
Cellular Senescence: Pathogenic Mechanisms in Lung Fibrosis


Abstract
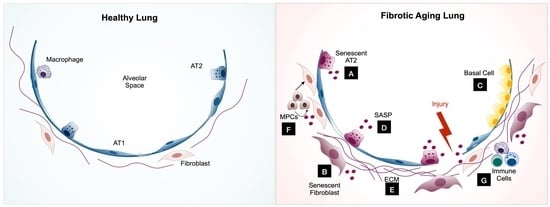
1. Introduction
1.1. Cellular Senescence and Lung Fibrosis
1.1.1. Cellular Senescence and Aging
1.1.2. Pulmonary Fibrosis
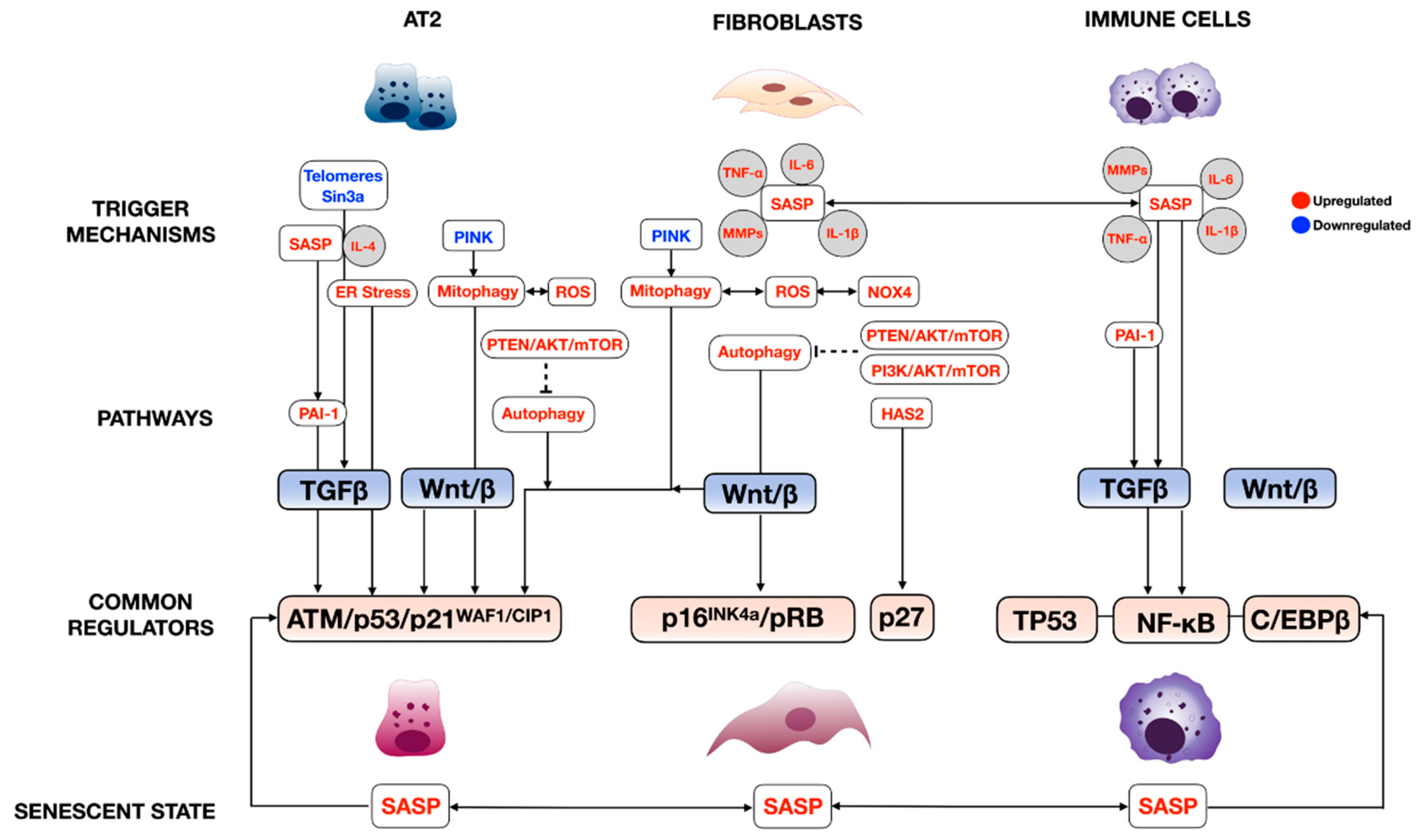
2. Senescence Regulatory Pathways
2.1. Cell Cycle Regulatory Pathway
2.2. Senescence-Associated Secretory Phenotype (SASP) Regulatory Pathway
3. The Role of Cellular Senescence in the Pathogenesis of Pulmonary Fibrosis
3.1. Epithelial Cells
3.1.1. Epithelial Senescence in IPF
3.1.2. AT2 Cell Cellular Senescence in Pulmonary Fibrosis
3.1.3. Basal Cells
3.2. Lung Fibroblasts
3.2.1. SASP
3.2.2. Mitochondrial Dysfunction
3.2.3. Autophagy
3.2.4. Apoptosis Resistance
3.3. Mesenchymal Progenitor Cells (MPCs)
3.4. Immune Cells
3.4.1. Immunosenescence
- Innate immunosenescence
- Adaptive immunosenescence
3.4.2. Senescence of Immune Cells
- Monocytes and macrophages
- T cells
- Other Immune Cells
3.5. Other Cells
Bone-Marrow Mesenchymal Stem Cells (B-MSCs)
4. Targeting Senescent Cells for the Treatment of Pulmonary Fibrosis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2017, 217, 65–77. [Google Scholar] [CrossRef]
- Loaiza, N.; Demaria, M. Cellular senescence and tumor promotion: Is aging the key? Biochim. Biophys. Acta Bioenerg. 2016, 1865, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Niccoli, T.; Partridge, L. Ageing as a Risk Factor for Disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef]
- Kirkwood, T.B. Understanding the Odd Science of Aging. Cell 2005, 120, 437–447. [Google Scholar] [CrossRef]
- North, B.J.; Sinclair, D.A. The Intersection Between Aging and Cardiovascular Disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- De Magalhães, J.P. How ageing processes influence cancer. Nat. Rev. Cancer 2013, 13, 357–365. [Google Scholar] [CrossRef]
- Gunasekaran, U.; Gannon, M. Type 2 Diabetes and the Aging Pancreatic Beta Cell. Aging 2011, 3, 565–575. [Google Scholar] [CrossRef]
- Castellani, R.J.; Rolston, R.K.; Smith, M.A. Alzheimer Disease. Dis. Month 2010, 56, 484–546. [Google Scholar] [CrossRef] [PubMed]
- Nalysnyk, L.; Cid-Ruzafa, J.; Rotella, P.; Esser, D. Incidence and prevalence of idiopathic pulmonary fibrosis: Review of the literature. Eur. Respir. Rev. 2012, 21, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Robles, S.J.; Adami, G.R. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene 1998, 16, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Rabeyrin, M.; Thivolet, F.; Ferretti, G.; Chalabreysse, L.; Jankowski, A.; Cottin, V.; Pison, C.; Cordier, J.-F.; Lantuejoul, S. Usual interstitial pneumonia end-stage features from explants with radiologic and pathological correlations. Ann. Diagn. Pathol. 2015, 19, 269–276. [Google Scholar] [CrossRef]
- American Thoracic Society. Idiopathic Pulmonary Fibrosis: Diagnosis and Treatment. Am. J. Respir. Crit. Care Med. 2000, 161, 646–664. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.-I.; Dauti, S.; Kim, H.J.; Park, S.H.; Park, J.S.; Lee, C.W. Risk factors for interstitial lung disease: A 9-year Nationwide population-based study. BMC Pulm. Med. 2018, 18, 96. [Google Scholar] [CrossRef]
- Raghu, G.; Chen, S.-Y.; Yeh, W.-S.; Maroni, B.; Li, Q.; Lee, Y.-C.; Collard, H.R. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: Incidence, prevalence, and survival, 2001–11. Lancet Respir. Med. 2014, 2, 566–572. [Google Scholar] [CrossRef]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Prim. 2017, 3, 17074. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Revealing the Pathogenic and Aging-related Mechanisms of the Enigmatic Idiopathic Pulmonary Fibrosis. An Integral Model. Am. J. Respir. Crit. Care Med. 2014, 189, 1161–1172. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Martínez-Zamudio, R.I.; Robinson, L.; Roux, P.-F.; Bischof, O. SnapShot: Cellular Senescence Pathways. Cell 2017, 170, 816–816.e1. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell. Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; De Los Santos, F.G.; Zhao, Y.; Wu, Z.; Rinke, A.E.; Kim, K.K.; Phan, S.H. Telomerase reverse transcriptase ameliorates lung fibrosis by protecting alveolar epithelial cells against senescence. J. Biol. Chem. 2019, 294, 8861–8871. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Guan, X.; Carraro, G.; Parimon, T.; Liu, X.; Huang, G.; Mulay, A.; Soukiasian, H.J.; David, G.; Weigt, S.S.; et al. Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 707–717. [Google Scholar] [CrossRef]
- Jiang, C.; Liu, G.; Luckhardt, T.; Antony, V.; Zhou, Y.; Carter, A.B.; Thannickal, V.J.; Liu, R.-M. Serpine 1 induces alveolar type II cell senescence through activating p53-p21-Rb pathway in fibrotic lung disease. Aging Cell 2017, 16, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, W.; Zheng, Z.; Wang, W.; Yuan, Y.; Hong, Q.; Lin, J.; Li, X.; Meng, Y. Cigarette smoke-inactivated SIRT1 promotes autophagy-dependent senescence of alveolar epithelial type 2 cells to induce pulmonary fibrosis. Free Radic. Biol. Med. 2021, 166, 116–127. [Google Scholar] [CrossRef]
- Young, A.R.; Narita, M. Connecting autophagy to senescence in pathophysiology. Curr. Opin. Cell Biol. 2010, 22, 234–240. [Google Scholar] [CrossRef]
