
Roles of Nuclear Receptors in Vascular Calcification



Abstract
1. Introduction
1.1. Nuclear Receptors
1.2. Vascular Calcification
1.2.1. Generalities
1.2.2. Bone Mineralization
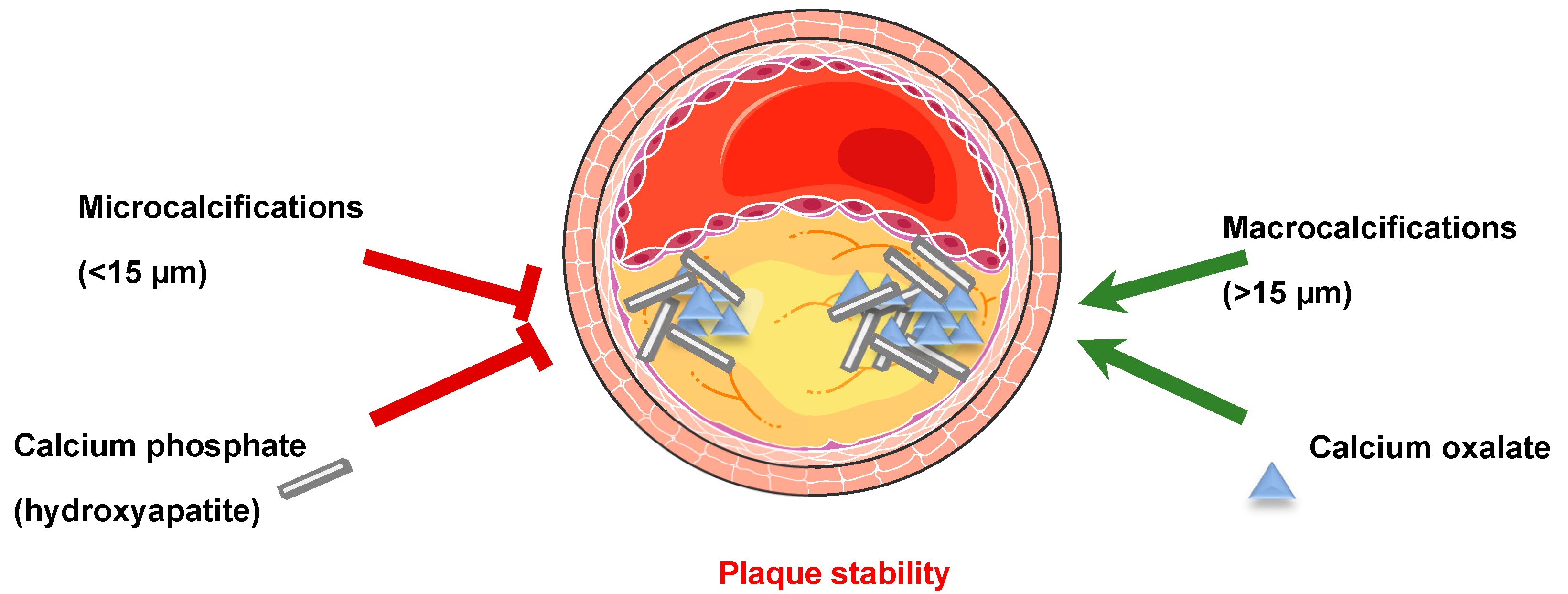
1.2.3. Soft Tissue Calcification
2. Role of Nuclear Receptors in Vascular Calcification
2.1. Vitamin D Receptor (VDR)
2.2. Human Steroid and Xenobiotic Receptor (SXR) and Its Rodent Homolog Pregnane X Receptor (PXR)
2.3. Sex Hormones and Receptors: Estrogen Receptors (ER), Androgen Receptor (AR) and Progesterone Receptor (PrR)
2.3.1. Estrogen Receptors
2.3.2. Androgen Receptor
2.3.3. Progesterone Receptor (PgR)
2.4. Peroxisome Proliferator-Activated Receptors (PPAR)
2.5. Liver X Receptors (LXR)
2.6. Farnesoid X Receptor (FXR)
2.7. Mineralocorticoid Receptor (MR) and Glucocorticoid Receptor (GR)
2.7.1. Mineralocorticoid Receptor (MR)
2.7.2. Glucocorticoid Receptor (GR)
2.8. Retinoic Acid Receptor (RAR)
2.9. Orphan Nuclear Receptors
3. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Weikum, E.R.; Liu, X.; Ortlund, E.A. The nuclear receptor superfamily: A structural perspective. Protein Sci. 2018, 27, 1876–1892. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.S.; Li, Y.; Zhang, H.; Zhang, D.; Zhang, X.B.; Wang, X.; Yu, Y. The ERalpha-miR-575-p27 feedback loop regulates tamoxifen sensitivity in ER-positive Breast Cancer. Theranostics 2020, 10, 10729–10742. [Google Scholar] [CrossRef]
- Shin, C.H.; Byun, J.; Lee, K.; Kim, B.; Noh, Y.K.; Tran, N.L.; Park, K.; Kim, S.H.; Kim, T.H.; Oh, S.J. Exosomal miRNA-19a and miRNA-614 induced by air pollutants promote proinflammatory M1 macrophage polarization via regulation of RORalpha expression in human respiratory mucosal microenvironment. J. Immunol. 2020, 205, 3179–3190. [Google Scholar] [CrossRef]
- Huang, J.T.; Welch, J.S.; Ricote, M.; Binder, C.J.; Willson, T.M.; Kelly, C.; Witztum, J.L.; Funk, C.D.; Conrad, D.; Glass, C.K. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature 1999, 400, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.S.; Czimmerer, Z.; Szanto, A.; Nagy, L. Pro-inflammatory cytokines negatively regulate PPARgamma mediated gene expression in both human and murine macrophages via multiple mechanisms. Immunobiology 2013, 218, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Malbeteau, L.; Pham, H.T.; Eve, L.; Stallcup, M.R.; Poulard, C.; Le Romancer, M. How protein methylation regulates steroid receptor function. Endocr. Rev. 2021. [Google Scholar] [CrossRef]
- Liu, W.; Zeng, M.; Fu, N. Functions of nuclear receptors SUMOylation. Clin. Chim. Acta 2021, 516, 27–33. [Google Scholar] [CrossRef]
- Demer, L.L.; Tintut, Y. Vascular calcification: Pathobiology of a multifaceted disease. Circulation 2008, 117, 2938–2948. [Google Scholar] [CrossRef]
- Schwarz, U.; Buzello, M.; Ritz, E.; Stein, G.; Raabe, G.; Wiest, G.; Mall, G.; Amann, K. Morphology of coronary atherosclerotic lesions in patients with end-stage renal failure. Nephrol. Dial. Transplant. 2000, 15, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Gao, J.; Lv, Q.; Cai, H.; Wang, F.; Ye, R.; Liu, X. Calcification in atherosclerotic plaque vulnerability: Friend or foe? Front. Physiol. 2020, 11, 56. [Google Scholar] [CrossRef]
- Barrett, H.E.; van der Heiden, K.; Farrell, E.; Gijsen, F.J.H.; Akyildiz, A.C. Calcifications in atherosclerotic plaques and impact on plaque biomechanics. J. Biomech. 2019, 87, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M. Vascular calcification mechanisms. J. Am. Soc. Nephrol. 2004, 15, 2959–2964. [Google Scholar] [CrossRef]
- Speer, M.Y.; Giachelli, C.M. Regulation of cardiovascular calcification. Cardiovasc. Pathol. 2004, 13, 63–70. [Google Scholar] [CrossRef]
- Niederhoffer, N.; Marque, V.; Lartaud-Idjouadiene, I.; Duvivier, C.; Peslin, R.; Atkinson, J. Vasodilators, aortic elasticity, and ventricular end-systolic stress in nonanesthetized unrestrained rats. Hypertension 1997, 30, 1169–1174. [Google Scholar] [CrossRef]
- Tsushima, M.; Terayama, Y.; Momose, A.; Funyu, T.; Ohyama, C.; Hada, R. Carotid intima media thickness and aortic calcification index closely relate to cerebro- and cardiovascular disorders in hemodialysis patients. Int. J. Urol. 2008, 15, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Jinnouchi, H.; Sato, Y.; Sakamoto, A.; Cornelissen, A.; Mori, M.; Kawakami, R.; Gadhoke, N.V.; Kolodgie, F.D.; Virmani, R.; Finn, A.V. Calcium deposition within coronary atherosclerotic lesion: Implications for plaque stability. Atherosclerosis 2020, 306, 85–95. [Google Scholar] [CrossRef]
- Cardoso, L.; Kelly-Arnold, A.; Maldonado, N.; Laudier, D.; Weinbaum, S. Effect of tissue properties, shape and orientation of microcalcifications on vulnerable cap stability using different hyperelastic constitutive models. J. Biomech. 2014, 47, 870–877. [Google Scholar] [CrossRef]
- Miralles, M.; Merino, J.; Busto, M.; Perich, X.; Barranco, C.; Vidal-Barraquer, F. Quantification and characterization of carotid calcium with multi-detector CT-angiography. Eur. J. Vasc. Endovasc. Surg. 2006, 32, 561–567. [Google Scholar] [CrossRef]
- Ehara, S.; Kobayashi, Y.; Yoshiyama, M.; Shimada, K.; Shimada, Y.; Fukuda, D.; Nakamura, Y.; Yamashita, H.; Yamagishi, H.; Takeuchi, K.; et al. Spotty calcification typifies the culprit plaque in patients with acute myocardial infarction: An intravascular ultrasound study. Circulation 2004, 110, 3424–3429. [Google Scholar] [CrossRef]
