
In Vitro and In Vivo Efficacies of the Linear and the Cyclic Version of an All-d-Enantiomeric Peptide Developed for the Treatment of Alzheimer’s Disease

 , , and
, , and
Abstract
1. Introduction
2. Results
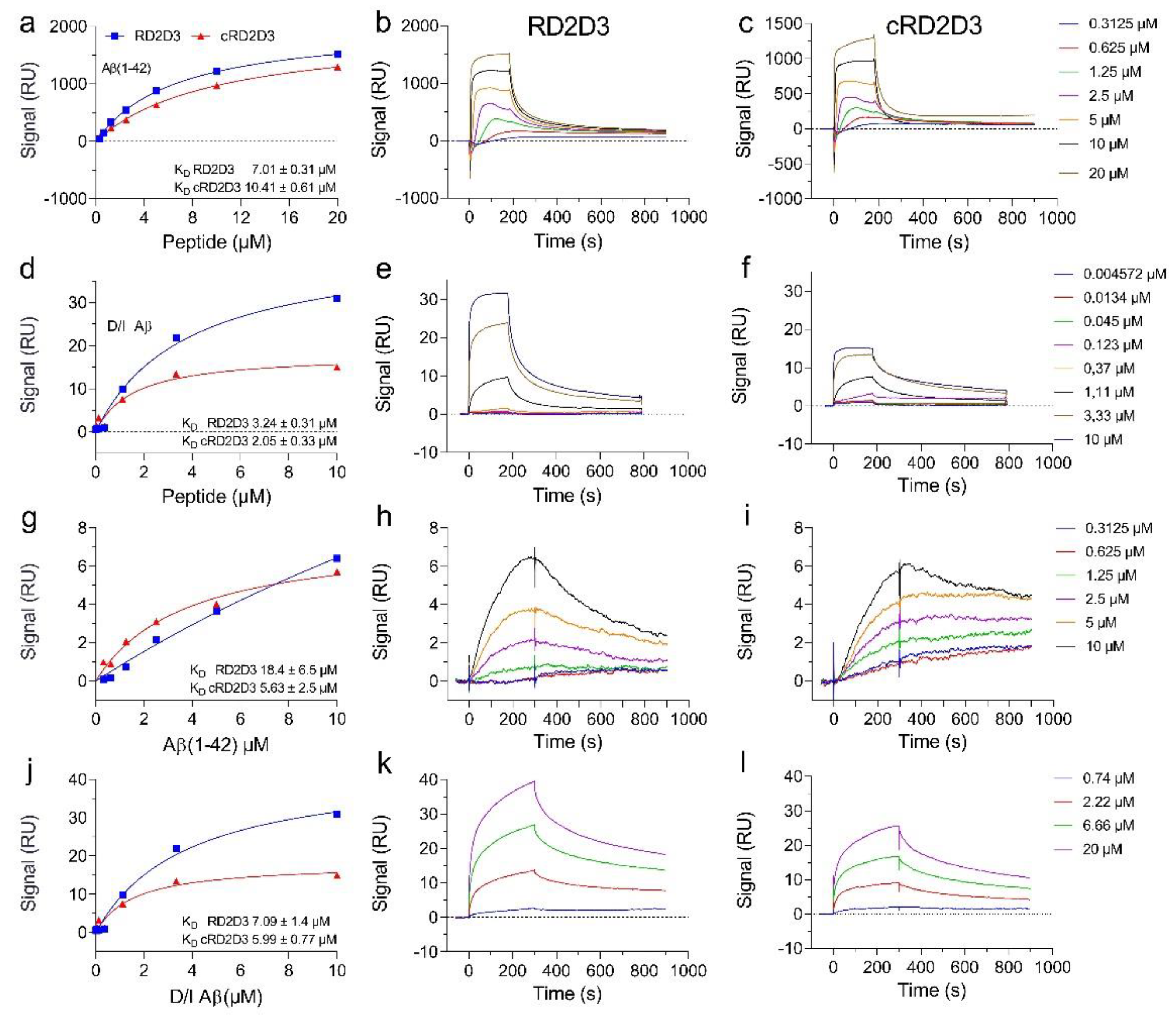
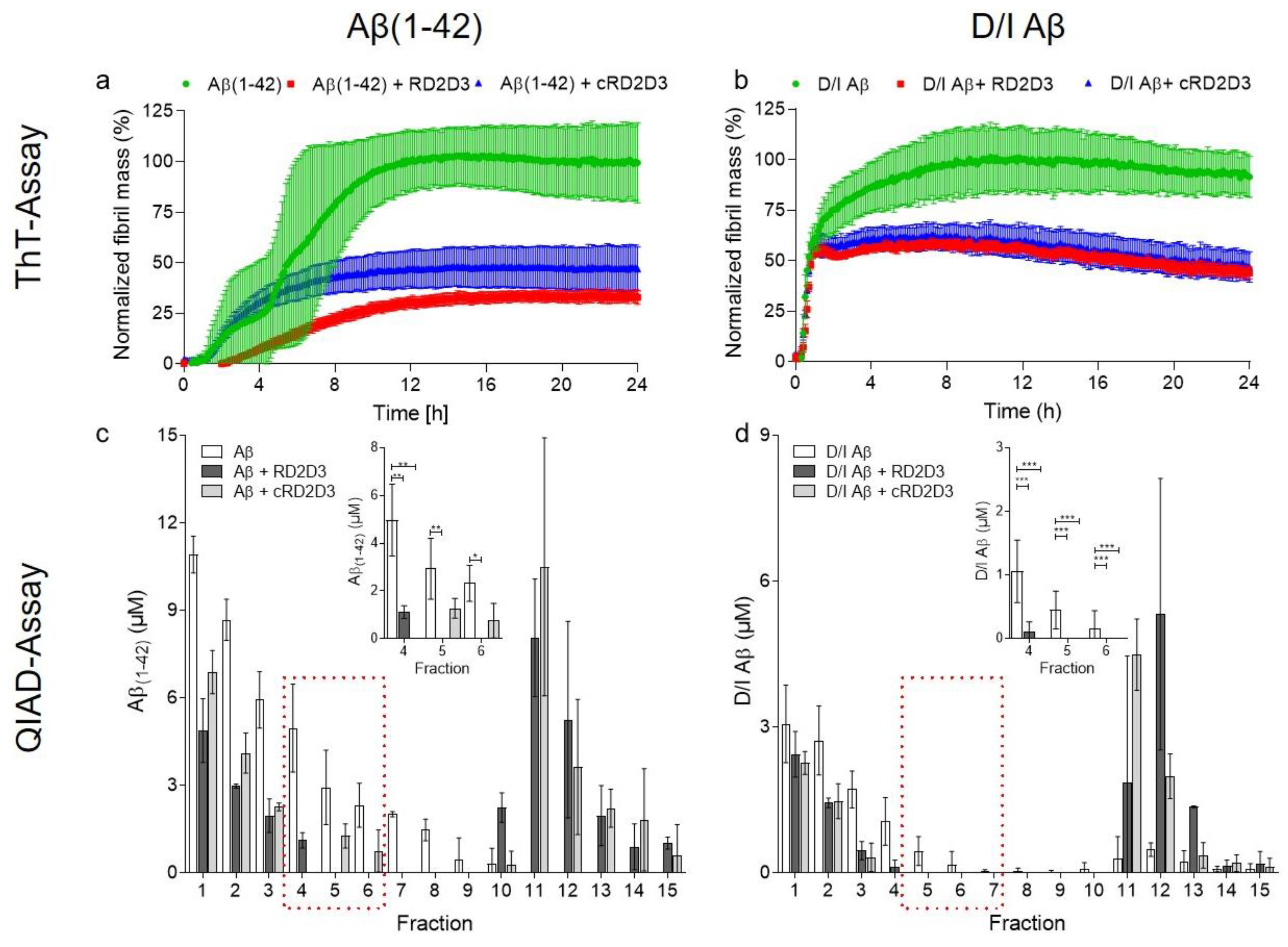
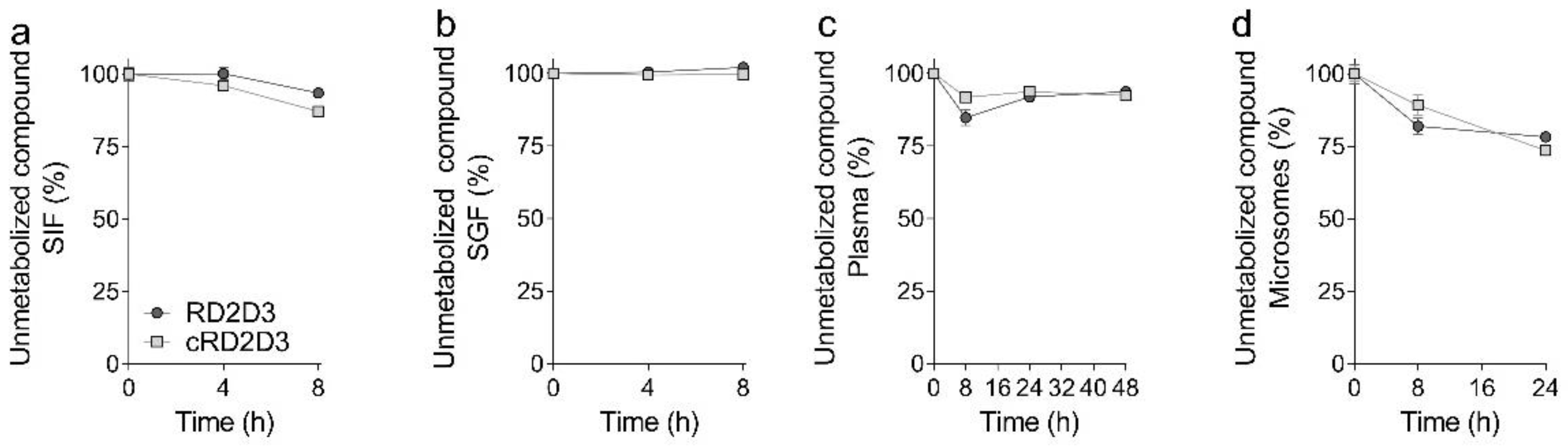
2.1. The Cyclic d-Peptide cRD2D3 Revealed Increased In Vitro Potency Compared to the Linear d-Peptide RD2D3 Without Differences in the Proteolytic Stability
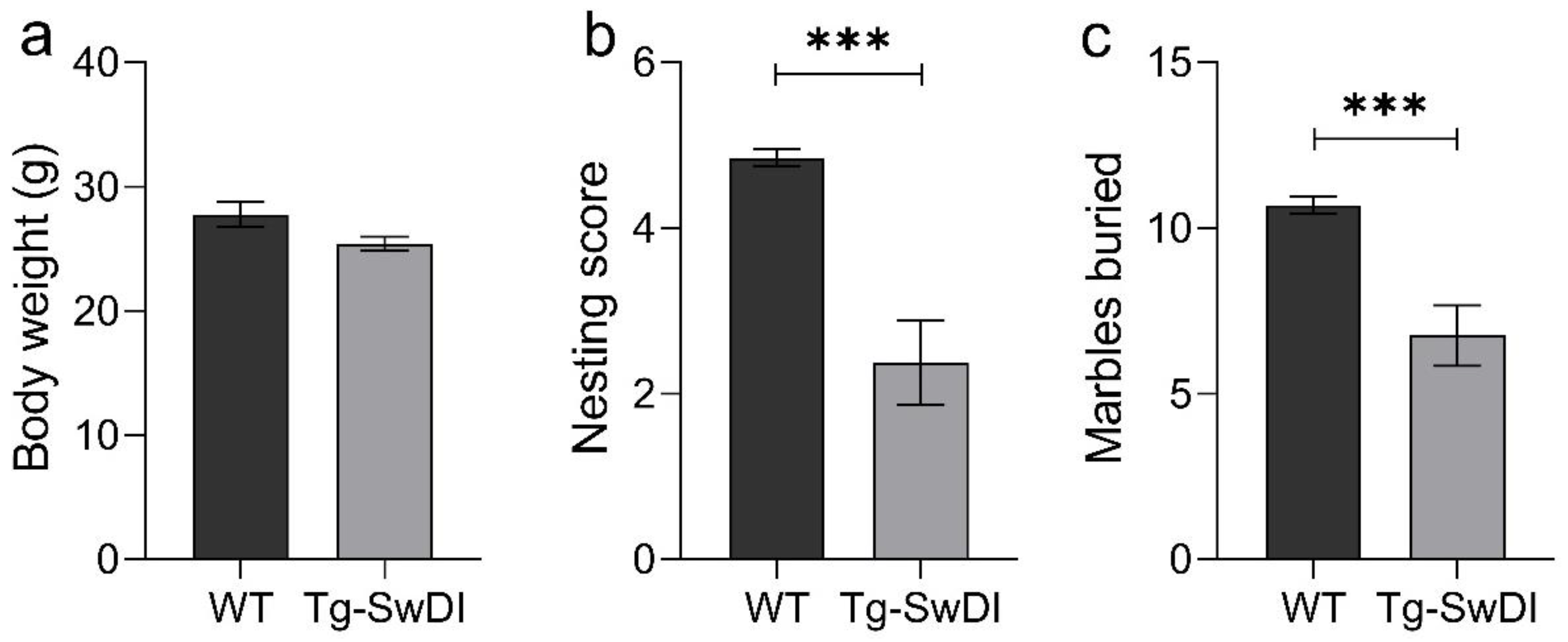
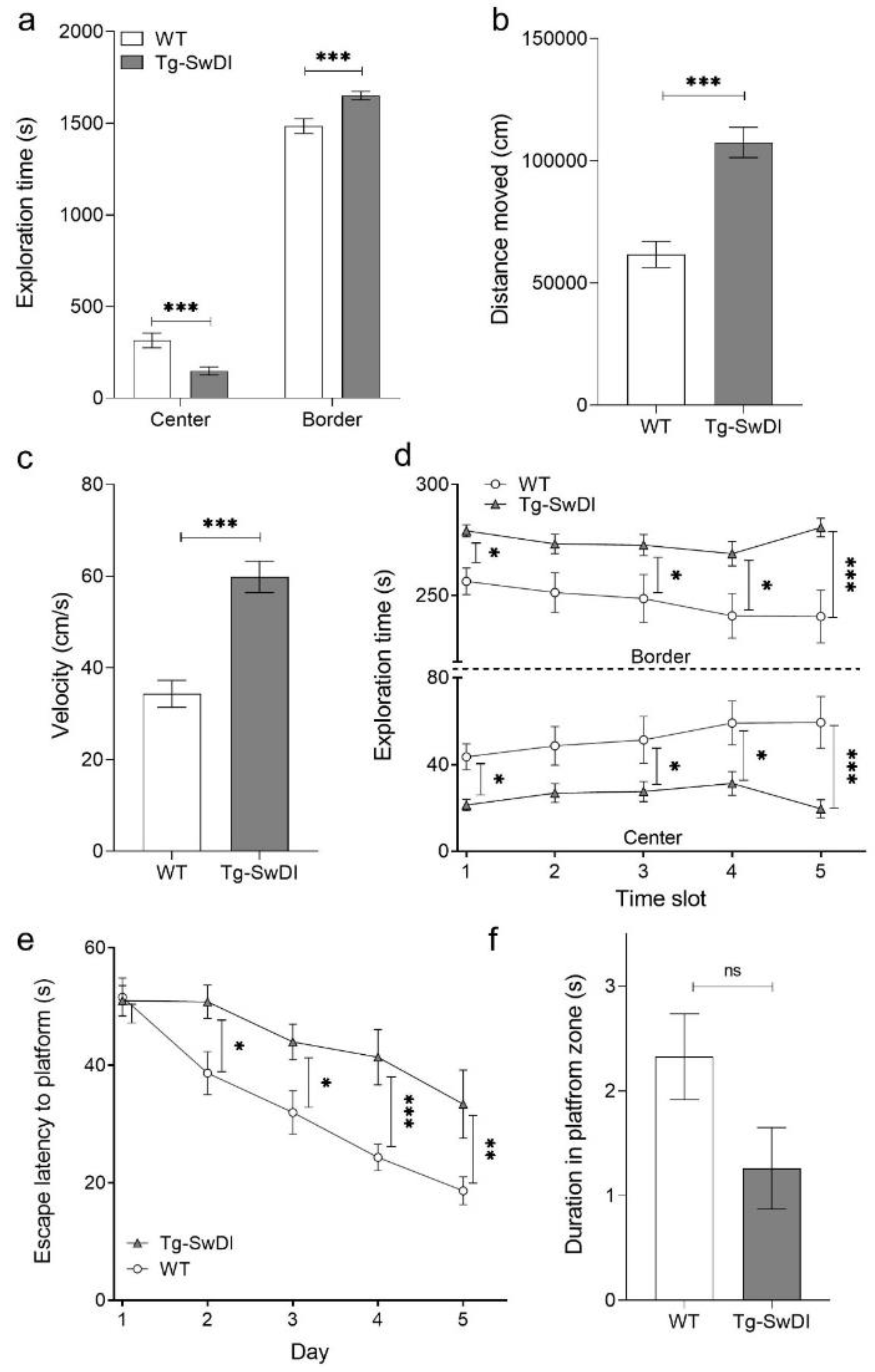
2.2. Tg-SwDI Mice Develop Cognitive Deficits at 12 Months of Age Compared to WT Mice
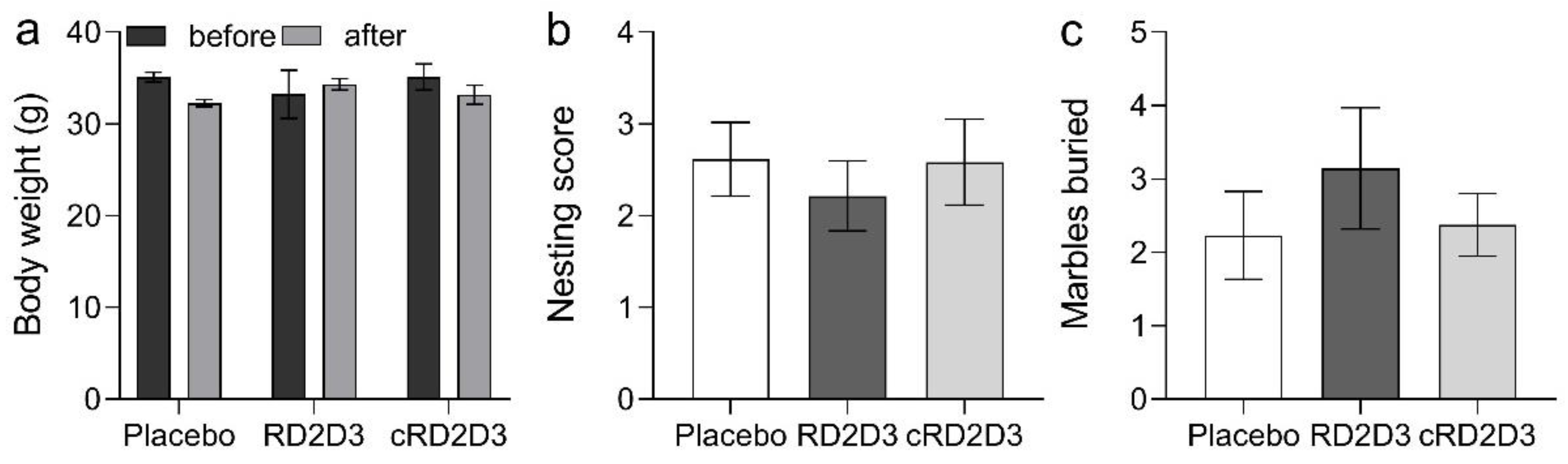
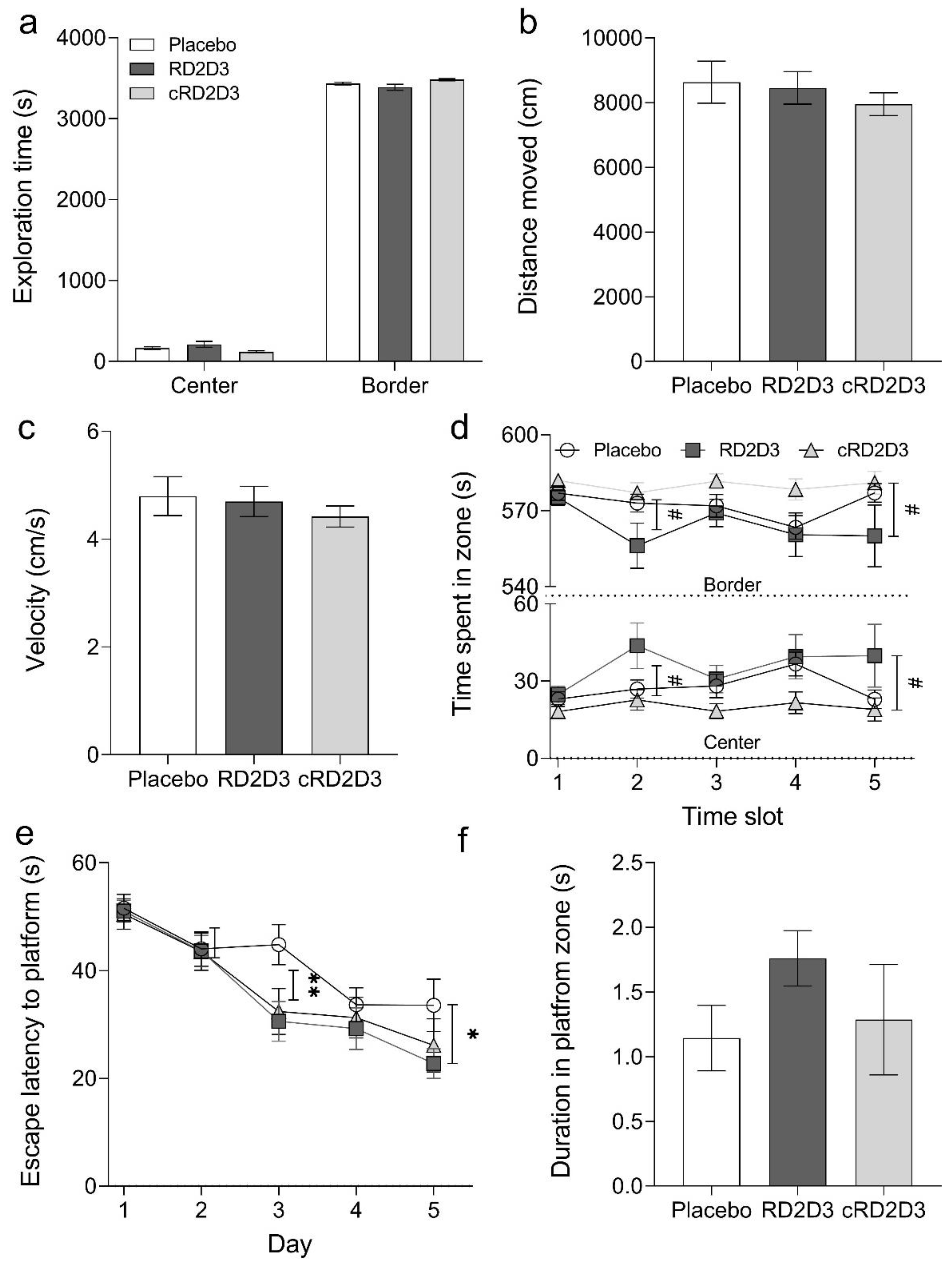
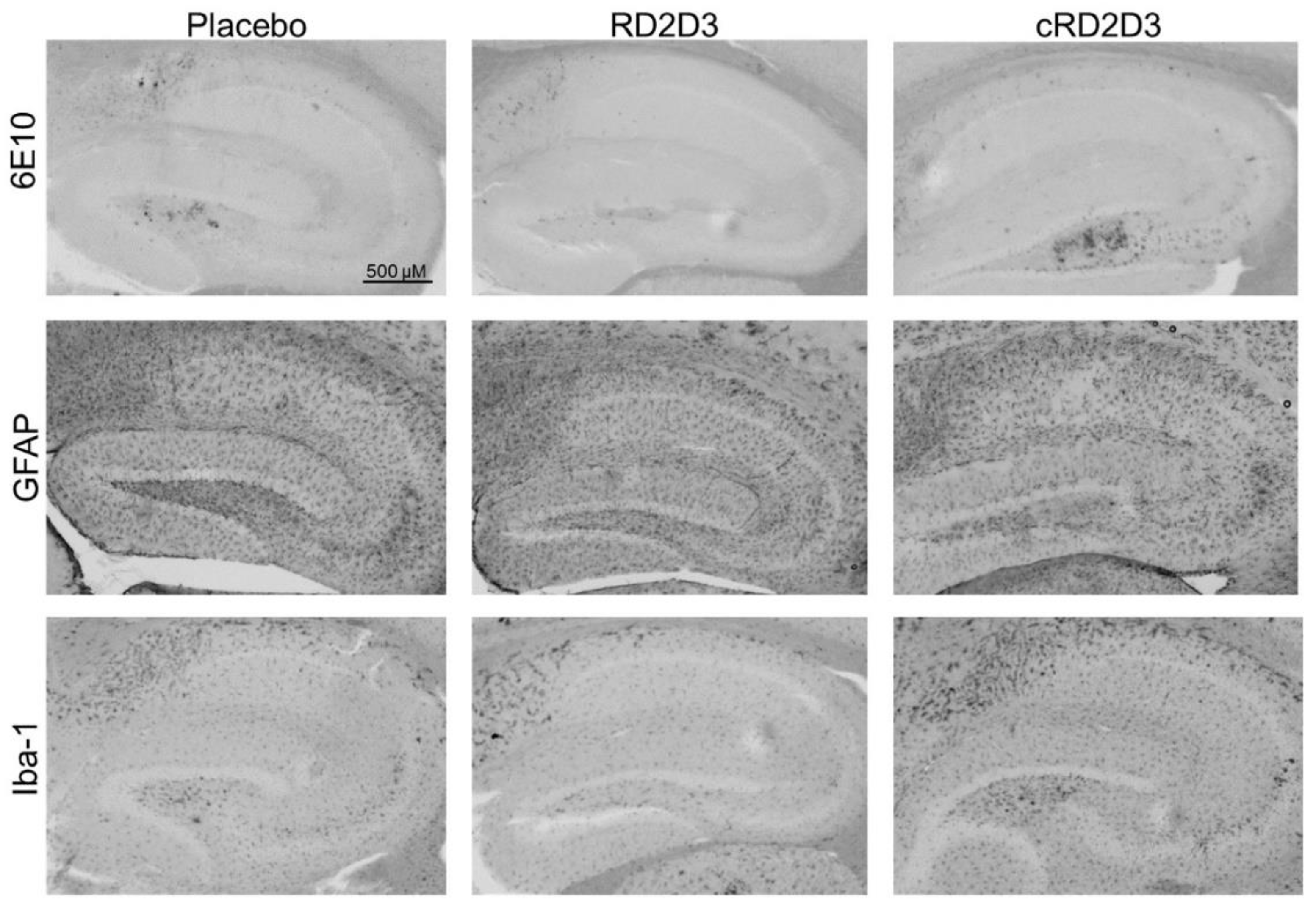
2.3. RD2D3 Showed Superior Efficacy to Improve Phenotypic Deficits over cRD2D3
3. Discussion
4. Materials and Methods
4.1. Peptides
4.2. In Vitro Potency
4.2.1. Binding Affinity
4.2.2. Thioflavin-T Assay
4.2.3. QIAD Assay
4.2.4. Proteolytic Stability
4.3. In Vivo Efficacy
4.3.1. Animals
4.3.2. Ethical Approval
4.3.3. Characterization
4.3.4. Nesting Behavior
4.3.5. Marble Burying
4.3.6. Open Field Test
4.3.7. Morris Water Maze
4.4. Treatment
4.5. Histology
