Li, L.-P.; Ye, J.-F.; Wang, R.-W.; Zeng, S.; Zhou, H.; Jiang, H.-D. Luteolin is a rare substrate of human catechol-O-methyltransferase favoring a para-methylation. Mol. Nutr. Food Res. 2013, 57, 877–885. [Google Scholar] [CrossRef]
- Rowland, A.; Miners, J.O.; Mackenzie, P.I. The UDP-glucuronosyltransferases. Their role in drug metabolism and detoxification. Int. J. Biochem. Cell Biol. 2013, 45, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Chen, Y.; Zhou, T.; Chen, G. Sulfation of dietary flavonoids by human sulfotransferases. Xenobiotica 2009, 39, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Basu, S.; Meng, S.; Wang, X.; Hu, M. Regioselective sulfation and glucuronidation of phenolics. Insights into the structural basis. Curr. Drug Metab. 2011, 12, 900–916. [Google Scholar] [CrossRef]
- Chen, Z.; Zheng, S.; Li, L.; Jiang, H. Metabolism of flavonoids in human. A comprehensive review. Curr. Drug Metab. 2014, 15, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Williamson, G.; Clifford, M.N. Colonic metabolites of berry polyphenols. The missing link to biological activity? Br. J. Nutr. 2010, 104 (Suppl. 3), S48–S66. [Google Scholar] [CrossRef]
- Vollmer, M.; Esders, S.; Farquharson, F.M.; Neugart, S.; Duncan, S.H.; Schreiner, M.; Louis, P.; Maul, R.; Rohn, S. Mutual interaction of phenolic compounds and microbiota. metabolism of complex phenolic apigenin-C- and kaempferol-O-derivatives by human fecal samples. J. Agric. Food Chem. 2018, 66, 485–497. [Google Scholar] [CrossRef]
- Miroddi, M.; Calapai, G.; Navarra, M.; Minciullo, P.L.; Gangemi, S. Passiflora incarnata L. Ethnopharmacology, clinical application, safety and evaluation of clinical trials. J. Ethnopharmacol. 2013, 150, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.Y.; Ling, A.P.K.; Koh, R.Y.; Wong, Y.P.; Say, Y.H. A Review on Medicinal Properties of Orientin. Adv. Pharmacol. Sci. 2016, 2016, 4104595. [Google Scholar] [CrossRef]
- An, F.; Yang, G.; Tian, J.; Wang, S. Antioxidant effects of the orientin and vitexin in Trollius chinensis Bunge in D-galactose-aged mice. Neural Regen. Res. 2012, 7, 2565–2575. [Google Scholar] [PubMed]
- Li, Y.-L.; Ma, S.-C.; Yang, Y.-T.; Ye, S.-M.; But, P.P.-H. Antiviral activities of flavonoids and organic acid from Trollius chinensis Bunge. J. Ethnopharmacol. 2002, 79, 365–368. [Google Scholar] [CrossRef]
- Ku, S.-K.; Kwak, S.; Bae, J.-S. Orientin inhibits high glucose-induced vascular inflammation in vitro and in vivo. Inflammation 2014, 37, 2164–2173. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Ku, S.-K.; Bae, J.-S. Vascular barrier protective effects of orientin and isoorientin in LPS-induced inflammation in vitro and in vivo. Vascul. Pharmacol. 2014, 62, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Ku, S.-K.; Lee, T.; Bae, J.-S. Orientin inhibits HMGB1-induced inflammatory responses in HUVECs and in murine polymicrobial sepsis. Inflammation 2014, 37, 1705–1717. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lan, N.; Ren, J.; Wu, Y.; Wang, S.-T.; Huang, X.-F.; Yu, Y. Orientin improves depression-like behavior and BDNF in chronic stressed mice. Mol. Nutr. Food Res. 2015, 59, 1130–1142. [Google Scholar] [CrossRef]
- Da Silva, R.Z.; Yunes, R.A.; Souza, M.M.; Monache, F.; Cechinel-Filho, V. Antinociceptive properties of conocarpan and orientin obtained from Piper solmsianum C. DC. var. solmsianum (Piperaceae). J. Nat. Med. 2010, 64, 402–408. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, B.C.; Kim, J.H.; Sim, G.S.; Lee, D.H.; Lee, K.E.; Yun, Y.P.; Pyo, H.B. The isolation and antioxidative effects of vitexin from Acer palmatum. Arch. Pharmacal Res. 2005, 28, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, D.; Yang, L.; Zhou, D.; Zhang, J. Purification and characterization of flavonoids from the leaves of Zanthoxylum bungeanum and correlation between their structure and antioxidant activity. PLoS ONE 2014, 9, e105725. [Google Scholar] [CrossRef]
- Guilhon, C.C.; Abdul Wahab, I.R.; Boylan, F.; Fernandes, P.D. Central antinociceptive and mechanism of action of Pereskia bleo Kunth leaves crude extract, fractions, and isolated compounds. Evid.-Based Complement. Altern. Med. 2015, 2015, 915927. [Google Scholar] [CrossRef]