- Mizukoshi, M.; Kubo, T.; Takarada, S.; Kitabata, H.; Ino, Y.; Tanimoto, T.; Komukai, K.; Tanaka, A.; Imanishi, T.; Akasaka, T. Coronary superficial and spotty calcium deposits in culprit coronary lesions of acute coronary syndrome as determined by optical coherence tomography. Am. J. Cardiol. 2013, 112, 34–40. [Google Scholar] [CrossRef]
- Karlof, E.; Seime, T.; Dias, N.; Lengquist, M.; Witasp, A.; Almqvist, H.; Kronqvist, M.; Gadin, J.R.; Odeberg, J.; Maegdefessel, L.; et al. Correlation of computed tomography with carotid plaque transcriptomes associates calcification with lesion-stabilization. Atherosclerosis 2019, 288, 175–185. [Google Scholar] [CrossRef]
- Fishbein, G.A.; Micheletti, R.G.; Currier, J.S.; Singer, E.; Fishbein, M.C. Atherosclerotic oxalosis in coronary arteries. Cardiovasc. Pathol. 2008, 17, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Bischetti, S.; Scimeca, M.; Bonanno, E.; Federici, M.; Anemona, L.; Menghini, R.; Casella, S.; Cardellini, M.; Ippoliti, A.; Mauriello, A. Carotid plaque instability is not related to quantity but to elemental composition of calcification. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Lampropoulos, C.E.; Papaioannou, I.; D’Cruz, D.P. Osteoporosis—A risk factor for cardiovascular disease? Nat. Rev. Rheumatol. 2012, 8, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Sprini, D.; Rini, G.B.; Di Stefano, L.; Cianferotti, L.; Napoli, N. Correlation between osteoporosis and cardiovascular disease. Clin. Cases Miner. Bone Metab. 2014, 11, 117–119. [Google Scholar] [CrossRef]
- Zhou, R.; Zhou, H.; Cui, M.; Chen, L.; Xu, J. The association between aortic calcification and fracture risk in postmenopausal women in China: The prospective Chongqing osteoporosis study. PLoS ONE 2014, 9, e93882. [Google Scholar] [CrossRef] [PubMed]
- Salari, P.; Abollahi, M. A comprehensive revieiw of the shared roles of inflammatory cytokines in osteoporosis an cardiovascular diseases as two commun old people problem; actions toward development of new drugs. Int. J. Pharmacol. 2011, 7, 552–567. [Google Scholar] [CrossRef]
- Kassem, M.; Abdallah, B.M.; Saeed, H. Osteoblastic cells: Differentiation and trans-differentiation. Arch. Biochem. Biophys. 2008, 473, 183–187. [Google Scholar] [CrossRef]
- Ciancaglini, P.; Yadav, M.C.; Simao, A.M.; Narisawa, S.; Pizauro, J.M.; Farquharson, C.; Hoylaerts, M.F.; Millan, J.L. Kinetic analysis of substrate utilization by native and TNAP-, NPP1-, or PHOSPHO1-deficient matrix vesicles. J. Bone Miner. Res. 2010, 25, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, D.E.; Siggelkow, H.; Ponce, L.M.; Viereck, V.; Wiese, K.G.; Peters, J.H. Alkaline phosphatase expression during monocyte differentiation. Overlapping markers as a link between monocytic cells, dendritic cells, osteoclasts and osteoblasts. Immunobiology 2000, 202, 68–81. [Google Scholar] [CrossRef]
- Fadini, G.P.; Albiero, M.; Menegazzo, L.; Boscaro, E.; Vigili de Kreutzenberg, S.; Agostini, C.; Cabrelle, A.; Binotto, G.; Rattazzi, M.; Bertacco, E.; et al. Widespread increase in myeloid calcifying cells contributes to ectopic vascular calcification in type 2 diabetes. Circ. Res. 2011, 108, 1112–1121. [Google Scholar] [CrossRef]
- Albiero, M.; Rattazzi, M.; Menegazzo, L.; Boscaro, E.; Cappellari, R.; Pagnin, E.; Bertacco, E.; Poncina, N.; Dyar, K.; Ciciliot, S.; et al. Myeloid calcifying cells promote atherosclerotic calcification via paracrine activity and allograft inflammatory factor-1 overexpression. Basic Res. Cardiol. 2013, 108, 368. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N.; Takahashi, N.; Akatsu, T.; Tanaka, H.; Sasaki, T.; Nishihara, T.; Koga, T.; Martin, T.J.; Suda, T. Origin of osteoclasts: Mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc. Natl. Acad. Sci. USA 1990, 87, 7260–7264. [Google Scholar] [CrossRef] [PubMed]
- Hayman, A.R. Tartrate-resistant acid phosphatase (TRAP) and the osteoclast/immune cell dichotomy. Autoimmunity 2008, 41, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Vattikuti, R.; Towler, D.A. Osteogenic regulation of vascular calcification: An early perspective. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E686–E696. [Google Scholar] [CrossRef]
- Tintut, Y.; Patel, J.; Territo, M.; Saini, T.; Parhami, F.; Demer, L.L. Monocyte/macrophage regulation of vascular calcification in vitro. Circulation 2002, 105, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Tintut, Y.; Alfonso, Z.; Saini, T.; Radcliff, K.; Watson, K.; Bostrom, K.; Demer, L.L. Multilineage potential of cells from the artery wall. Circulation 2003, 108, 2505–2510. [Google Scholar] [CrossRef]
- Jono, S.; Nishizawa, Y.; Shioi, A.; Morii, H. 1,25-Dihydroxyvitamin D3 increases in vitro vascular calcification by modulating secretion of endogenous parathyroid hormone-related peptide. Circulation 1998, 98, 1302–1306. [Google Scholar] [CrossRef]
- Oksala, N.; Levula, M.; Pelto-Huikko, M.; Kytomaki, L.; Soini, J.T.; Salenius, J.; Kahonen, M.; Karhunen, P.J.; Laaksonen, R.; Parkkila, S.; et al. Carbonic anhydrases II and XII are up-regulated in osteoclast-like cells in advanced human atherosclerotic plaques—Tampere Vascular Study. Ann. Med. 2010, 42, 360–370. [Google Scholar] [CrossRef]
- Qiao, J.H.; Mishra, V.; Fishbein, M.C.; Sinha, S.K.; Rajavashisth, T.B. Multinucleated giant cells in atherosclerotic plaques of human carotid arteries: Identification of osteoclast-like cells and their specific proteins in artery wall. Exp. Mol. Pathol. 2015, 99, 654–662. [Google Scholar] [CrossRef]
- Saponaro, F.; Saba, A.; Zucchi, R. An update on vitamin D metabolism. Int. J. Mol. Sci. 2020, 21, 6573. [Google Scholar] [CrossRef] [PubMed]
