
TMB in NSCLC: A Broken Dream?



{kind=link}
Abstract
1. Introduction
1.1. Etiopathogenesis of Lung Cancer
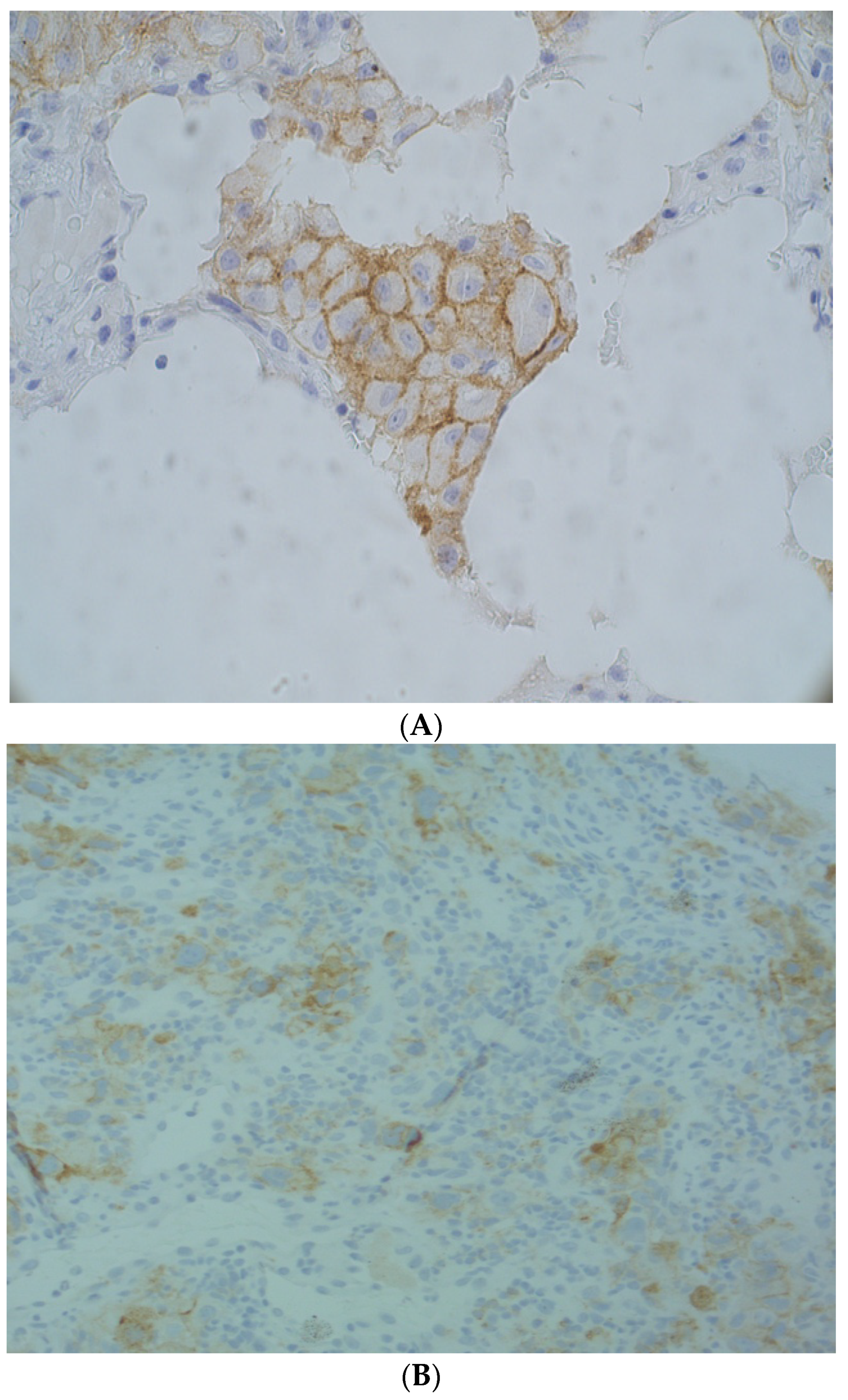
1.2. Biomarkers Used in Patient Selection for Treatment with Immune Checkpoint Inhibitors
1.3. Role of Tumor Mutation Burden (TMB)
1.3.1. TMB Definition
1.3.2. TMB Detection
1.3.3. TMB Pitfalls
How Many Genes Need to Be Analyzed for “a Perfect” TMB?