- Qiu, T.; Tian, Y.; Gao, Y.; Ma, M.; Li, H.; Liu, X.; Wu, H.; Zhang, Y.; Ding, H.; Cao, M.; et al. PTEN loss regulates alveolar epithelial cell senescence in pulmonary fibrosis depending on Akt activation. Aging 2019, 11, 7492–7509. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Hu, Q.; Hu, Y.; Hafner, K.; Costa, R.; Berg, A.V.D.; Königshoff, M. Chronic WNT/β-catenin signaling induces cellular senescence in lung epithelial cells. Cell. Signal. 2020, 70, 109588. [Google Scholar] [CrossRef]
- Li, Y.; Liang, J.; Yang, T.; Mena, J.M.; Huan, C.; Xie, T.; Kurkciyan, A.; Liu, N.; Jiang, D.; Noble, P.W. Hyaluronan synthase 2 regulates fibroblast senescence in pulmonary fibrosis. Matrix Biol. 2016, 55, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Orjalo, A.V.; Desprez, P.-Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.-C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Brumwell, A.N.; Davis, S.S.; Jackson, J.R.; Valdovinos, A.; Calhoun, C.; Alimirah, F.; Castellanos, C.A.; Ruan, R.; Wei, Y.; et al. Secretion of leukotrienes by senescent lung fibroblasts promotes pulmonary fibrosis. JCI Insight 2019, 4, e130056. [Google Scholar] [CrossRef]
- Rana, T.; Jiang, C.; Liu, G.; Miyata, T.; Antony, V.; Thannickal, V.J.; Liu, R.-M. PAI-1 Regulation of TGF-β1–induced Alveolar Type II Cell Senescence, SASP Secretion, and SASP-mediated Activation of Alveolar Macrophages. Am. J. Respir. Cell Mol. Biol. 2020, 62, 319–330. [Google Scholar] [CrossRef]
- Hu, B.; Ullenbruch, M.R.; Jin, H.; Gharaee-Kermani, M.; Phan, S.; Phan, S. An essential role for CCAAT/enhancer binding protein β in bleomycin-induced pulmonary fibrosis. J. Pathol. 2006, 211, 455–462. [Google Scholar] [CrossRef] [PubMed]
- DeMaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Mutze, K.; Korfei, M.; Klee, S.; Wagner, D.; Costa, R.; Schiller, H.; Günther, A.; Königshoff, M. LSC–2017–Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur. Respir. J. 2017, 50, 1602367. [Google Scholar] [CrossRef]
- Fingerlin, E.T.; Murphy, E.; Zhang, W.; Peljto, A.L.; Brown, K.K.; Steele, M.P.; Loyd, J.; Cosgrove, G.P.; Lynch, D.; Groshong, S.; et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013, 45, 613–620. [Google Scholar] [CrossRef]
- Moore, C.; Blumhagen, R.Z.; Yang, I.V.; Walts, A.; Powers, J.; Walker, T.; Bishop, M.; Russell, P.; Vestal, B.; Cardwell, J.; et al. Resequencing Study Confirms That Host Defense and Cell Senescence Gene Variants Contribute to the Risk of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 199–208. [Google Scholar] [CrossRef]
- Kropski, J.A.; Blackwell, T.S.; Loyd, J.E. The genetic basis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1717–1727. [Google Scholar] [CrossRef]
- Schwartz, D.A. Idiopathic Pulmonary Fibrosis Is a Genetic Disease Involving Mucus and the Peripheral Airways. Ann. Am. Thorac. Soc. 2018, 15, S192–S197. [Google Scholar] [CrossRef]
- Allen, R.J.; Guillen-Guio, B.; Oldham, J.M.; Ma, S.-F.; Dressen, A.; Paynton, M.L.; Kraven, L.M.; Obeidat, M.; Li, X.; Ng, M.; et al. Genome-Wide Association Study of Susceptibility to Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.-I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.-K.T.; Funari, V.A.; Gokey, J.J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016, 1, e90558. [Google Scholar] [CrossRef]
- Povedano, J.M.; Martinez, P.; Flores, J.M.; Mulero, F.; Blasco, M.A. Mice with Pulmonary Fibrosis Driven by Telomere Dysfunction. Cell Rep. 2015, 12, 286–299. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Shi, Y.; Liu, Y.; Pan, X.-H.; Zhang, K.-X. Telomere shortening activates TGF-β/Smads signaling in lungs and enhances both lipopolysaccharide and bleomycin-induced pulmonary fibrosis. Acta Pharmacol. Sin. 2018, 39, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Barkauskas, C.E.; Limjunyawong, N.; Stanley, S.E.; Kembou, F.; Tuder, R.M.; Hogan, B.L.M.; Mitzner, W.; Armanios, M. Telomere dysfunction causes alveolar stem cell failure. Proc. Natl. Acad. Sci. USA 2015, 112, 5099–5104. [Google Scholar] [CrossRef]
- Yu, T.-Y.; Kao, Y.-W.; Lin, J.-J. Telomeric transcripts stimulate telomere recombination to suppress senescence in cells lacking telomerase. Proc. Natl. Acad. Sci. USA 2014, 111, 3377–3382. [Google Scholar] [CrossRef] [PubMed]
- Höhn, A.; Weber, D.; Jung, T.; Ott, C.; Hugo, M.; Kochlik, B.; Kehm, R.; König, J.; Grune, T.; Castro, J.P. Happily (n)ever after: Aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol. 2017, 11, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Katzen, J.; Wagner, B.D.; Venosa, A.; Kopp, M.; Tomer, Y.; Russo, S.J.; Headen, A.C.; Basil, M.C.; Stark, J.M.; Mulugeta, S.; et al. A SFTPC BRICHOS mutant links epithelial ER stress and spontaneous lung fibrosis. JCI Insight 2019, 4, e126125. [Google Scholar] [CrossRef]
- Nureki, S.-I.; Tomer, Y.; Venosa, A.; Katzen, J.; Russo, S.J.; Jamil, S.; Barrett, M.; Nguyen, V.; Kopp, M.; Mulugeta, S.; et al. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J. Clin. Investig. 2018, 128, 4008–4024. [Google Scholar] [CrossRef] [PubMed]
- Borok, Z.; Horie, M.; Flodby, P.; Wang, H.; Liu, Y.; Ganesh, S.; Firth, A.L.; Minoo, P.; Li, C.; Beers, M.F.; et al. Grp78 Loss in Epithelial Progenitors Reveals an Age-linked Role for Endoplasmic Reticulum Stress in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B.; Futcher, A.B.; Greider, C. Telomeres shorten during ageing of human fibroblasts. Nat. Cell Biol. 1990, 345, 458–460. [Google Scholar] [CrossRef]