- Saccone, D.; Asani, F.; Bornman, L. Regulation of the vitamin D receptor gene by environment, genetics and epigenetics. Gene 2015, 561, 171–180. [Google Scholar] [CrossRef]
- Demer, L.L.; Hsu, J.J.; Tintut, Y. Steroid hormone vitamin D: Implications for cardiovascular disease. Circ. Res. 2018, 122, 1576–1585. [Google Scholar] [CrossRef]
- Schmidt, N.; Brandsch, C.; Schutkowski, A.; Hirche, F.; Stangl, G.I. Dietary vitamin D inadequacy accelerates calcification and osteoblast-like cell formation in the vascular system of LDL receptor knockout and wild-type mice. J. Nutr. 2014, 144, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Ellam, T.; Hameed, A.; ul Haque, R.; Muthana, M.; Wilkie, M.; Francis, S.E.; Chico, T.J. Vitamin D deficiency and exogenous vitamin D excess similarly increase diffuse atherosclerotic calcification in apolipoprotein E knockout mice. PLoS ONE 2014, 9, e88767. [Google Scholar]
- Schmidt, N.; Brandsch, C.; Kuhne, H.; Thiele, A.; Hirche, F.; Stangl, G.I. Vitamin D receptor deficiency and low vitamin D diet stimulate aortic calcification and osteogenic key factor expression in mice. PLoS ONE 2012, 7, e35316. [Google Scholar] [CrossRef]
- Shamsuzzaman, S.; Onal, M.; St John, H.C.; Jeffery, J.J.; Pike, J.W. Absence of the vitamin D receptor inhibits atherosclerotic plaque calcification in female hypercholesterolemic mice. J. Cell. Biochem. 2017, 118, 1050–1064. [Google Scholar] [CrossRef]
- Han, M.S.; Che, X.; Cho, G.H.; Park, H.R.; Lim, K.E.; Park, N.R.; Jin, J.S.; Jung, Y.K.; Jeong, J.H.; Lee, I.K.; et al. Functional cooperation between vitamin D receptor and Runx2 in vitamin D-induced vascular calcification. PLoS ONE 2013, 8, e83584. [Google Scholar] [CrossRef] [PubMed]
- Sallam, T.; Tintut, Y.; Demer, L.L. Regulation of calcific vascular and valvular disease by nuclear receptors. Curr. Opin. Lipidol. 2019, 30, 357–363. [Google Scholar] [CrossRef]
- Acar, S.; Demir, K.; Shi, Y. Genetic causes of rickets. J. Clin. Res. Pediatr. Endocrinol. 2017, 9 (Suppl. 2), 88–105. [Google Scholar] [CrossRef]
- Lomashvili, K.A.; Wang, X.; O’Neill, W.C. Role of local versus systemic vitamin D receptors in vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Fusaro, M.; Ciceri, P.; Gasperoni, L.; Cianciolo, G. The role of vitamin K in vascular calcification. Adv. Chronic. Kidney Dis. 2019, 26, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Roumeliotis, S.; Dounousi, E.; Salmas, M.; Eleftheriadis, T.; Liakopoulos, V. Vascular calcification in chronic kidney disease: The role of vitamin K-dependent matrix Gla protein. Front. Med. 2020, 7, 154. [Google Scholar] [CrossRef]
- Jiang, X.; Tao, H.; Qiu, C.; Ma, X.; Li, S.; Guo, X.; Lv, A.; Li, H. Vitamin K2 regression aortic calcification induced by warfarin via Gas6/Axl survival pathway in rats. Eur. J. Pharmacol. 2016, 786, 10–18. [Google Scholar] [CrossRef]
- Brandenburg, V.M.; Reinartz, S.; Kaesler, N.; Kruger, T.; Dirrichs, T.; Kramann, R.; Peeters, F.; Floege, J.; Keszei, A.; Marx, N.; et al. Slower progress of aortic valve calcification with vitamin K supplementation: Results from a prospective interventional proof-of-concept study. Circulation 2017, 135, 2081–2083. [Google Scholar] [CrossRef]
- Mandatori, D.; Pipino, C.; di Tomo, P.; Schiavone, V.; Ranieri, A.; Pantalone, S.; di Silvestre, S.; di Pietrantonio, N.; Ucci, M.; Palmerini, C.; et al. Osteogenic transdifferentiation of vascular smooth muscle cells isolated from spontaneously hypertensive rats and potential menaquinone-4 inhibiting effect. J. Cell. Physiol. 2019, 234, 19761–19773. [Google Scholar] [CrossRef]
- Scheiber, D.; Veulemans, V.; Horn, P.; Chatrou, M.L.; Potthoff, S.A.; Kelm, M.; Schurgers, L.J.; Westenfeld, R. High-dose menaquinone-7 supplementation reduces cardiovascular calcification in a murine model of extraosseous calcification. Nutrients 2015, 7, 6991–7011. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Z.; Zhu, J.; Long, X.; Yan, J. Vitamin K2 can suppress the expression of Toll-like receptor 2 (TLR2) and TLR4, and inhibit calcification of aortic intima in ApoE(−/−) mice as well as smooth muscle cells. Vascular 2018, 26, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Zheng, H.; Tao, H.; Yu, W.; Jiang, X.; Li, A.; Jin, H.; Lv, A.; Li, H. Vitamin K2 inhibits rat vascular smooth muscle cell calcification by restoring the Gas6/Axl/Akt anti-apoptotic pathway. Mol. Cell. Biochem. 2017, 433, 149–159. [Google Scholar] [CrossRef]
- Saito, E.; Wachi, H.; Sato, F.; Sugitani, H.; Seyama, Y. Treatment with vitamin k(2) combined with bisphosphonates synergistically inhibits calcification in cultured smooth muscle cells. J. Atheroscler. Thromb. 2007, 14, 317–324. [Google Scholar] [CrossRef]
- Cui, L.; Xu, J.; Zhang, J.; Zhang, M.; Zhang, S.; Bai, Y. Menaquinone-4 modulates the expression levels of calcification-associated factors to inhibit calcification of rat aortic vascular smooth muscle cells in a dose-dependent manner. Exp. Ther. Med. 2018, 16, 3172–3178. [Google Scholar] [CrossRef] [PubMed]
- Spronk, H.M.; Soute, B.A.; Schurgers, L.J.; Thijssen, H.H.; de Mey, J.G.; Vermeer, C. Tissue-specific utilization of menaquinone-4 results in the prevention of arterial calcification in warfarin-treated rats. J. Vasc. Res. 2003, 40, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Elango, K.; Javaid, A.; Khetarpal, B.K.; Ramalingam, S.; Kolandaivel, K.P.; Gunasekaran, K.; Ahsan, C. The effects of warfarin and direct oral anticoagulants on systemic vascular calcification: A review. Cells 2021, 10, 773. [Google Scholar] [CrossRef]