1.3.4. TMB in Clinical Practice
1.4. Data on Lung Cancer Trials
2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Früh, M.; Rolland, E.; Pignon, J.P.; Seymour, L.; Ding, K.; Tribodet, H.; Winton, T.; Le Chevalier, T.; Scagliotti, G.V.; Douillard, J.Y.; et al. Pooled Analysis of the Effect of Age on Adjuvant Cisplatin-Based Chemotherapy for Completely Resected Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2008, 26, 3573–3581. [Google Scholar] [CrossRef]
- Liu, X.; Conner, H.; Kobayashi, T.; Kim, H.; Wen, F.; Abe, S.; Fang, Q.; Wang, X.; Hashimoto, M.; Bitterman, P.; et al. Cigarette smoke extract induces DNA damage but not apoptosis in human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2005, 33, 121–129. [Google Scholar] [CrossRef]
- Field, R.W.; Withers, B.L. Occupational and Environmental Causes of Lung Cancer. Clin. Chest. Med. 2012, 33, 681–703. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. WHO Panel. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances since the 2004 Classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef]
- D’Arcangelo, M.; D’Incecco, A.; Ligorio, C.; Damiani, S.; Puccetti, M.; Bravaccini, S.; Terracciano, L.; Bennati, C.; Minuti, G.; Vecchiarelli, S.; et al. Programmed death ligand 1 expression in early stage, resectable non-small cell lung cancer. Oncotarget 2019, 10, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Melosky, B.; Chu, Q.; Juergens, R.; Leighl, N.; McLeod, D.; Hirsh, V. Pointed Progress in Second-Line Advanced Non-Small-Cell Lung Cancer: The Rapidly Evolving Field of Checkpoint Inhibition. J. Clin. Oncol. 2016, 34, 1676–1688. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. KEYNOTE-024 Investigators. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. KEYNOTE-001 Investigators.Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; OAK Study Group.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Bravaccini, S.; Ulivi, P. What’s the best modality for patient selection for predicting response to PD-1/PD-L1 inhibitors? Transl. Lung Cancer Res. 2020, 9, 158–159. [Google Scholar] [CrossRef]
- Sharabi, A.; Kim, S.S.; Kato, S.; Sanders, P.D.; Patel, S.P.; Sanghvi, P.; Weihe, E.; Kurzrock, R. Exceptional Response to Nivolumab and Stereotactic Body Radiation Therapy (SBRT) in Neuroendocrine Cervical Carcinoma with High Tumor Mutational Burden: Management Considerations from the Center for Personalized Cancer Therapy at UC San Diego Moores Cancer Center. Oncologist 2017, 22, 631–637. [Google Scholar] [CrossRef]
- Campesato, L.F.; Barroso-Sousa, R.; Jimenez, L.; Correa, B.R.; Sabbaga, J.; Hoff, P.M.; Reis, L.F.; Galante, P.A.; Camargo, A.A. Comprehensive cancer-gene panels can be used to estimate mutational load and predict clinical benefit to PD-1 blockade in clinical practice. Oncotarget 2015, 6, 34221–34227. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Lam, V.K.; Zhang, J. Blood-based tumor mutation burden: Continued progress toward personalizing immunotherapy in non-small cell lung cancer. J. Thorac. Dis. 2019, 11, 2208–2211. [Google Scholar] [CrossRef]
- Qiu, P.; Poehlein, C.H.; Marton, M.J.; Laterza, O.F.; Levitan, D. Measuring Tumor Mutational Burden (TMB) in Plasma from mCRPC Patients Using Two Commercial NGS Assays. Sci. Rep. 2019, 9, 114. [Google Scholar] [CrossRef]
- Fumet, J.D.; Truntzer, C.; Yarchoan, M.; Ghiringhelli, F. Tumour mutational burden as a biomarker for immunotherapy: Current data and emerging concepts. Eur. J. Cancer 2020, 131, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Viveiros, P.; Lopes, G.; Sukhadia, B.; Sheikh, M.M.; Saravia, D.; Florou, V.; Sokol, E.S.; Frampton, G.M.; Chalmers, Z.R.; et al. Clinical and Immunological Implications of Frameshift Mutations in Lung Cancer. J. Thorac. Oncol. 2019, 14, 1807–1817. [Google Scholar] [CrossRef]
- Shen, L.; Zhang, J.; Lee, H.; Batista, M.T.; Johnston, S.A. RNA Transcription and Splicing Errors as a Source of Cancer Frameshift Neoantigens for Vaccines. Sci. Rep. 2019, 9, 14184. [Google Scholar] [CrossRef]
- Slansky, J.E.; Spellman, P.T. Alternative Splicing in Tumors-A Path to Immunogenicity? N. Engl. J. Med. 2019, 380, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Büttner, R.; Longshore, J.W.; López-Ríos, F.; Merkelbach-Bruse, S.; Normanno, N.; Rouleau, E.; Penault-Llorca, F. Implementing TMB measurement in clinical practice: Considerations on assay requirements. ESMO Open. 2019, 4, e000442. [Google Scholar] [CrossRef] [PubMed]