4.6. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Grande, G.; Qiu, C.; Fratiglioni, L. Prevention of dementia in an ageing world: Evidence and biological rationale. Ageing Res. Rev. 2020, 64, 101045. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimer’s Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [PubMed]
- Willbold, D.; Kutzsche, J. Do We Need Anti-Prion Compounds to Treat Alzheimer’s Disease? Molecules 2019, 24, 2237. [Google Scholar] [CrossRef]
- van Groen, T.; Wiesehan, K.; Funke, S.A.; Kadish, I.; Nagel-Steger, L.; Willbold, D. Reduction of Alzheimer’s disease amyloid plaque load in transgenic mice by D3, A D-enantiomeric peptide identified by mirror image phage display. ChemMedChem 2008, 3, 1848–1852. [Google Scholar] [CrossRef] [PubMed]
- Van Regenmortel, M.H.; Muller, S. D-peptides as immunogens and diagnostic reagents. Curr. Opin. Biotechnol. 1998, 9, 377–382. [Google Scholar] [CrossRef]
- Dintzis, H.M.; Symer, D.E.; Dintzis, R.Z.; Zawadzke, L.E.; Berg, J.M. A comparison of the immunogenicity of a pair of enantiomeric proteins. Proteins Struct. Funct. Bioinform. 1993, 16, 306–308. [Google Scholar] [CrossRef]
- Kutzsche, J.; Jürgens, D.; Willuweit, A.; Adermann, K.; Fuchs, C.; Simons, S.; Windisch, M.; Hümpel, M.; Rossberg, W.; Wolzt, M.; et al. Safety and pharmacokinetics of the orally available antiprionic compound PRI-002: A single and multiple ascending dose phase I study. Alzheimer Dement. Transl. Res. Clin. Interv. 2020, 6, e12001. [Google Scholar] [CrossRef]