- Rulcova, A.; Prokopova, I.; Krausova, L.; Bitman, M.; Vrzal, R.; Dvorak, Z.; Blahos, J.; Pavek, P. Stereoselective interactions of warfarin enantiomers with the pregnane X nuclear receptor in gene regulation of major drug-metabolizing cytochrome P450 enzymes. J. Thromb. Haemost. 2010, 8, 2708–2717. [Google Scholar] [CrossRef]
- Yu, Z.; Seya, K.; Chiyoya, M.; Daitoku, K.; Motomura, S.; Imaizumi, T.; Fukuda, I.; Furukawa, K.I. Warfarin calcifies human aortic valve interstitial cells at high-phosphate conditions via pregnane X receptor. J. Bone Miner. Metab. 2019, 37, 944–956. [Google Scholar] [CrossRef]
- Beazley, K.E.; Deasey, S.; Lima, F.; Nurminskaya, M.V. Transglutaminase 2-mediated activation of beta-catenin signaling has a critical role in warfarin-induced vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 123–130. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tabb, M.M.; Sun, A.; Zhou, C.; Grun, F.; Errandi, J.; Romero, K.; Pham, H.; Inoue, S.; Mallick, S.; Lin, M.; et al. Vitamin K2 regulation of bone homeostasis is mediated by the steroid and xenobiotic receptor SXR. J. Biol. Chem. 2003, 278, 43919–43927. [Google Scholar] [CrossRef]
- Azuma, K.; Ouchi, Y.; Inoue, S. Vitamin K: Novel molecular mechanisms of action and its roles in osteoporosis. Geriatr. Gerontol. Int. 2014, 14, 1–7. [Google Scholar] [CrossRef]
- Yang, W.; Yu, Z.; Chiyoya, M.; Liu, X.; Daitoku, K.; Motomura, S.; Imaizumi, T.; Fukuda, I.; Furukawa, K.I.; Tsuji, M.; et al. Menaquinone-4 accelerates calcification of human aortic valve interstitial cells in high-phosphate medium through PXR. J. Pharmacol. Exp. Ther. 2020, 372, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Horie-Inoue, K.; Ikeda, K.; Blumberg, B.; Inoue, S. Steroid and xenobiotic receptor SXR mediates vitamin K2-activated transcription of extracellular matrix-related genes and collagen accumulation in osteoblastic cells. J. Biol. Chem. 2006, 281, 16927–16934. [Google Scholar] [CrossRef]
- Azuma, K.; Casey, S.C.; Ito, M.; Urano, T.; Horie, K.; Ouchi, Y.; Kirchner, S.; Blumberg, B.; Inoue, S. Pregnane X receptor knockout mice display osteopenia with reduced bone formation and enhanced bone resorption. J. Endocrinol. 2010, 207, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Assem, M.; Tay, J.C.; Watkins, P.B.; Blumberg, B.; Schuetz, E.G.; Thummel, K.E. Steroid and xenobiotic receptor and vitamin D receptor crosstalk mediates CYP24 expression and drug-induced osteomalacia. J. Clin. Investig. 2006, 116, 1703–1712. [Google Scholar] [CrossRef]
- Bolego, C.; Vegeto, E.; Pinna, C.; Maggi, A.; Cignarella, A. Selective agonists of estrogen receptor isoforms: New perspectives for cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2192–2199. [Google Scholar] [CrossRef]
- Nakamura, T.; Imai, Y.; Matsumoto, T.; Sato, S.; Takeuchi, K.; Igarashi, K.; Harada, Y.; Azuma, Y.; Krust, A.; Yamamoto, Y.; et al. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell 2007, 130, 811–823. [Google Scholar] [CrossRef]
- Hofbauer, L.C.; Khosla, S.; Dunstan, C.R.; Lacey, D.L.; Spelsberg, T.C.; Riggs, B.L. Estrogen stimulates gene expression and protein production of osteoprotegerin in human osteoblastic cells. Endocrinology 1999, 140, 4367–4370. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.E.; Allison, M.A.; Rossouw, J.E.; Carr, J.J.; Langer, R.D.; Hsia, J.; Kuller, L.H.; Cochrane, B.B.; Hunt, J.R.; Ludlam, S.E.; et al. Estrogen therapy and coronary-artery calcification. N. Engl. J. Med. 2007, 356, 2591–2602. [Google Scholar] [CrossRef] [PubMed]
- Budoff, M.J.; Chen, G.P.; Hunter, C.J.; Takasu, J.; Agrawal, N.; Sorochinsky, B.; Mao, S. Effects of hormone replacement on progression of coronary calcium as measured by electron beam tomography. J. Women’s Health 2005, 14, 410–417. [Google Scholar] [CrossRef]
- Christian, R.C.; Harrington, S.; Edwards, W.D.; Oberg, A.L.; Fitzpatrick, L.A. Estrogen status correlates with the calcium content of coronary atherosclerotic plaques in women. J. Clin. Endocrinol. Metab. 2002, 87, 1062–1067. [Google Scholar] [CrossRef]
- Jeon, G.H.; Kim, S.H.; Yun, S.C.; Chae, H.D.; Kim, C.H.; Kang, B.M. Association between serum estradiol level and coronary artery calcification in postmenopausal women. Menopause 2010, 17, 902–907. [Google Scholar] [CrossRef]
- El Khoudary, S.R.; Zhao, Q.; Venugopal, V.; Manson, J.E.; Brooks, M.M.; Santoro, N.; Black, D.M.; Harman, S.M.; Cedars, M.I.; Hopkins, P.N.; et al. Effects of hormone therapy on heart fat and coronary artery calcification progression: Secondary analysis from the KEEPS trial. J. Am. Heart Assoc. 2019, 8, e012763. [Google Scholar] [CrossRef]
- Tse, J.; Martin-McNaulty, B.; Halks-Miller, M.; Kauser, K.; DelVecchio, V.; Vergona, R.; Sullivan, M.E.; Rubanyi, G.M. Accelerated atherosclerosis and premature calcified cartilaginous metaplasia in the aorta of diabetic male Apo E knockout mice can be prevented by chronic treatment with 17 beta-estradiol. Atherosclerosis 1999, 144, 303–313. [Google Scholar] [CrossRef]
- Osako, M.K.; Nakagami, H.; Koibuchi, N.; Shimizu, H.; Nakagami, F.; Koriyama, H.; Shimamura, M.; Miyake, T.; Rakugi, H.; Morishita, R. Estrogen inhibits vascular calcification via vascular RANKL system: Common mechanism of osteoporosis and vascular calcification. Circ. Res. 2010, 107, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhao, Q.; Chen, Z.; Geng, Y.J.; Zhang, W.; Zhou, Q.; Yang, W.; Liu, Q.; Liu, H. Estrogen inhibits vascular calcification in rats via hypoxia-induced factor-1alpha signaling. Vascular 2020, 28, 465–474. [Google Scholar] [CrossRef]