- Jiang, J.; Miracco, E.J.; Hong, K.; Eckert, B.; Chan, H.; Cash, D.D.; Min, B.; Zhou, Z.H.; Collins, K.; Feigon, J. The architecture of Tetrahymena telomerase holoenzyme. Nat. Cell Biol. 2013, 496, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Degryse, A.L.; Xu, X.C.; Newman, J.L.; Mitchell, D.B.; Tanjore, H.; Polosukhin, V.V.; Jones, B.R.; McMahon, F.B.; Gleaves, L.A.; Phillips, J.A.; et al. Telomerase deficiency does not alter bleomycin-induced fibrosis in mice. Exp. Lung Res. 2012, 38, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Arish, N.; Petukhov, D.; Wallach-Dayan, S.B. The Role of Telomerase and Telomeres in Interstitial Lung Diseases: From Molecules to Clinical Implications. Int. J. Mol. Sci. 2019, 20, 2996. [Google Scholar] [CrossRef]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef]
- Naikawadi, R.P.; Disayabutr, S.; Mallavia, B.; Donne, M.L.; Green, G.; La, J.L.; Rock, J.R.; Looney, M.R.; Wolters, P.J. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight 2016, 1, e86704. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, J.; Liu, Y.; Zhang, S.; Wang, Y.; Liu, B.; Liu, H.; Li, R.; Lv, C.; Song, X. Regulation of TERRA on telomeric and mitochondrial functions in IPF pathogenesis. BMC Pulm. Med. 2017, 17, 163. [Google Scholar] [CrossRef] [PubMed]
- Disayabutr, S.; Kim, E.K.; Cha, S.-I.; Green, G.; Naikawadi, R.P.; Jones, K.D.; Golden, J.A.; Schroeder, A.; Matthay, M.A.; Kukreja, J.; et al. miR-34 miRNAs Regulate Cellular Senescence in Type II Alveolar Epithelial Cells of Patients with Idiopathic Pulmonary Fibrosis. PLoS ONE 2016, 11, e0158367. [Google Scholar] [CrossRef]
- Denoyelle, C.; Abou-Rjaily, G.; Bezrookove, V.; Verhaegen, M.; Johnson, T.M.; Fullen, D.R.; Pointer, J.N.; Gruber, S.B.; Su, L.D.; Nikiforov, M.A.; et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat. Cell Biol. 2006, 8, 1053–1063. [Google Scholar] [CrossRef]
- Lawson, W.E.; Cheng, D.-S.; Degryse, A.L.; Tanjore, H.; Polosukhin, V.V.; Xu, X.C.; Newcomb, D.C.; Jones, B.R.; Roldan, J.; Lane, K.B.; et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc. Natl. Acad. Sci. USA 2011, 108, 10562–10567. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Trabucco, S.E.; Zhang, H. Oxidative Stress, Mitochondrial Dysfunction and the Mitochondria Theory of Aging. Interdiscip. Top Gerontol. 2014, 39, 86–107. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Lai, Y.-C.; Romero, Y.; Brands, J.; Croix, C.M.S.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Brands, J.; Voltz, L.; Fiedler, K.; Mays, B.; Croix, C.S.; Sembrat, J.; Mallampalli, R.K.; Rojas, M.; Mora, A.L. ATF3 represses PINK1 gene transcription in lung epithelial cells to control mitochondrial homeostasis. Aging Cell 2018, 17, e12720. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.K.; Tiwari, N.; Marudamuthu, A.S.; Puthusseri, B.; Bhandary, Y.P.; Fu, J.; Levin, J.; Idell, S.; Shetty, S. p53 and miR-34a Feedback Promotes Lung Epithelial Injury and Pulmonary Fibrosis. Am. J. Pathol. 2017, 187, 1016–1034. [Google Scholar] [CrossRef]
- Marudamuthu, A.S.; Bhandary, Y.P.; Fan, L.; Radhakrishnan, V.; MacKenzie, B.; Maier, E.; Shetty, S.K.; Nagaraja, M.; Gopu, V.; Tiwari, N.; et al. Caveolin-1–derived peptide limits development of pulmonary fibrosis. Sci. Transl. Med. 2019, 11, eaat2848. [Google Scholar] [CrossRef]
- Cheng, H.-L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef]
- Chu, H.; Jiang, S.; Liu, Q.; Ma, Y.; Zhu, X.; Liang, M.; Shi, X.; Ding, W.; Zhou, X.; Zou, H.; et al. Sirtuin1 Protects against Systemic Sclerosis–related Pulmonary Fibrosis by Decreasing Proinflammatory and Profibrotic Processes. Am. J. Respir. Cell Mol. Biol. 2018, 58, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Mise, N.; Fernandez, I.E.; Eickelberg, O. Resveratrol regulates ECM remodeling in lung fibrosis. Eur. Respir. J. 2014, 44, P3914. [Google Scholar]
- Yao, C.; Carraro, G.; Konda, B.; Guan, X.; Mizuno, T.; Chiba, N.; Kostelny, M.; Kurkciyan, A.; David, G.; McQualter, J.L.; et al. Sin3a regulates epithelial progenitor cell fate during lung development. Development 2017, 144, 2618–2628. [Google Scholar] [CrossRef] [PubMed]
- Dannenberg, J.-H.; David, G.; Zhong, S.; Van Der Torre, J.; Wong, W.H.; Depinho, R.A. mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genes Dev. 2005, 19, 1581–1595. [Google Scholar] [CrossRef] [PubMed]
- Chilosi, M.; Carloni, A.; Rossi, A.; Poletti, V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl. Res. 2013, 162, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, N.F.; Schamberger, A.C.; Nayakanti, S.; Hatz, R.; Behr, J.; Eickelberg, O. Detection and quantification of epithelial progenitor cell populations in human healthy and IPF lungs. Respir. Res. 2016, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Carraro, G.; Mulay, A.; Yao, C.; Mizuno, T.; Konda, B.; Petrov, M.; Lafkas, D.; Arron, J.R.; Hogaboam, C.M.; Chen, P.; et al. Single-Cell Reconstruction of Human Basal Cell Diversity in Normal and Idiopathic Pulmonary Fibrosis Lungs. Am. J. Respir. Crit. Care Med. 2020, 202, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- DePianto, D.J.; Heiden, J.A.V.; Morshead, K.B.; Sun, K.-H.; Modrusan, Z.; Teng, G.; Wolters, P.J.; Arron, J.R. Molecular mapping of interstitial lung disease reveals a phenotypically distinct senescent basal epithelial cell population. JCI Insight 2021, 6, e143626. [Google Scholar] [CrossRef]