- van Groen, T.; Schemmert, S.; Brener, O.; Gremer, L.; Ziehm, T.; Tusche, M.; Nagel-Steger, L.; Kadish, I.; Schartmann, E.; Elfgen, A.; et al. The Abeta oligomer eliminating D-enantiomeric peptide RD2 improves cognition without changing plaque pathology. Sci. Rep. 2017, 7, 16275. [Google Scholar] [CrossRef]
- Schemmert, S.; Schartmann, E.; Zafiu, C.; Kass, B.; Hartwig, S.; Lehr, S.; Bannach, O.; Langen, K.; Shah, N.J.; Kutzsche, J.; et al. Abeta Oligomer Elimination Restores Cognition in Transgenic Alzheimer’s Mice with Full-blown Pathology. Mol. Neurobiol. 2019, 56, 2211–2223. [Google Scholar] [CrossRef]
- Kutzsche, J.; Schemmert, S.; Tusche, M.; Neddens, J.; Rabl, R.; Jürgens, D.; Brener, O.; Willuweit, A.; Hutter-Paier, B.; Willbold, D. Large-Scale Oral Treatment Study with the Four Most Promising D3-Derivatives for the Treatment of Alzheimer’s Disease. Molecules 2017, 22, 1693. [Google Scholar] [CrossRef]
- Schemmert, S.; Schartmann, E.; Honold, D.; Zafiu, C.; Ziehm, T.; Langen, K.-J.; Shah, N.J.; Kutzsche, J.; Willuweit, A.; Willbold, D. Deceleration of the neurodegenerative phenotype in pyroglutamate-Abeta accumulating transgenic mice by oral treatment with the Abeta oligomer eliminating compound RD2. Neurobiol. Dis. 2019, 124, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Cavini, I.A.; Munte, C.E.; Erlach, M.B.; van Groen, T.; Kadish, I.; Zhang, T.; Ziehm, T.; Nagel-Steger, L.; Kutzsche, J.; Kremer, W.; et al. Inhibition of amyloid Aβ aggregation by high pressures or specific d-enantiomeric peptides. Chem. Commun. 