- Choi, B.G.; Vilahur, G.; Zafar, M.U.; Cardoso, L.; Yadegar, D.; Ibanez, B.; Tunstead, J.; Viles-Gonzalez, J.F.; Schaffler, M.B.; Fuster, V.; et al. Selective estrogen receptor modulation influences atherosclerotic plaque composition in a rabbit menopause model. Atherosclerosis 2008, 201, 76–84. [Google Scholar] [CrossRef] [PubMed]
- McRobb, L.S.; McGrath, K.C.Y.; Tsatralis, T.; Liong, E.C.; Tan, J.T.M.; Hughes, G.; Handelsman, D.J.; Heather, A.K. Estrogen receptor control of atherosclerotic calcification and smooth muscle cell osteogenic differentiation. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1127–1137. [Google Scholar] [CrossRef]
- Hodis, H.N.; Mack, W.J.; Shoupe, D.; Azen, S.P.; Stanczyk, F.Z.; Hwang-Levine, J.; Budoff, M.J.; Henderson, V.W. Methods and baseline cardiovascular data from the Early versus Late Intervention Trial with Estradiol testing the menopausal hormone timing hypothesis. Menopause 2015, 22, 391–401. [Google Scholar] [CrossRef]
- Rosenfeld, M.E.; Kauser, K.; Martin-McNulty, B.; Polinsky, P.; Schwartz, S.M.; Rubanyi, G.M. Estrogen inhibits the initiation of fatty streaks throughout the vasculature but does not inhibit intra-plaque hemorrhage and the progression of established lesions in apolipoprotein E deficient mice. Atherosclerosis 2002, 164, 251–259. [Google Scholar] [CrossRef]
- Christian, R.C.; Liu, P.Y.; Harrington, S.; Ruan, M.; Miller, V.M.; Fitzpatrick, L.A. Intimal estrogen receptor (ER)beta, but not ERalpha expression, is correlated with coronary calcification and atherosclerosis in pre- and postmenopausal women. J. Clin. Endocrinol. Metab. 2006, 91, 2713–2720. [Google Scholar] [CrossRef]
- Balica, M.; Bostrom, K.; Shin, V.; Tillisch, K.; Demer, L.L. Calcifying subpopulation of bovine aortic smooth muscle cells is responsive to 17 beta-estradiol. Circulation 1997, 95, 1954–1960. [Google Scholar] [CrossRef]
- Rzewuska-Lech, E.; Jayachandran, M.; Fitzpatrick, L.A.; Miller, V.M. Differential effects of 17beta-estradiol and raloxifene on VSMC phenotype and expression of osteoblast-associated proteins. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E105–E112. [Google Scholar] [CrossRef]
- Karwowski, W.; Lekesiz, K.; Koc-Zorawska, E.; Wnuczko, K.; Borysewicz-Sanczyk, H.; Naumnik, B. Effects of 17beta-estradioland raloxifene on endothelial OPG and RANKL secretion. Ginekol. Pol. 2017, 88, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Kapustin, A.N.; Shanahan, C.M. Osteocalcin: A novel vascular metabolic and osteoinductive factor? Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2169–2171. [Google Scholar] [CrossRef]
- Nanao-Hamai, M.; Son, B.K.; Hashizume, T.; Ogawa, S.; Akishita, M. Protective effects of estrogen against vascular calcification via estrogen receptor alpha-dependent growth arrest-specific gene 6 transactivation. Biochem. Biophys. Res. Commun. 2016, 480, 429–435. [Google Scholar] [CrossRef]
- Son, B.K.; Kozaki, K.; Iijima, K.; Eto, M.; Nakano, T.; Akishita, M.; Ouchi, Y. Gas6/Axl-PI3K/Akt pathway plays a central role in the effect of statins on inorganic phosphate-induced calcification of vascular smooth muscle cells. Eur. J. Pharmacol. 2007, 556, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vanderschueren, D.; Vandenput, L.; Boonen, S.; Lindberg, M.K.; Bouillon, R.; Ohlsson, C. Androgens and bone. Endocr. Rev. 2004, 25, 389–425. [Google Scholar] [CrossRef] [PubMed]
- Allison, M.A.; Wright, C.M. Age and gender are the strongest clinical correlates of prevalent coronary calcification (R1). Int. J. Cardiol. 2005, 98, 325–330. [Google Scholar] [CrossRef]
- Zhu, D.; Hadoke, P.W.; Wu, J.; Vesey, A.T.; Lerman, D.A.; Dweck, M.R.; Newby, D.E.; Smith, L.B.; MacRae, V.E. Ablation of the androgen receptor from vascular smooth muscle cells demonstrates a role for testosterone in vascular calcification. Sci. Rep. 2016, 6, 24807. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.Y.; Christian, R.C.; Ruan, M.; Miller, V.M.; Fitzpatrick, L.A. Correlating androgen and estrogen steroid receptor expression with coronary calcification and atherosclerosis in men without known coronary artery disease. J. Clin. Endocrinol. Metab. 2005, 90, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- McRobb, L.; Handelsman, D.J.; Heather, A.K. Androgen-induced progression of arterial calcification in apolipoprotein E-null mice is uncoupled from plaque growth and lipid levels. Endocrinology 2009, 150, 841–848. [Google Scholar] [CrossRef]
- Hak, A.E.; Witteman, J.C.; de Jong, F.H.; Geerlings, M.I.; Hofman, A.; Pols, H.A. Low levels of endogenous androgens increase the risk of atherosclerosis in elderly men: The Rotterdam study. J. Clin. Endocrinol. Metab. 2002, 87, 3632–3639. [Google Scholar] [CrossRef]
- Son, B.K.; Akishita, M.; Iijima, K.; Ogawa, S.; Maemura, K.; Yu, J.; Takeyama, K.; Kato, S.; Eto, M.; Ouchi, Y. Androgen receptor-dependent transactivation of growth arrest-specific gene 6 mediates inhibitory effects of testosterone on vascular calcification. J. Biol. Chem. 2010, 285, 7537–7544. [Google Scholar] [CrossRef] [PubMed]
- Nanao-Hamai, M.; Son, B.K.; Komuro, A.; Asari, Y.; Hashizume, T.; Takayama, K.I.; Ogawa, S.; Akishita, M. Ginsenoside Rb1 inhibits vascular calcification as a selective androgen receptor modulator. Eur. J. Pharmacol. 2019, 859, 172546. [Google Scholar] [CrossRef]
- Pang, H.; Xiao, L.; Lu, Z.; Chen, H.; Shang, Z.; Jiang, N.; Wang, X.; Wei, F.; Jiang, A.; Chen, Y.; et al. Targeting androgen receptor in macrophages inhibits phosphate-induced vascular smooth muscle cell calcification by decreasing IL-6 expression. Vasc. Pharmacol. 2020, 130, 106681. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Rodriguez-Manzaneque, J.C.; Lydon, J.P.; Edwards, D.P.; O’Malley, B.W.; Iruela-Arispe, M.L. Progesterone regulates proliferation of endothelial cells. J. Biol. Chem. 1999, 274, 2185–2192. [Google Scholar] [CrossRef]