- Zhu, Q.; Mao, L.-N.; Liu, C.-P.; Sun, Y.-H.; Jiang, B.; Zhang, W.; Li, J.-X. Antinociceptive effects of vitexin in a mouse model of postoperative pain. Sci. Rep. 2016, 6, 19266. [Google Scholar] [CrossRef] [PubMed]
- Dos Reis, G.O.; Vicente, G.; Carvalho, F.K. de; Heller, M.; Micke, G.A.; Pizzolatti, M.G.; Fröde, T.S. Croton antisyphiliticus Mart. attenuates the inflammatory response to carrageenan-induced pleurisy in mice. Inflammopharmacology 2014, 22, 115–126. [Google Scholar] [CrossRef]
- Flores, G.; Dastmalchi, K.; Dabo, A.J.; Whalen, K.; Pedraza-Peñalosa, P.; Foronjy, R.F.; D’Armiento, J.M.; Kennelly, E.J. Antioxidants of therapeutic relevance in COPD from the neotropical blueberry Anthopterus wardii. Food Chem. 2012, 131, 119–125. [Google Scholar] [CrossRef]
- Basile, A.; Giordano, S.; López-Sáez, J.A.; Cobianchi, R.C. Antibacterial activity of pure flavonoids isolated from mosses. Phytochemistry 1999, 52, 1479–1482. [Google Scholar] [CrossRef]
- Quílez, A.; Berenguer, B.; Gilardoni, G.; Souccar, C.; de Mendonça, S.; Oliveira, L.F.S.; Martín-Calero, M.J.; Vidari, G. Anti-secretory, anti-inflammatory and anti-Helicobacter pylori activities of several fractions isolated from Piper carpunya Ruiz & Pav. J. Ethnopharmacol. 2010, 128, 583–589. [Google Scholar] [PubMed]
- Knipping, K.; Garssen, J.; van’t Land, B. An evaluation of the inhibitory effects against rotavirus infection of edible plant extracts. Virol. J. 2012, 9, 137. [Google Scholar] [CrossRef]
- He, M.; Min, J.-W.; Kong, W.-L.; He, X.-H.; Li, J.-X.; Peng, B.-W. A review on the pharmacological effects of vitexin and isovitexin. Fitoterapia 2016, 115, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Gadioli, I.L.; da Cunha, M.d.S.B.; Carvalho, M.V.O.; de Costa, A.M.; Pineli, L.d.L.d.O. A systematic review on phenolic compounds in Passiflora plants. Exploring biodiversity for food, nutrition, and popular medicine. Crit. Rev. Food Sci. Nutr. 2018, 58, 785–807. [Google Scholar] [CrossRef]
- Hostetler, G.L.; Ralston, R.A.; Schwartz, S.J. Flavones. Food Sources, Bioavailability, Metabolism, and Bioactivity. Adv. Nutr. 2017, 8, 423–435. [Google Scholar] [CrossRef]
- Kaldas, M.I.; Walle, U.K.; Walle, T. Resveratrol transport and metabolism by human intestinal Caco-2 cells. J. Pharm. Pharmacol. 2003, 55, 307–312. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RT (min) | (M − H)− exp. (m/z) | Fragment Ions (m/z) | RT (min) Aglycone a.e.h. | RT (min) Aglycone r.c. | Possible Metabolite | Initial Substance |
|---|---|---|---|---|---|---|
| 5.134 | 477.0674 | 301.0300 (-GlucA), 176.0107 (-Quercetin) | 12.402 | 12.342 | Quercetin-glucuronide | O, IO |
| 5.476 | 461.0724 | n.d. | 12.437 | 12.425 | Luteolin-glucuronide | O, IO |
| 6.204 | 299.0550 | 285.0325 (-CH2) | n.e. | n.e. | Methoxy-luteolin | IV |
| 11.658 | 364.9978 | 285.0403 (-SO3) | 12.437 | 12.425 | Luteolin-sulfate | IV |
| 11.909 | 379.0135 | 299.0566 (-SO3), 285.0350 (-CH2, -SO3) | n.e. | n.e. | Methoxy-luteolin-sulfate | IV |
| 3.146 | 527.0499 | 327,0485 (-C6H12O6, -SO3) | 5.754 | 5.758 | O-sulfate | O |
| 2.514 5.107 | 527.0501 | 327.0485 (--C6H12O6, -SO3), 79.9563 (-IO) | 5.926 | 5.947 | IO-sulfate | IO |
| 5.739 6.503 | 541.06574 | 461.1075 (-SO3) | n.e. | n.e. | Methoxy-IO-sulfate | IO |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tremmel, M.; Kiermaier, J.; Heilmann, J. In Vitro Metabolism of Six C-Glycosidic Flavonoids from Passiflora incarnata L. Int. J. Mol. Sci. 2021, 22, 6566. https://doi.org/10.3390/ijms22126566
Tremmel M, Kiermaier J, Heilmann J. In Vitro Metabolism of Six C-Glycosidic Flavonoids from Passiflora incarnata L. International Journal of Molecular Sciences. 2021; 22(12):6566. https://doi.org/10.3390/ijms22126566
Chicago/Turabian StyleTremmel, Martina, Josef Kiermaier, and Jörg Heilmann. 2021. "In Vitro Metabolism of Six C-Glycosidic Flavonoids from Passiflora incarnata L." International Journal of Molecular Sciences 22, no. 12: 6566. https://doi.org/10.3390/ijms22126566
APA StyleTremmel, M., Kiermaier, J., & Heilmann, J. (2021). In Vitro Metabolism of Six C-Glycosidic Flavonoids from Passiflora incarnata L. International Journal of Molecular Sciences, 22(12), 6566. https://doi.org/10.3390/ijms22126566