- Jaeger, B.; Schupp, J.C.; Plappert, L.; Terwolbeck, O.; Kayser, G.; Engelhard, P.; Adams, T.S.; Zweigerdt, R.; Kempf, H.; Lienenklaus, S.; et al. Airway Basal Cells show a dedifferentiated KRT17highPhenotype and promote Fibrosis in Idiopathic Pulmonary Fibrosis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; Deiuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef]
- Valenzi, E.; Tabib, T.; Papazoglou, A.; Sembrat, J.; Bittar, H.E.T.; Rojas, M.; Lafyatis, R. Disparate Interferon Signaling and Shared Aberrant Basaloid Cells in Single-Cell Profiling of Idiopathic Pulmonary Fibrosis and Systemic Sclerosis-Associated Interstitial Lung Disease. Front. Immunol. 2021, 12, 595811. [Google Scholar] [CrossRef] [PubMed]
- Strunz, M.; Simon, L.M.; Ansari, M.; Kathiriya, J.J.; Angelidis, I.; Mayr, C.H.; Tsidiridis, G.; Lange, M.; Mattner, L.F.; Yee, M.; et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 2020, 11, 1–20. [Google Scholar] [CrossRef]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.-I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.; Hodwin, B.; Ramanujam, D.P.; Engelhardt, S.; Sarikas, A. Essential Role for Premature Senescence of Myofibroblasts in Myocardial Fibrosis. J. Am. Coll. Cardiol. 2016, 67, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef]
- Hohmann, M.S.; Habiel, D.M.; Coelho, A.L.; Verri, W.; Hogaboam, C.M. Quercetin Enhances Ligand-induced Apoptosis in Senescent Idiopathic Pulmonary Fibrosis Fibroblasts and Reduces Lung Fibrosis In Vivo. Am. J. Respir. Cell Mol. Biol. 2019, 60, 28–40. [Google Scholar] [CrossRef]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of Persistent Fibrosis in Aging by Targeting Nox4-Nrf2 Redox Imbalance. Sci. Transl. Med. 2014, 6, 231ra47. [Google Scholar] [CrossRef]
- Álvarez, D.; Cárdenes, N.; Sellarés, J.; Bueno, M.; Corey, C.; Hanumanthu, V.S.; Peng, Y.; D’Cunha, H.; Sembrat, J.; Nouraie, M.; et al. IPF lung fibroblasts have a senescent phenotype. Am. J. Physiol. Cell. Mol. Physiol. 2017, 313, L1164–L1173. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.; Montaño, M.; García-Alvarez, J.; Ruiz, V.; Uhal, B.D.; Selman, M.; Pardo, A. Fibroblasts from Idiopathic Pulmonary Fibrosis and Normal Lungs Differ in Growth Rate, Apoptosis, and Tissue Inhibitor of Metalloproteinases Expression. Am. J. Respir. Cell Mol. Biol. 2001, 24, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Cruz, T.; Jia, M.; Tabib, T.; Sembrat, J.; Liu, J.; Bondonese, A.; Kavanagh, J.; Nayra, C.; Bruno, T.; Mora, A.; et al. SASP from lung senescent fibroblasts induces immunesenescence and fibrosis. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Zank, D.C.; Bueno, M.; Mora, A.L.; Rojas, M. Idiopathic Pulmonary Fibrosis: Aging, Mitochondrial Dysfunction, and Cellular Bioenergetics. Front. Med. 2018, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef]
- Mora, A.L.; Bueno, M.; Rojas, M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J. Clin. Investig. 2017, 127, 405–414. [Google Scholar] [CrossRef]
- Xie, N.; Tan, Z.; Banerjee, S.; Cui, H.; Ge, J.; Liu, R.-M.; Bernard, K.; Thannickal, V.J.; Liu, G. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1462–1474. [Google Scholar] [CrossRef] [PubMed]
- Caporarello, N.; A Meridew, J.; Jones, D.L.; Tan, Q.; Haak, A.J.; Choi, K.M.; Manlove, L.J.; Prakash, Y.S.; Tschumperlin, D.J.; Ligresti, G. PGC1α repression in IPF fibroblasts drives a pathologic metabolic, secretory and fibrogenic state. Thorax 2019, 74, 749–760. [Google Scholar] [CrossRef]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Müller, F.L.; Guo, M.; Cooper, M.; Kotton, D.N.; Fabian, A.J.; Walkley, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nat. Cell Biol. 2011, 470, 359–365. [Google Scholar] [CrossRef]
- Bernard, K.; Logsdon, N.J.; Miguel, V.; Benavides, G.A.; Zhang, J.; Carter, A.B.; Darley-Usmar, V.; Thannickal, V.J. NADPH Oxidase 4 (Nox4) Suppresses Mitochondrial Biogenesis and Bioenergetics in Lung Fibroblasts via a Nuclear Factor Erythroid-derived 2-like 2 (Nrf2)-dependent Pathway. J. Biol. Chem. 2017, 292, 3029–3038. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Araya, J.; Minagawa, S.; Hara, H.; Saito, N.; Kadota, T.; Sato, N.; Yoshida, M.; Tsubouchi, K.; Kurita, Y.; et al. Involvement of PARK2-Mediated Mitophagy in Idiopathic Pulmonary Fibrosis Pathogenesis. J. Immunol. 2016, 197, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Sosulski, M.L.; Gongora, R.; Danchuk, S.; Dong, C.; Luo, F.; Sanchez, C.G. Deregulation of selective autophagy during aging and pulmonary fibrosis: The role of TGF β1. Aging Cell 2015, 14, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Waghray, M.; Cui, Z.; Horowitz, J.C.; Subramanian, I.M.; Martinez, F.J.; Toews, G.B.; Thannickal, V.J. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J. 2005, 19, 854–856. [Google Scholar] [CrossRef]
- Lerner, C.; Bitto, A.; Pulliam, D.; Nacarelli, T.; Konigsberg, M.; Van Remmen, H.; Torres, C.; Sell, C. Reduced mammalian target of rapamycin activity facilitates mitochondrial retrograde signaling and increases life span in normal human fibroblasts. Aging Cell 2013, 12, 966–977. [Google Scholar] [CrossRef]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial Reactive Oxygen Species Regulate Transforming Growth Factor-β Signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef]