2018, 54, 3294–3297. [Google Scholar] [CrossRef] [PubMed]
- Leithold, L.H.E.; Jiang, N.; Post, J.; Niemietz, N.; Schartmann, E.; Ziehm, T.; Kutzsche, J.; Shah, N.J.; Breitkreutz, J.; Langen, K.-J.; et al. Pharmacokinetic properties of tandem d-peptides designed for treatment of Alzheimer’s disease. Eur. J. Pharm. Sci. 2016, 89, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Ziehm, T.; Brener, O.; Van Groen, T.; Kadish, I.; Frenzel, D.; Tusche, M.; Kutzsche, J.; Reiß, K.; Gremer, L.; Nagel-Steger, L.; et al. Increase of Positive Net Charge and Conformational Rigidity Enhances the Efficacy of d-Enantiomeric Peptides Designed to Eliminate Cytotoxic Aβ Species. ACS Chem. Neurosci. 2016, 7, 1088–1096. [Google Scholar] [CrossRef]
- Schartmann, E.; Schemmert, S.; Niemietz, N.; Honold, D.; Ziehm, T.; Tusche, M.; Elfgen, A.; Gering, I.; Brener, O.; Shah, N.J.; et al. In Vitro Potency and Preclinical Pharmacokinetic Comparison of All-D-Enantiomeric Peptides Developed for the Treatment of Alzheimer’s Disease. J. Alzheimer Dis. 2018, 64, 859–873. [Google Scholar] [CrossRef]