- Cutini, P.H.; Massheimer, V.L. In vitro effects of progesterone and the synthetic progestin medroxyprogesterone acetate on vascular remodeling. Mol. Cell. Endocrinol. 2019, 498, 110543. [Google Scholar] [CrossRef] [PubMed]
- Dixon, E.D.; Nardo, A.D.; Claudel, T.; Trauner, M. The role of lipid sensing nuclear receptors (PPARs and LXR) and metabolic lipases in obesity, diabetes and NAFLD. Genes 2021, 12, 645. [Google Scholar] [CrossRef]
- Finck, B.N.; Chinetti, G.; Staels, B. PPARs/RXRs in cardiovascular physiology and disease. PPAR Res. 2008, 2008, 173780. [Google Scholar] [CrossRef] [PubMed]
- Woldt, E.; Terrand, J.; Mlih, M.; Matz, R.L.; Bruban, V.; Coudane, F.; Foppolo, S.; El Asmar, Z.; Chollet, M.E.; Ninio, E.; et al. The nuclear hormone receptor PPARgamma counteracts vascular calcification by inhibiting Wnt5a signalling in vascular smooth muscle cells. Nat. Commun. 2012, 3, 1077. [Google Scholar] [CrossRef]
- Morello, R.; Bertin, T.K.; Schlaubitz, S.; Shaw, C.A.; Kakuru, S.; Munivez, E.; Hermanns, P.; Chen, Y.; Zabel, B.; Lee, B. Brachy-syndactyly caused by loss of Sfrp2 function. J. Cell. Physiol. 2008, 217, 127–137. [Google Scholar] [CrossRef]
- Gao, M.; Chen, T.; Wu, L.; Zhao, X.; Mao, H.; Xing, C. Effect of pioglitazone on the calcification of rat vascular smooth muscle cells through the downregulation of the Wnt/betacatenin signaling pathway. Mol. Med. Rep. 2017, 16, 6208–6213. [Google Scholar] [CrossRef]
- Cheng, L.; Zhang, L.; Yang, J.; Hao, L. Activation of peroxisome proliferator-activated receptor gamma inhibits vascular calcification by upregulating Klotho. Exp. Ther. Med. 2017, 13, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Kuro-o, M. Klotho. Pflug. Arch. 2010, 459, 333–343. [Google Scholar] [CrossRef]
- Lim, K.; Lu, T.S.; Molostvov, G.; Lee, C.; Lam, F.T.; Zehnder, D.; Hsiao, L.L. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation 2012, 125, 2243–2255. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, Y.; Zhang, Y.; Bi, X.; Nie, L.; Liu, C.; Xiong, J.; He, T.; Xu, X.; Yu, Y.; et al. High phosphate-induced downregulation of PPARgamma contributes to CKD-associated vascular calcification. J. Mol. Cell. Cardiol. 2018, 114, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.X.; Duan, D.; O’Neill, K.D.; Moe, S.M. High glucose increases the expression of Cbfa1 and BMP-2 and enhances the calcification of vascular smooth muscle cells. Nephrol. Dial. Transplant. 2006, 21, 3435–3442. [Google Scholar] [CrossRef]
- Zhou, Y.B.; Zhang, J.; Peng, D.Q.; Chang, J.R.; Cai, Y.; Yu, Y.R.; Jia, M.Z.; Wu, W.; Guan, Y.F.; Tang, C.S.; et al. Peroxisome proliferator-activated receptor gamma ligands retard cultured vascular smooth muscle cells calcification induced by high glucose. Cell Biochem. Biophys. 2013, 66, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Etherton, T.D.; Martin, K.R.; Vanden Heuvel, J.P.; Gillies, P.J.; West, S.G.; Kris-Etherton, P.M. Anti-inflammatory effects of polyunsaturated fatty acids in THP-1 cells. Biochem. Biophys. Res. Commun. 2005, 336, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Abedin, M.; Tintut, Y.; Demer, L.L. Vascular calcification: Mechanisms and clinical ramifications. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1161–1170. [Google Scholar] [CrossRef]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef]
- Hsu, J.J.; Lu, J.; Huang, M.S.; Geng, Y.; Sage, A.P.; Bradley, M.N.; Tontonoz, P.; Demer, L.L.; Tintut, Y. T0901317, an LXR agonist, augments PKA-induced vascular cell calcification. FEBS Lett. 2009, 583, 1344–1348. [Google Scholar] [CrossRef]
- Ting, T.C.; Miyazaki-Anzai, S.; Masuda, M.; Levi, M.; Demer, L.L.; Tintut, Y.; Miyazaki, M. Increased lipogenesis and stearate accelerate vascular calcification in calcifying vascular cells. J. Biol. Chem. 2011, 286, 23938–23949. [Google Scholar] [CrossRef]
- Ma, C.; Feng, K.; Yang, X.; Yang, Z.; Wang, Z.; Shang, Y.; Fan, G.; Liu, L.; Yang, S.; Li, X.; et al. Targeting macrophage liver X receptors by hydrogel-encapsulated T0901317 reduces atherosclerosis without effect on hepatic lipogenesis. Br. J. Pharmacol. 2021, 178, 1620–1638. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, Z.; Feng, Q.; Chen, W.D.; Wang, Y.D. Farnesoid X receptor: A potential therapeutic target in multiple organs. Histol. Histopathol. 2020, 35, 1403–1414. [Google Scholar] [PubMed]
- He, F.; Li, J.; Mu, Y.; Kuruba, R.; Ma, Z.; Wilson, A.; Alber, S.; Jiang, Y.; Stevens, T.; Watkins, S.; et al. Downregulation of endothelin-1 by farnesoid X receptor in vascular endothelial cells. Circ. Res. 2006, 98, 192–199. [Google Scholar] [CrossRef]
- Zhang, Q.; He, F.; Kuruba, R.; Gao, X.; Wilson, A.; Li, J.; Billiar, T.R.; Pitt, B.R.; Xie, W.; Li, S. FXR-mediated regulation of angiotensin type 2 receptor expression in vascular smooth muscle cells. Cardiovasc. Res. 2008, 77, 560–569. [Google Scholar] [CrossRef]
- Miyazaki-Anzai, S.; Levi, M.; Kratzer, A.; Ting, T.C.; Lewis, L.B.; Miyazaki, M. Farnesoid X receptor activation prevents the development of vascular calcification in ApoE−/− mice with chronic kidney disease. Circ. Res. 2010, 106, 1807–1817. [Google Scholar] [CrossRef]
- Li, C.; Zhang, S.; Chen, X.; Ji, J.; Yang, W.; Gui, T.; Gai, Z.; Li, Y. Farnesoid X receptor activation inhibits TGFBR1/TAK1-mediated vascular inflammation and calcification via miR-135a-5p. Commun. Biol. 2020, 3, 327. [Google Scholar] [CrossRef]