- Amara, N.; Goven, D.; Prost, F.; Muloway, R.; Crestani, B.; Boczkowski, J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGF 1-induced fibroblast differentiation into myofibroblasts. Thorax 2010, 65, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Rajawat, Y.S.; Hilioti, Z.; Bossis, I. Aging: Central role for autophagy and the lysosomal degradative system. Ageing Res. Rev. 2009, 8, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Kuwano, K.; Araya, J.; Hara, H.; Minagawa, S.; Takasaka, N.; Ito, S.; Kobayashi, K.; Nakayama, K. Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). Respir. Investig. 2016, 54, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Araya, J.; Kojima, J.; Takasaka, N.; Ito, S.; Fujii, S.; Hara, H.; Yanagisawa, H.; Kobayashi, K.; Tsurushige, C.; Kawaishi, M.; et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2013, 304, L56–L69. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Wang, X.; Yu, H.; Wang, C.; Liu, Y.; Zhao, R.; Zhang, G. Beclin 1, LC3, and p62 expression in paraquat-induced pulmonary fibrosis. Hum. Exp. Toxicol. 2019, 38, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.S.; Lin, L.; Geyer, A.; Haspel, J.A.; An, C.H.; Cao, J.; Rosas, I.O.; Morse, D. Autophagy in Idiopathic Pulmonary Fibrosis. PLoS ONE 2012, 7, e41394. [Google Scholar] [CrossRef]
- Ricci, A.; Cherubini, E.; Scozzi, D.; Pietrangeli, V.; Tabbì, L.; Raffa, S.; Leone, L.; Visco, V.; Torrisi, M.R.; Bruno, P.; et al. Decreased expression of autophagic beclin 1 protein in idiopathic pulmonary fibrosis fibroblasts. J. Cell. Physiol. 2013, 228, 1516–1524. [Google Scholar] [CrossRef]
- Romero, Y.; Bueno, M.; Ramirez, R.; Álvarez, D.; Sembrat, J.C.; Goncharova, E.A.; Rojas, M.; Selman, M.; Mora, A.L.; Pardo, A. mTORC1 activation decreases autophagy in aging and idiopathic pulmonary fibrosis and contributes to apoptosis resistance in IPF fibroblasts. Aging Cell 2016, 15, 1103–1112. [Google Scholar] [CrossRef]
- Nho, R.S.; Hergert, P. IPF Fibroblasts Are Desensitized to Type I Collagen Matrix-Induced Cell Death by Suppressing Low Autophagy via Aberrant Akt/mTOR Kinases. PLoS ONE 2014, 9, e94616. [Google Scholar] [CrossRef]
- Surolia, R.; Li, F.J.; Wang, Z.; Li, H.; Dsouza, K.; Thomas, V.; Mirov, S.; Pérez-Sala, D.; Athar, M.; Thannickal, V.J.; et al. Vimentin intermediate filament assembly regulates fibroblast invasion in fibrogenic lung injury. JCI Insight 2019, 4, e123253. [Google Scholar] [CrossRef]
- Cha, S.-I.; Groshong, S.D.; Frankel, S.K.; Edelman, B.L.; Cosgrove, G.P.; Terry-Powers, J.L.; Remigio, L.K.; Curran-Everett, U.; Brown, K.K.; Cool, C.D.; et al. Compartmentalized Expression of c-FLIP in Lung Tissues of Patients with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 42, 140–148. [Google Scholar] [CrossRef]
- Yanai, H.; Shteinberg, A.; Porat, Z.; Budovsky, A.; Braiman, A.; Zeische, R.; Fraifeld, V.E. Cellular senescence-like features of lung fibroblasts derived from idiopathic pulmonary fibrosis patients. Aging 2015, 7, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-T.; Akhter, H.; Jiang, C.; MacEwen, M.; Ding, Q.; Antony, V.; Thannickal, V.J.; Liu, R.-M. Plasminogen activator inhibitor 1, fibroblast apoptosis resistance, and aging-related susceptibility to lung fibrosis. Exp. Gerontol. 2015, 61, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Hernandez, G.; Ballester, B.; Morell, A.; Roger, I.; Montero, P.; Escrivá, J.; Lloris, J.M.; Molina-Molina, M.; Morcillo, E.; et al. The JAK2 pathway is activated in idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 24. [Google Scholar] [CrossRef]
- Rehan, M.; Kurundkar, D.; Kurundkar, A.R.; Logsdon, N.J.; Smith, S.R.; Chanda, D.; Bernard, K.; Sanders, Y.Y.; Deshane, J.S.; Dsouza, K.G.; et al. Restoration of SIRT3 gene expression by airway delivery resolves age-associated persistent lung fibrosis in mice. Nat. Aging 2021, 1, 205–217. [Google Scholar] [CrossRef]
- Xia, H.; Khalil, W.; Kahm, J.; Jessurun, J.; Kleidon, J.; Henke, C.A. Pathologic Caveolin-1 Regulation of PTEN in Idiopathic Pulmonary Fibrosis. Am. J. Pathol. 2010, 176, 2626–2637. [Google Scholar] [CrossRef] [PubMed]
- Nho, R.S.; Peterson, M.; Hergert, P.; Henke, C.A. FoxO3a (Forkhead Box O3a) Deficiency Protects Idiopathic Pulmonary Fibrosis (IPF) Fibroblasts from Type I Polymerized Collagen Matrix-Induced Apoptosis via Caveolin-1 (cav-1) and Fas. PLoS ONE 2013, 8, e61017. [Google Scholar] [CrossRef]
- Im, J.; Kim, K.; Hergert, P.; Nho, R.S. Idiopathic pulmonary fibrosis fibroblasts become resistant to Fas ligand-dependent apoptosis via the alteration of decoy receptor 3. J. Pathol. 2016, 240, 25–37. [Google Scholar] [CrossRef]
- Xia, H.; Bodempudi, V.; Benyumov, A.; Hergert, P.; Tank, D.; Herrera, J.; Braziunas, J.; Larsson, O.; Parker, M.; Rossi, D.; et al. Identification of a Cell-of-Origin for Fibroblasts Comprising the Fibrotic Reticulum in Idiopathic Pulmonary Fibrosis. Am. J. Pathol. 2014, 184, 1369–1383. [Google Scholar] [CrossRef]
- Habiel, D.M.; Hohmann, M.S.; Espindola, M.S.; Coelho, A.L.; Jones, I.; Jones, H.; Carnibella, R.; Pinar, I.; Werdiger, F.; Hogaboam, C.M. DNA-PKcs modulates progenitor cell proliferation and fibroblast senescence in idiopathic pulmonary fibrosis. BMC Pulm. Med. 2019, 19, 1–16. [Google Scholar] [CrossRef]
- Yang, L.; Xia, H.; Smith, K.A.; Gilbertsen, A.; Beisang, D.; Kuo, J.; Bitterman, P.B.; Henke, C.A. A CD44/Brg1 nuclear complex confers mesenchymal progenitor cells with enhanced fibrogenicity in idiopathic pulmonary fibrosis. JCI Insight 2021, 6, 144652. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Fleming, T.; Terjung, S.; Gorzelanny, C.; Gebhardt, C.; Agrawal, R.; Mall, M.A.; Ranzinger, J.; Zeier, M.; Madhusudhan, T.; et al. Homeostatic nuclear RAGE–ATM interaction is essential for efficient DNA repair. Nucleic Acids Res. 2017, 45, 10595–10613. [Google Scholar] [CrossRef]