- Heydt, C.; Rehker, J.; Pappesch, R.; Buhl, T.; Ball, M.; Siebolts, U.; Haak, A.; Lohneis, P.; Büttner, R.; Hillmer, A.M.; et al. Analysis of tumor mutational burden: Correlation of five large gene panels with whole exome sequencing. Sci. Rep. 2020, 10, 11387. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.; Hamilton, P.T.; Zhang, A.W.; Pattnaik, S.; Becht, E.; Mezheyeuski, A.; Bruun, J.; Micke, P.; de Reynies, A.; Nelson, B.H. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9020–9029. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.; Bartok, O.; Patkar, S.; Eli, G.B.; Cohen, S.; Litchfield, K.; Levy, R.; Jiménez-Sánchez, A.; Trabish, S.; Lee, J.S.; et al. UVB-Induced Tumor Heterogeneity Diminishes Immune Response in Melanoma. Cell 2019, 179, 219–235. [Google Scholar] [CrossRef]
- Lu, S.; Stein, J.E.; Rimm, D.L.; Wang, D.W.; Bell, J.M.; Johnson, D.B.; Sosman, J.A.; Schalper, K.A.; Anders, R.A.; Wang, H.; et al. Comparison of Biomarker Modalities for Predicting Response to PD-1/PD-L1 Checkpoint Blockade: A Systematic Review and Meta-analysis. JAMA Oncol. 2019, 5, 1195–1204. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Meng, Y.; Blank, C.; Brown, I.; Kacha, A.; Kline, J.; Harlin, H. Immune resistance orchestrated by the tumor microenvironment. Immunol. Rev. 2006, 213, 131–145. [Google Scholar] [CrossRef]
- Strickler, J.H.; Hanks, B.A.; Khasraw, M. Tumor Mutational Burden as a Predictor of Immunotherapy Response: Is More Always Better? Clin. Cancer Res. 2021, 27, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haanen, J.B.; Ribas, A.; Schumacher, T.N. The “cancer immunogram”. Science 2016, 352, 658–660. [Google Scholar] [CrossRef]
- Merino, D.M.; McShane, L.M.; Fabrizio, D.; Funari, V.; Chen, S.J.; White, J.R.; Wenz, P.; Baden, J.; Barrett, J.C.; TMB Harmonization Consortium.; et al. Establishing guidelines to harmonize tumor mutational burden (TMB): In silico assessment of variation in TMB quantification across diagnostic platforms: Phase I of the Friends of Cancer Research TMB Harmonization Project. J. ImmunoTher. Cancer 2020, 8, e000147. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Yao, L.; Yang, Q.; Chi, T. Association of MSH2 Expression With Tumor Mutational Burden and the Immune Microenvironment in Lung Adenocarcinoma. Front. Oncol. 2020, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef]
- Dudnik, E.; Peled, N.; Nechushtan, H.; Wollner, M.; Onn, A.; Agbarya, A.; Moskovitz, M.; Keren, S.; Popovits-Hadari, N.; Israel Lung Cancer Group.; et al. BRAF Mutant Lung Cancer: Programmed Death Ligand 1 Expression, Tumor Mutational Burden, Microsatellite Instability Status, and Response to Immune Check-Point Inhibitors. J. Thorac. Oncol. 2018, 13, 1128–1137. [Google Scholar] [CrossRef]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.E.; Badin, F.; et al. CheckMate 026 Investigators. First-Line Nivolumab in Stage IV or Recurrent Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S..; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; POPLAR Study Group.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.B.; Abu-Akeel, M.; et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 2018, 33, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F.; Bunn, P.A.; Minna, J.D. Small-cell lung cancer: What we know, what we need to know and the path forward. Nat. Rev. Cancer 2017, 17, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lee, H.; Lee, B.; Lee, S.H.; Sun, J.M.; Park, W.Y.; Ahn, J.S.; Ahn, M.J.; Park, K. DNA Damage Response and Repair Pathway Alteration and Its Association With Tumor Mutation Burden and Platinum-Based Chemotherapy in SCLC. J. Thorac. Oncol. 2019, 14, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Callahan, M.K.; Awad, M.M.; Calvo, E.; Ascierto, P.A.; Atmaca, A.; Rizvi, N.A.; Hirsch, F.R.; Selvaggi, G.; Szustakowski, J.D.; et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell 2018, 33, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. Impower 133 Study Group. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bravaccini, S.; Bronte, G.; Ulivi, P. TMB in NSCLC: A Broken Dream? Int. J. Mol. Sci. 2021, 22, 6536. https://doi.org/10.3390/ijms22126536
Bravaccini S, Bronte G, Ulivi P. TMB in NSCLC: A Broken Dream? International Journal of Molecular Sciences. 2021; 22(12):6536. https://doi.org/10.3390/ijms22126536
Chicago/Turabian StyleBravaccini, Sara, Giuseppe Bronte, and Paola Ulivi. 2021. "TMB in NSCLC: A Broken Dream?" International Journal of Molecular Sciences 22, no. 12: 6536. https://doi.org/10.3390/ijms22126536
APA StyleBravaccini, S., Bronte, G., & Ulivi, P. (2021). TMB in NSCLC: A Broken Dream? International Journal of Molecular Sciences, 22(12), 6536. https://doi.org/10.3390/ijms22126536