- Brener, O.; Dunkelmann, T.; Gremer, L.; Van Groen, T.; Mirecka, E.A.; Kadish, I.; Willuweit, A.; Kutzsche, J.; Jürgens, D.; Rudolph, S.; et al. QIAD assay for quantitating a compound’s efficacy in elimination of toxic Aβ oligomers. Sci. Rep. 2015, 5, 13222. [Google Scholar] [CrossRef]
- Alzheimer, A. Concerning unsual medical cases in old age. Z. Gesamte Neurol. Psychiatr. 1911, 4, 356–385. [Google Scholar] [CrossRef]
- Scheltens, P.P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Li, S.; Selkoe, D.J. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer’s brain. J. Neurochem. 2020, 154, 583–597. [Google Scholar] [CrossRef]
- Schartmann, E.; Schemmert, S.; Ziehm, T.; Leithold, L.H.E.; Jiang, N.; Tusche, M.; Shah, N.J.; Langen, K.-J.; Kutzsche, J.; Willbold, D.; et al. Comparison of blood-brain barrier penetration efficiencies between linear and cyclic all-d-enantiomeric peptides developed for the treatment of Alzheimer’s disease. Eur. J. Pharm. Sci. 2018, 114, 93–102. [Google Scholar] [CrossRef]
- Klein, A.N.; Ziehm, T.; van Groen, T.; Kadish, I.; Elfgen, A.; Tusche, M.; Thomaier, M.; Reiss, K.; Brener, O.; Gremer, L.; et al. Optimization of d-Peptides for Abeta Monomer Binding Specificity Enhances Their Potential to Eliminate Toxic Abeta Oligomers. ACS Chem. Neurosci. 2017, 8, 1889–1900. [Google Scholar] [CrossRef]