- Habiel, D.M.; Camelo, A.; Espindola, M.; Burwell, T.; Hanna, R.; Miranda, E.; Carruthers, A.; Bell, M.; Coelho, A.L.; Liu, H.; et al. Divergent roles for Clusterin in Lung Injury and Repair. Sci. Rep. 2017, 7, 15444. [Google Scholar] [CrossRef]
- Xia, H.; Gilbertsen, A.; Herrera, J.; Racila, E.; Smith, K.; Peterson, M.; Griffin, T.; Benyumov, A.; Yang, L.; Bitterman, P.; et al. Calcium-binding protein S100A4 confers mesenchymal progenitor cell fibrogenicity in idiopathic pulmonary fibrosis. J. Clin. Investig. 2017, 127, 2586–2597. [Google Scholar] [CrossRef]
- Yang, L.; Herrera, J.; Gilbertsen, A.; Xia, H.; Smith, K.; Benyumov, A.; Bitterman, P.; Henke, C.A. IL-8 mediates idiopathic pulmonary fibrosis mesenchymal progenitor cell fibrogenicity. Am. J. Physiol. Cell. Mol. Physiol. 2018, 314, L127–L136. [Google Scholar] [CrossRef]
- Hohmann, M.S.; Habiel, D.M.; Espindola, M.S.; Huang, G.; Jones, I.; Narayanan, R.; Coelho, A.L.; Oldham, J.M.; Noth, I.; Ma, S.-F.; et al. Antibody-mediated depletion of CCR10+ EphA3+ cells ameliorates fibrosis in IPF. JCI Insight 2021, 141061. [Google Scholar] [CrossRef]
- Vicente, R.; Mausset-Bonnefont, A.-L.; Jorgensen, C.; Louis-Plence, P.; Brondello, J.-M. Cellular senescence impact on immune cell fate and function. Aging Cell 2016, 15, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.A.; Chotirmall, S.H. The Impact of Immunosenescence on Pulmonary Disease. Mediat. Inflamm. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shenderov, K.; Collins, S.L.; Powell, J.D.; Horton, M.R. Immune dysregulation as a driver of idiopathic pulmonary fibrosis. J. Clin. Investig. 2021, 131, e143226. [Google Scholar] [CrossRef] [PubMed]
- Faner, R.; Rojas, M.; MacNee, W.; Agustí, A. Abnormal Lung Aging in Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 306–313. [Google Scholar] [CrossRef]
- Linton, P.-J.; Thoman, M.L. Immunosenescence in monocytes, macrophages, and dendritic cells: Lessons learned from the lung and heart. Immunol. Lett. 2014, 162, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247. [Google Scholar] [CrossRef] [PubMed]
- Desai, O.; Winkler, J.; Minasyan, M.; Herzog, E.L. The Role of Immune and Inflammatory Cells in Idiopathic Pulmonary Fibrosis. Front. Med. 2018, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Aw, D.; Silva, A.B.; Palmer, D.B. Immunosenescence: Emerging challenges for an ageing population. Immunology 2007, 120, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Feehan, J.; Tripodi, N.; Apostolopoulos, V. The twilight of the immune system: The impact of immunosenescence in aging. Maturitas 2021, 147, 7–13. [Google Scholar] [CrossRef]
- Nikolich-Žugich, J. The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 2018, 19, 10–19. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Hirokawa, K.; Cohen, A.A.; Witkowski, J.M. Immunosenescence is both functional/adaptive and dysfunctional/maladaptive. Semin. Immunopathol. 2020, 42, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Ventura, M.T.; Casciaro, M.; Gangemi, S.; Buquicchio, R. Immunosenescence in aging: Between immune cells depletion and cytokines up-regulation. Clin. Mol. Allergy 2017, 15, 1–8. [Google Scholar] [CrossRef]
- Kaszubowska, L. Telomere shortening and ageing of the immune system. J Physiol Pharmacol 2008, 59, 169–186. [Google Scholar] [PubMed]
- Zhou, D.; Borsa, M.; Simon, A.K. Hallmarks and detection techniques of cellular senescence and cellular ageing in immune cells. Aging Cell 2021, 20, e13316. [Google Scholar] [CrossRef]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kale, A.; Sharma, A.; Stolzing, A.; Desprez, P.-Y.; Campisi, J. Role of immune cells in the removal of deleterious senescent cells. Immun. Ageing 2020, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.; Arjona, A.; Sapey, E.; Bai, F.; Fikrig, E.; Montgomery, R.; Lord, J.M.; Shaw, A.C. Human innate immunosenescence: Causes and consequences for immunity in old age. Trends Immunol. 2009, 30, 325–333. [Google Scholar] [CrossRef]
- O’Dwyer, D.N.; Ashley, S.L.; Moore, B.B. Influences of innate immunity, autophagy, and fibroblast activation in the pathogenesis of lung fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2016, 311, L590–L601. [Google Scholar] [CrossRef]
- Marchal-Sommé, J.; Uzunhan, Y.; Marchand-Adam, S.; Kambouchner, M.; Valeyre, M.; Crestani, B.; Soler, P. Dendritic Cells Accumulate in Human Fibrotic Interstitial Lung Disease. Am. J. Respir. Crit. Care Med. 2007, 176, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Tsoumakidou, M.; Karagiannis, K.; Bouloukaki, I.; Zakynthinos, S.; Tzanakis, N.; Siafakas, N. Increased Bronchoalveolar Lavage Fluid CD1c Expressing Dendritic Cells in Idiopathic Pulmonary Fibrosis. Respiration 2009, 78, 446–452. [Google Scholar] [CrossRef]
- Agrawal, A.; Sridharan, A.; Prakash, S.; Agrawal, H. Dendritic cells and aging: Consequences for autoimmunity. Expert Rev. Clin. Immunol. 2012, 8, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Valera, I. Plasmacytoid dendritic cells contribute to pro-inflammatory and pro-fibrotic milieu in lung fibrosis. J. Immunol. 2019, 202, 176–182. [Google Scholar]