- Miyazaki-Anzai, S.; Masuda, M.; Shiozaki, Y.; Keenan, A.L.; Chonchol, M.; Kremoser, C.; Miyazaki, M. Free deoxycholic acid exacerbates vascular calcification in chronic kidney disease through ER stress-mediated ATF4 activation. Kidney360 2021, 2, 557–868. [Google Scholar] [CrossRef]
- Id Boufker, H.; Lagneaux, L.; Fayyad-Kazan, H.; Badran, B.; Najar, M.; Wiedig, M.; Ghanem, G.; Laurent, G.; Body, J.J.; Journe, F. Role of farnesoid X receptor (FXR) in the process of differentiation of bone marrow stromal cells into osteoblasts. Bone 2011, 49, 1219–1231. [Google Scholar] [CrossRef]
- Lombes, M.; Oblin, M.E.; Gasc, J.M.; Baulieu, E.E.; Farman, N.; Bonvalet, J.P. Immunohistochemical and biochemical evidence for a cardiovascular mineralocorticoid receptor. Circ. Res. 1992, 71, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Fagart, J.; Wurtz, J.M.; Souque, A.; Hellal-Levy, C.; Moras, D.; Rafestin-Oblin, M.E. Antagonism in the human mineralocorticoid receptor. EMBO J. 1998, 17, 3317–3325. [Google Scholar] [CrossRef]
- Chapman, K.; Holmes, M.; Seckl, J. 11beta-hydroxysteroid dehydrogenases: Intracellular gate-keepers of tissue glucocorticoid action. Physiol. Rev. 2013, 93, 1139–1206. [Google Scholar] [CrossRef]
- Hatakeyama, H.; Inaba, S.; Takeda, R.; Miyamori, I. 11beta-hydroxysteroid dehydrogenase in human vascular cells. Kidney Int. 2000, 57, 1352–1357. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhu, D.; Rashdan, N.A.; Chapman, K.E.; Hadoke, P.W.; MacRae, V.E. A novel role for the mineralocorticoid receptor in glucocorticoid driven vascular calcification. Vascul. Pharmacol. 2016, 86, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Barrera-Chimal, J.; Jaisser, F. Vascular mineralocorticoid receptor activation and disease. Exp. Eye Res. 2019, 188, 107796. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, I.Z.; Mendelsohn, M.E. Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ. Res. 2005, 96, 643–650. [Google Scholar] [CrossRef]
- Jaffe, I.Z.; Tintut, Y.; Newfell, B.G.; Demer, L.L.; Mendelsohn, M.E. Mineralocorticoid receptor activation promotes vascular cell calcification. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Tatsumoto, N.; Yamada, S.; Tokumoto, M.; Eriguchi, M.; Noguchi, H.; Torisu, K.; Tsuruya, K.; Kitazono, T. Spironolactone ameliorates arterial medial calcification in uremic rats: The role of mineralocorticoid receptor signaling in vascular calcification. Am. J. Physiol. Renal Physiol. 2015, 309, F967–F979. [Google Scholar] [CrossRef]
- Voelkl, J.; Alesutan, I.; Leibrock, C.B.; Quintanilla-Martinez, L.; Kuhn, V.; Feger, M.; Mia, S.; Ahmed, M.S.; Rosenblatt, K.P.; Kuro, O.M.; et al. Spironolactone ameliorates PIT1-dependent vascular osteoinduction in klotho-hypomorphic mice. J. Clin. Investig. 2013, 123, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, H.Y.; Giachelli, C.M. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ. Res. 2006, 98, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.W.; He, W.B.; Xie, C.M.; Gao, M.; Feng, L.Y.; Liu, Z.Y.; Wang, J.F.; Huang, H.; Liu, P.M. Aldosterone enhances high phosphate-induced vascular calcification through inhibition of AMPK-mediated autophagy. J. Cell. Mol. Med. 2020, 24, 13648–13659. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Zhang, L.; Cong, G.; Ren, L.; Hao, L. MicroRNA-34b/c inhibits aldosterone-induced vascular smooth muscle cell calcification via a SATB2/Runx2 pathway. Cell Tissue Res. 2016, 366, 733–746. [Google Scholar] [CrossRef]
- Kirsch, T.; Wang, W.; Pfander, D. Functional differences between growth plate apoptotic bodies and matrix vesicles. J. Bone Miner. Res. 2003, 18, 1872–1881. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.E.; DuPont, J.J.; Iyer, S.L.; McGraw, A.P.; Jaffe, I.Z. No significant role for smooth muscle cell mineralocorticoid receptors in atherosclerosis in the apolipoprotein-E knockout mouse model. Front. Cardiovasc. Med. 2018, 5, 81. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Shioi, A.; Jono, S.; Nishizawa, Y.; Morii, H. Dexamethasone enhances in vitro vascular calcification by promoting osteoblastic differentiation of vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2112–2118. [Google Scholar] [CrossRef]
- Kirton, J.P.; Wilkinson, F.L.; Canfield, A.E.; Alexander, M.Y. Dexamethasone downregulates calcification-inhibitor molecules and accelerates osteogenic differentiation of vascular pericytes: Implications for vascular calcification. Circ. Res. 2006, 98, 1264–1272. [Google Scholar] [CrossRef]
- Preusch, M.R.; Rattazzi, M.; Albrecht, C.; Merle, U.; Tuckermann, J.; Schutz, G.; Blessing, E.; Zoppellaro, G.; Pauletto, P.; Krempien, R.; et al. Critical role of macrophages in glucocorticoid driven vascular calcification in a mouse-model of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2158–2164. [Google Scholar] [CrossRef]
- Terkeltaub, R. Macrophage glucocorticoid receptors join the intercellular dialogue in atherosclerotic lesion calcification. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2096–2098. [Google Scholar] [CrossRef]
- Sun, F.; Shi, J.; Chen, S.; Deng, C.; Hu, X.; Li, H.; Li, G.; Liu, Y.; Dong, N. Lazaroid U-74389G inhibits the osteoblastic differentiation of IL-1beta-indcued aortic valve interstitial cells through glucocorticoid receptor and inhibition of NF-kappaB pathway. J Steroid Biochem. Mol. Biol. 2015, 152, 114–123. [Google Scholar] [CrossRef]