- Davis, J.; Xu, F.; Deane, R.; Romanov, G.; Previti, M.L.; Zeigler, K.; Zlokovic, B.V.; van Nostrand, W.E. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J. Biol. Chem. 2004, 279, 20296–20306. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Xu, F.; Davis, J.; Otte-Höller, I.; Verbeek, M.M.; van Nostrand, W.E. Cerebral microvascular amyloid beta protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid beta precursor protein. Am. J. Pathol. 2005, 167, 505–515. [Google Scholar] [CrossRef]
- Xu, F.; Grande, A.M.; Robinson, J.K.; Previti, M.L.; Vasek, M.; Davis, J.; van Nostrand, W.E. Early-onset subicular microvascular amyloid and neuroinflammation correlate with behavioral deficits in vasculotropic mutant amyloid beta-protein precursor transgenic mice. Neuroscience 2007, 146, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Robison, L.S.; Francis, N.; Popescu, D.L.; Anderson, M.E.; Hatfield, J.; Xu, F.; Anderson, B.J.; Van Nostrand, W.E.; Robinson, J.K. Environmental Enrichment: Disentangling the Influence of Novelty, Social, and Physical Activity on Cerebral Amyloid Angiopathy in a Transgenic Mouse Model. Int. J. Mol. Sci. 2020, 21, 843. [Google Scholar] [CrossRef] [PubMed]
- Elfgen, A.; Santiago-Schübel, B.; Gremer, L.; Kutzsche, J.; Willbold, D. Surprisingly high stability of the Aβ oligomer eliminating all-d-enantiomeric peptide D3 in media simulating the route of orally administered drugs. Eur. J. Pharm. Sci. 2017, 107, 203–207. [Google Scholar] [CrossRef]