- Trujillo, G.; Meneghin, A.; Flaherty, K.R.; Sholl, L.M.; Myers, J.L.; Kazerooni, E.A.; Gross, B.H.; Oak, S.R.; Coelho, A.L.; Evanoff, H.; et al. TLR9 Differentiates Rapidly from Slowly Progressing Forms of Idiopathic Pulmonary Fibrosis. Sci. Transl. Med. 2010, 2, 57ra82. [Google Scholar] [CrossRef] [PubMed]
- Yamanouchi, H.; Ohtsuki, Y.; Fujita, J.; Bandoh, S.; Yoshinouchi, T.; Ishida, T. The distribution and number of Leu-7 (CD57) positive cells in lung tissue from patients with pulmonary fibrosis. Acta Med. Okayama 2002, 56, 83–89. [Google Scholar]
- Listì, F.; Candore, G.; Modica, M.A.; Russo, M.; Di Lorenzo, G.; Esposito-Pellitteri, M.; Colonna-Romano, G.; Aquino, A.; Bulati, M.; Lio, D.; et al. A Study of Serum Immunoglobulin Levels in Elderly Persons That Provides New Insights into B Cell Immunosenescence. Ann. N. Y. Acad. Sci. 2006, 1089, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, S. T Cells in Fibrosis and Fibrotic Diseases. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.K.; Delaney, C.; Toubai, T.; Ghosh, A.; Reddy, P.; Banerjee, R.; Yung, R. Aging is associated with increased regulatory T-cell function. Aging Cell 2014, 13, 441–448. [Google Scholar] [CrossRef]
- Song, L.; Weng, D.; Liu, F.; Chen, Y.; Li, C.; Dong, L.; Tang, W.; Chen, J. Tregs Promote the Differentiation of Th17 Cells in Silica-Induced Lung Fibrosis in Mice. PLoS ONE 2012, 7, e37286. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, D.; Wang, L.; Wang, S.; Roden, A.C.; Zhao, H.; Li, X.; Prakash, Y.S.; Matteson, E.L.; Tschumperlin, D.J.; et al. Profibrotic effect of IL-17A and elevated IL-17RA in idiopathic pulmonary fibrosis and rheumatoid arthritis-associated lung disease support a direct role for IL-17A/IL-17RA in human fibrotic interstitial lung disease. Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L487–L497. [Google Scholar] [CrossRef] [PubMed]
- van Duin, D.; Mohanty, S.; Thomas, V.; Ginter, S.; Montgomery, R.R.; Fikrig, E.; Allore, H.G.; Medzhitov, R.; Shaw, A.C. Age-Associated Defect in Human TLR-1/2 Function. J. Immunol. 2007, 178, 970–975. [Google Scholar] [CrossRef]
- van Beek, A.A.; Bossche, J.V.D.; Mastroberardino, P.G.; de Winther, M.P.; Leenen, P.J. Metabolic Alterations in Aging Macrophages: Ingredients for Inflammaging? Trends Immunol. 2019, 40, 113–127. [Google Scholar] [CrossRef]
- Greiffo, F.; Fernandez, I.; Frankenberger, M.; Behr, J.; Eickelberg, O. Circulating monocytes from interstitial lung disease patients show an activated phenotype. Eur. Respir. J. 2016, 48, pa3894. [Google Scholar] [CrossRef]
- Venosa, A. Senescence in Pulmonary Fibrosis: Between Aging and Exposure. Front. Med. 2020, 7. [Google Scholar] [CrossRef]
- Scott, M.K.D.; Quinn, K.; Li, Q.; Carroll, R.; Warsinske, H.; Vallania, F.; Chen, S.; A Carns, M.; Aren, K.; Sun, J.; et al. Increased monocyte count as a cellular biomarker for poor outcomes in fibrotic diseases: A retrospective, multicentre cohort study. Lancet Respir. Med. 2019, 7, 497–508. [Google Scholar] [CrossRef]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.; et al. Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging 2016, 8, 1294–1315. [Google Scholar] [CrossRef]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.I.; et al. p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging 2017, 9, 1867–1884. [Google Scholar] [CrossRef] [PubMed]
- Larson-Casey, J.L.; Deshane, J.S.; Ryan, A.J.; Thannickal, V.J.; Carter, A.B. Macrophage Akt1 Kinase-Mediated Mitophagy Modulates Apoptosis Resistance and Pulmonary Fibrosis. Immunology 2016, 44, 582–596. [Google Scholar] [CrossRef] [PubMed]
- Tsitoura, E.; Vasarmidi, E.; Bibaki, E.; Trachalaki, A.; Koutoulaki, C.; Papastratigakis, G.; Papadogiorgaki, S.; Chalepakis, G.; Tzanakis, N.; Antoniou, K.M. Accumulation of damaged mitochondria in alveolar macrophages with reduced OXPHOS related gene expression in IPF. Respir. Res. 2019, 20, 264. [Google Scholar] [CrossRef]
- Vasileiou, P.V.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.I.; Chronopoulos, E.; Passias, P.-G.; Kouloukoussa, M.; Gorgoulis, V.G.; Havaki, S. Mitochondrial Homeostasis and Cellular Senescence. Cells 2019, 8, 686. [Google Scholar] [CrossRef] [PubMed]
- Stout-Delgado, H.W.; Cho, S.J.; Chu, S.G.; Mitzel, D.N.; Villalba, J.; El-Chemaly, S.; Ryter, S.W.; Choi, A.M.K.; Rosas, I.O. Age-Dependent Susceptibility to Pulmonary Fibrosis Is Associated with NLRP3 Inflammasome Activation. Am. J. Respir. Cell Mol. Biol. 2016, 55, 252–263. [Google Scholar] [CrossRef]
- Chou, J.P.; Effros, R.B. T Cell Replicative Senescence in Human Aging. Curr. Pharm. Des. 2013, 19, 1680–1698. [Google Scholar] [CrossRef]
- Xu, W.; Larbi, A. Markers of T Cell Senescence in Humans. Int. J. Mol. Sci. 2017, 18, 1742. [Google Scholar] [CrossRef]
- Huff, W.X.; Kwon, J.H.; Henriquez, M.; Fetcko, K.; Dey, M. The Evolving Role of CD8+CD28- Immunosenescent T Cells in Cancer Immunology. Int. J. Mol. Sci. 2019, 20, 2810. [Google Scholar] [CrossRef]
- Gilani, S.R.; Vuga, L.J.; Lindell, K.O.; Gibson, K.F.; Xue, J.; Kaminski, N.; Valentine, V.G.; Lindsay, E.K.; George, M.P.; Steele, C.; et al. CD28 Down-Regulation on Circulating CD4 T-Cells Is Associated with Poor Prognoses of Patients with Idiopathic Pulmonary Fibrosis. PLoS ONE 2010, 5, e8959. [Google Scholar] [CrossRef]