- Peacock, J.D.; Levay, A.K.; Gillaspie, D.B.; Tao, G.; Lincoln, J. Reduced sox9 function promotes heart valve calcification phenotypes in vivo. Circ. Res. 2010, 106, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Huk, D.J.; Hammond, H.L.; Kegechika, H.; Lincoln, J. Increased dietary intake of vitamin A promotes aortic valve calcification in vivo. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 285–293. [Google Scholar] [CrossRef]
- Shimono, K.; Tung, W.E.; Macolino, C.; Chi, A.H.; Didizian, J.H.; Mundy, C.; Chandraratna, R.A.; Mishina, Y.; Enomoto-Iwamoto, M.; Pacifici, M.; et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-gamma agonists. Nat. Med. 2011, 17, 454–460. [Google Scholar] [CrossRef]
- Fu, X.; Wang, X.D.; Mernitz, H.; Wallin, R.; Shea, M.K.; Booth, S.L. 9-Cis retinoic acid reduces 1alpha,25-dihydroxycholecalciferol-induced renal calcification by altering vitamin K-dependent gamma-carboxylation of matrix gamma-carboxyglutamic acid protein in A/J male mice. J. Nutr. 2008, 138, 2337–2341. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.A.; Chen, J.; Nallamshetty, S.; Pham, T.; Goto, S.; Muehlschlegel, J.D.; Libby, P.; Aikawa, M.; Aikawa, E.; Plutzky, J. Retinoids repress human cardiovascular cell calcification with evidence for distinct selective retinoid modulator effects. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 656–669. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Choi, Y.K.; Do, J.Y.; Ha, C.M.; Lee, S.J.; Jeon, J.H.; Lee, W.K.; Choi, H.S.; Park, K.G.; Lee, I.K. Estrogen-related receptor gamma plays a key role in vascular calcification through the upregulation of BMP2 expression. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2384–2390. [Google Scholar] [CrossRef] [PubMed]
- Van Tiel, C.M.; de Vries, C.J. NR4All in the vessel wall. J. Steroid Biochem. Mol. Biol. 2012, 130, 186–193. [Google Scholar] [CrossRef]
- Zhu, Y.; Han, X.Q.; Sun, X.J.; Yang, R.; Ma, W.Q.; Liu, N.F. Lactate accelerates vascular calcification through NR4A1-regulated mitochondrial fission and BNIP3-related mitophagy. Apoptosis 2020, 25, 321–340. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| NR | Short Description of Findings | + or − Role * | Refs |
|---|---|---|---|
| VDR | U-shaped dualistic role for vitamin D; both low and high levels linked to VC. Contradictory VDR−/− mouse studies but mostly supporting inducing role. Human data: VDR mutations do not lead to VC. | + | [43,44,45,46,47,48,50] |
| SXR/PXR | Several studies support a protective role for vitamin K in VC. However, MK4 can induce calcification through SXR/PXR. PXR−/− mice have osteopenia but VC was not studied. Crosstalk SXR/PXR and vitamin D metabolism. No clear-cut conclusion regarding role SXP/PXR in VC. | ? | [54,55,56,57,58,59,60,61,62,67,69,70,71,72] |
| ER | Rodent and human studies demonstrate protective role for estradiol in VC. However, other rodent studies show opposite. Contradictory in vitro results. | ? | [76,77,78,79,81,82,83,84,85,89,90,91,93] |
| AR | Androgen treatment was shown to induce VC but low androgen has been associated with increased VC. Contradictory in vitro effects. | ? | [97,99,100,101,102] |
| PgR | In vitro studies show that PgR activation has beneficial effects for both VC and osteoporosis. | − | [105] |
| PPARγ | SMC knockout leads to increase in VC. Involves LRP1-Wnt pathway and other mechanisms through Klotho. | − | [108,109,110,111,113,114] |
| LXR | Activation induces mineralization in vitro. However, in vivo studies show the opposite. | ? | [120,121,122] |
| FXR | In vitro and in vivo data show that FXR activation leads to a decrease in VC. Multiple mechanisms proposed. | − | [126,127,128] |
| MR | Not sure whether aldosterone or cortisol activates MR in vasculature. MR activation by aldosterone leads to increase in VC. MR inhibitors can potentially prevent/treat VC. | + | [132,133,134,135,137,138,139,141,142] |
| GR | GR activation leads to increased VC. Opposite results with non-glucocorticoid steroid that binds to GR. Still, natural ligand leads to increased VC but in vivo implication not sure. | +? | [134,145,146,149] |
| RAR | Contradictory in vitro and in vivo results. In mice it induces VC while in humans it inhibits. | ? | [150,151,152,153,154] |
| Orphan | ERRγ: Activation increases VC in vitro and in vivo. NR4A1: Activation increases VC. | + | [155] [157] |
| Gene/Protein Involved in Vascular Calcification | NRs with Inducing Effect | NRs with Reducing Effect |
|---|---|---|
| TNAP/ALP | SXR/PXR [65,69,70] | PgR [105] PPARγ [108] |
| ER [89] AR [95,97] LXR [120] | FXR [126] | |
| MR [137] | RAR [154] | |
| BMP2 | SXR/PXR [65,69] ER [84] | ER [82] |
| MR [137] ERRγ [155] | PPARγ [114] | |
| Runx-2 | ERRγ [155] | PPARγ [108,114] |
| FXR [127] RAR [150] | ||
| Osteocalcin | ER [89] | PPARγ [108] |
| MGP | SXR/PXR [67,70] ER [82] | ER [85] |
| PPARγ [116] RAR [154] | ||
| OPN | SXR/PXR [67,70] | ER [85] |
| ERRγ [155] | LXR [120] | |
| OPG | SXR/PXR [67,70] | ER [90,91] |
| ER [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chinetti, G.; Neels, J.G. Roles of Nuclear Receptors in Vascular Calcification. Int. J. Mol. Sci. 2021, 22, 6491. https://doi.org/10.3390/ijms22126491
Chinetti G, Neels JG. Roles of Nuclear Receptors in Vascular Calcification. International Journal of Molecular Sciences. 2021; 22(12):6491. https://doi.org/10.3390/ijms22126491
Chicago/Turabian StyleChinetti, Giulia, and Jaap G. Neels. 2021. "Roles of Nuclear Receptors in Vascular Calcification" International Journal of Molecular Sciences 22, no. 12: 6491. https://doi.org/10.3390/ijms22126491
APA StyleChinetti, G., & Neels, J. G. (2021). Roles of Nuclear Receptors in Vascular Calcification. International Journal of Molecular Sciences, 22(12), 6491. https://doi.org/10.3390/ijms22126491