- Deacon, R.M.J. Assessing nest building in mice. Nat. Protoc. 2006, 1, 1117–1119. [Google Scholar] [CrossRef] [PubMed]
- Deacon, R.M.J. Digging and marble burying in mice: Simple methods for in vivo identification of biological impacts. Nat. Protoc. 2006, 1, 122–124. [Google Scholar] [CrossRef]
- Archer, J. Tests for emotionality in rats and mice: A review. Anim. Behav. 1973, 21, 205–235. [Google Scholar] [CrossRef]
- Paulson, J.B.; Ramsden, M.; Forster, C.; Sherman, M.A.; McGowan, E.; Ashe, K.H. Amyloid plaque and neurofibrillary tangle pathology in a regulatable mouse model of Alzheimer’s disease. Am. J. Pathol. 2008, 173, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.G.M.; Garrud, P.; Rawlins, J.N.P.; O’Keefe, J. Place navigation impaired in rats with hippocampal lesions. Nature 1982, 297, 681–683. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 6E10 IR (%) | GFAP IR (%) | Iba-1 IR (%) | ||||
|---|---|---|---|---|---|---|
| Cortex | Hippocampus | Cortex | Hippocampus | Cortex | Hippocampus | |
| Placebo | 1.8 ± 0.7 | 2.5 ± 0.7 | 43.8 ± 2.0 | 39.8 ± 1.0 | 8.7 ± 1.7 | 11. 5 ± 1.1 |
| RD2D3 | 1.0 ± 0.2 | 2.5 ± 0.5 | 40.6 ± 2.2 | 38.0 ± 1.2 | 8.1 ± 1.6 | 8.6 ± 1.3 |