- Habiel, D.M.; Espindola, M.S.; Kitson, C.; Azzara, A.V.; Coelho, A.L.; Stripp, B.; Hogaboam, C.M. Characterization of CD28null T cells in idiopathic pulmonary fibrosis. Mucosal Immunol. 2018, 12, 212–222. [Google Scholar] [CrossRef]
- Ni, K.; Liu, M.; Zheng, J.; Wen, L.; Chen, Q.; Xiang, Z.; Lam, K.-T.; Liu, Y.; Chan, G.C.-F.; Lau, Y.-L.; et al. PD-1/PD-L1 Pathway Mediates the Alleviation of Pulmonary Fibrosis by Human Mesenchymal Stem Cells in Humanized Mice. Am. J. Respir. Cell Mol. Biol. 2018, 58, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Liu, X.; Liang, J.; Habiel, D.M.; Vrishika, K.; Coelho, A.L.; Deng, N.; Xie, T.; Wang, Y.; Liu, N.; et al. PD-L1 on invasive fibroblasts drives fibrosis in a humanized model of idiopathic pulmonary fibrosis. JCI Insight 2019, 4, e125326. [Google Scholar] [CrossRef] [PubMed]
- Cronkhite, J.T.; Xing, C.; Raghu, G.; Chin, K.M.; Torres, F.; Rosenblatt, R.L.; Garcia, C.K. Telomere Shortening in Familial and Sporadic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Galati, D.; De Martino, M.; Trotta, A.M.; Rea, G.; Bruzzese, D.; Cicchitto, G.; Stanziola, A.A.; Napolitano, M.; Sanduzzi, A.; Bocchino, M. Peripheral depletion of NK cells and imbalance of the Treg/Th17 axis in idiopathic pulmonary fibrosis patients. Cytokine 2014, 66, 119–126. [Google Scholar] [CrossRef]
- Papanikolaou, I.C.; Boki, K.A.; Giamarellos-Bourboulis, E.J.; Kotsaki, A.; Kagouridis, K.; Karagiannidis, N.; Polychronopoulos, V.S. Innate immunity alterations in idiopathic interstitial pneumonias and rheumatoid arthritis-associated interstitial lung diseases. Immunol. Lett. 2015, 163, 179–186. [Google Scholar] [CrossRef]
- Esposito, I.; Perna, F.; Ponticiello, A.; Perrella, M.; Gilli, M.; Sanduzzi, A. Natural Killer Cells in Bal and Peripheral Blood of Patients with Idiopathic Pulmonary Fibrosis (IPF). Int. J. Immunopathol. Pharmacol. 2005, 18, 541–545. [Google Scholar] [CrossRef]
- Song, P.; An, J.; Zou, M.-H. Immune Clearance of Senescent Cells to Combat Ageing and Chronic Diseases. Cells 2020, 9, 671. [Google Scholar] [CrossRef]
- Schneider, J.L.; Rowe, J.H.; Garcia-De-Alba, C.; Kim, C.F.; Sharpe, A.H.; Haigis, M.C. The aging lung: Physiology, disease, and immunity. Cell 2021, 184, 1990–2019. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Discher, D.E.; Péault, B.M.; Phinney, D.G.; Hare, J.M.; Caplan, A.I. Mesenchymal stem cell perspective: Cell biology to clinical progress. NPJ Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef]
- Li, X.; An, G.; Wang, Y.; Liang, D.; Zhu, Z.; Tian, L. Targeted migration of bone marrow mesenchymal stem cells inhibits silica-induced pulmonary fibrosis in rats. Stem Cell Res. Ther. 2018, 9, 335. [Google Scholar] [CrossRef]
- Cárdenes, N.; Álvarez, D.; Sellarés, J.; Peng, Y.; Corey, C.; Wecht, S.; Nouraie, S.M.; Shanker, S.; Sembrat, J.; Bueno, M.; et al. Senescence of bone marrow-derived mesenchymal stem cells from patients with idiopathic pulmonary fibrosis. Stem Cell Res. Ther. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Erratum: Corrigendum: Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1384. [Google Scholar] [CrossRef]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; Van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nat. Cell Biol. 2018, 562, 578–582. [Google Scholar] [CrossRef]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Sen, J.M.; Gorospe, M.; et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- Pan, J.; Li, D.; Xu, Y.; Zhang, J.; Wang, Y.; Chen, M.; Lin, S.; Huang, L.; Chung, E.J.; Citrin, D.E.; et al. Inhibition of Bcl-2/xl With ABT-263 Selectively Kills Senescent Type II Pneumocytes and Reverses Persistent Pulmonary Fibrosis Induced by Ionizing Radiation in Mice. Int. J. Radiat. Oncol. 2017, 99, 353–361. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; Lebrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Animal Models | Spontaneous Fibrosis | Induced Fibrosis | References |
|---|---|---|---|
| Tert−/− | No | 4th-generation inbred, reduced required dose of bleomycin to induce fibrosis | [51] |
| Terc−/− | No | 3rd-generation enhanced fibrosis upon liposaccharide and bleomycin | [52] |
| SftpcCerER; Tert flox/flox | No | Bleomycin induced enhanced fibrosis | [26] |
| SftpcCerER; Trf1flox/flox | Yes | [51,53] | |
| SftpcCerER; Trf2flox/flox | No | Increased susceptibility to bleomycin | [54] |
| SftpcCerER; Grp78flox/flox | Yes | [55] | |
| SftpcCerER; Sin3aflox/flox | Yes | [25] | |
| mSFTPC.rtTA; SFTPCL188Q | No | Bleomycin induced exaggerated lung fibrosis | [56] |
| SftpcCerER; SFTPCI73T | Yes | [57] | |
| SftpcCerER; SFTPCC121G | Yes | [58] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parimon, T.; Hohmann, M.S.; Yao, C. Cellular Senescence: Pathogenic Mechanisms in Lung Fibrosis. Int. J. Mol. Sci. 2021, 22, 6214. https://doi.org/10.3390/ijms22126214
Parimon T, Hohmann MS, Yao C. Cellular Senescence: Pathogenic Mechanisms in Lung Fibrosis. International Journal of Molecular Sciences. 2021; 22(12):6214. https://doi.org/10.3390/ijms22126214
Chicago/Turabian StyleParimon, Tanyalak, Miriam S. Hohmann, and Changfu Yao. 2021. "Cellular Senescence: Pathogenic Mechanisms in Lung Fibrosis" International Journal of Molecular Sciences 22, no. 12: 6214. https://doi.org/10.3390/ijms22126214
APA StyleParimon, T., Hohmann, M. S., & Yao, C. (2021). Cellular Senescence: Pathogenic Mechanisms in Lung Fibrosis. International Journal of Molecular Sciences, 22(12), 6214. https://doi.org/10.3390/ijms22126214