| cRD2D3 | 1.1 ± 0.2 | 4.2 ± 0.7 | 43.1 ± 1.8 | 39.5 ± 1.0 | 8.1 ± 1.6 | 10.2 ± 1.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schemmert, S.; Camargo, L.C.; Honold, D.; Gering, I.; Kutzsche, J.; Willuweit, A.; Willbold, D. In Vitro and In Vivo Efficacies of the Linear and the Cyclic Version of an All-d-Enantiomeric Peptide Developed for the Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 6553. https://doi.org/10.3390/ijms22126553
Schemmert S, Camargo LC, Honold D, Gering I, Kutzsche J, Willuweit A, Willbold D. In Vitro and In Vivo Efficacies of the Linear and the Cyclic Version of an All-d-Enantiomeric Peptide Developed for the Treatment of Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(12):6553. https://doi.org/10.3390/ijms22126553
Chicago/Turabian StyleSchemmert, Sarah, Luana Cristina Camargo, Dominik Honold, Ian Gering, Janine Kutzsche, Antje Willuweit, and Dieter Willbold. 2021. "In Vitro and In Vivo Efficacies of the Linear and the Cyclic Version of an All-d-Enantiomeric Peptide Developed for the Treatment of Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 12: 6553. https://doi.org/10.3390/ijms22126553
APA StyleSchemmert, S., Camargo, L. C., Honold, D., Gering, I., Kutzsche, J., Willuweit, A., & Willbold, D. (2021). In Vitro and In Vivo Efficacies of the Linear and the Cyclic Version of an All-d-Enantiomeric Peptide Developed for the Treatment of Alzheimer’s Disease. International Journal of Molecular Sciences, 22(12), 6553. https://doi.org/10.3390/ijms22126553