
Bilirubin: A Promising Therapy for Parkinson’s Disease



Abstract
1. Introduction
2. Parkinson’s Disease: From Pathogenesis to Management
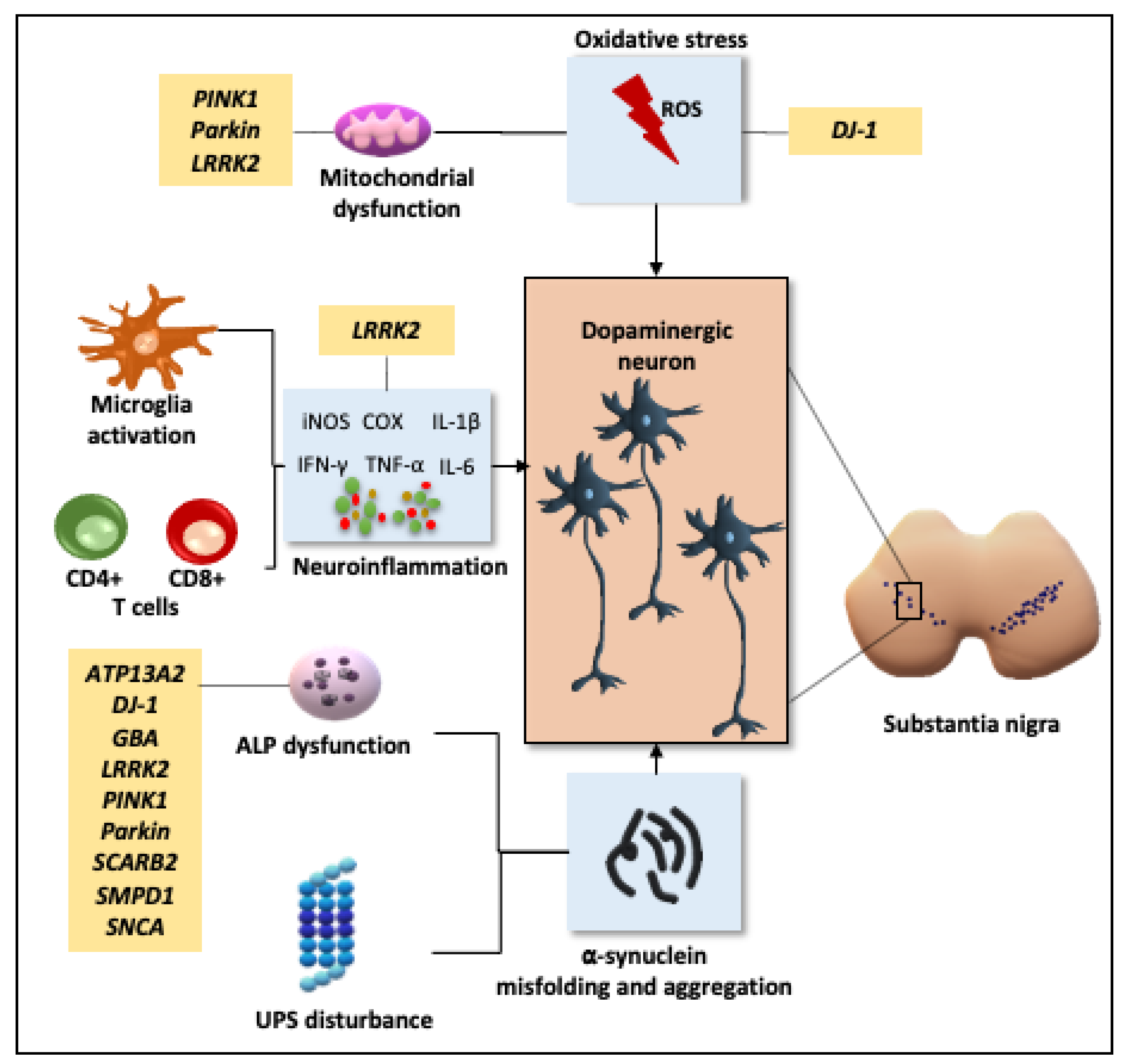
2.1. Pathogenesis
2.1.1. Oxidative Stress and Mitochondrial Dysfunction
2.1.2. Neuroinflammation
2.1.3. Disruption of Cellular Proteostasis
2.1.4. Genetic Influence
2.2. Challenges in the Management of PD
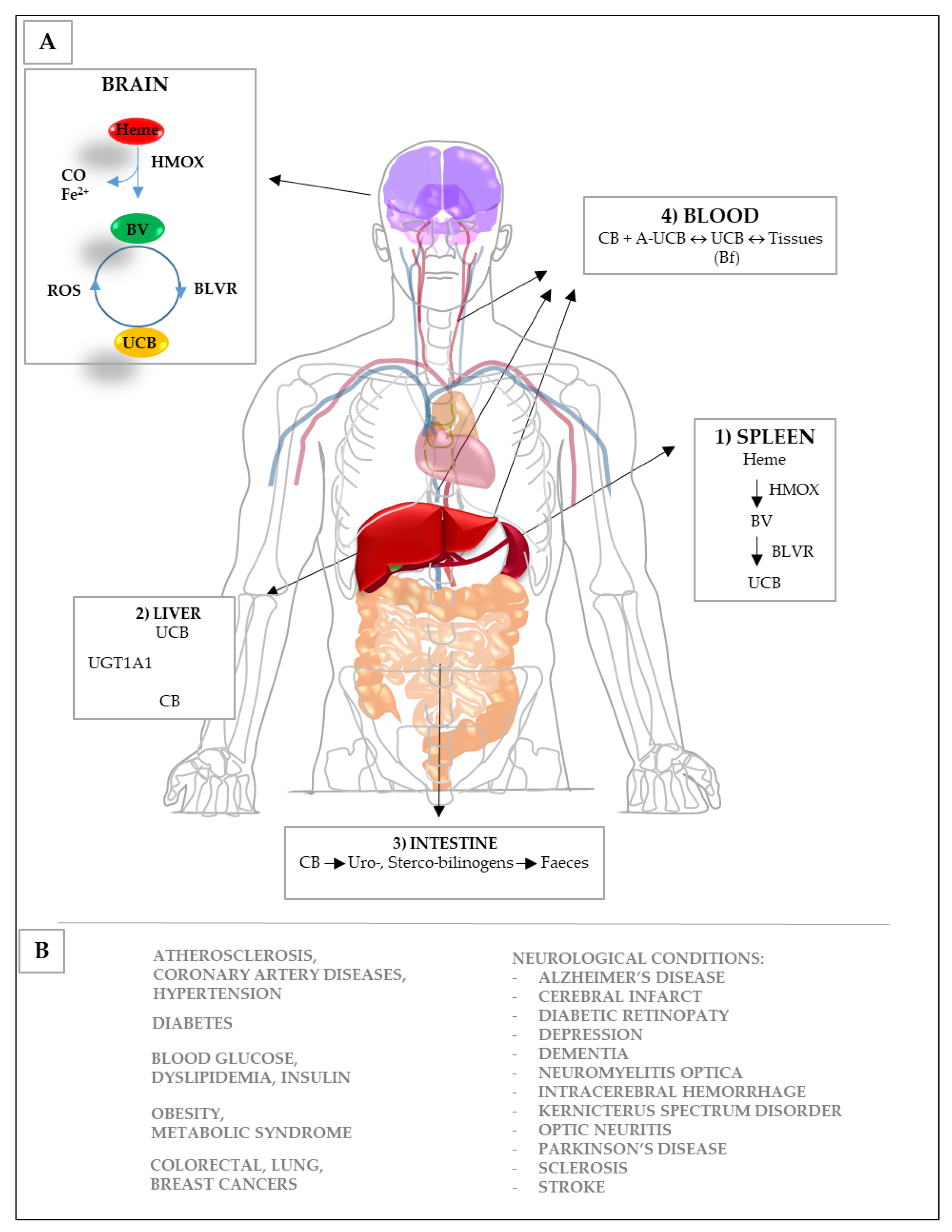
3. Bilirubin and the Yellow Players in Neurological Diseases
3.1. Potential Mechanisms of Action
3.1.1. Oxidation
3.1.2. Inflammation

{kind=link}
{kind=link}
{kind=link}
| Pathological Mechanism in PD | YPs (Protective Effect) | Ref. |
|---|---|---|
| REDOX | UCB (↓) Heme (↓) BV (↓) | [95,96,97,103] [106,107,108] [104] |
| Anti-oxidant enzymes | UCB (↑) Heme (↑) BLVRA (↑) | [104,105] [106,107] [14,105] |
| Carbonylation and lipid peroxidation | Membrane protection by scavenging lipophilic radicals (↑) BV (↓) | [97,98,99] [105] |
| DNA damage | BV (↓) | [108,133] |
| Mitochondrial disfunction | Heme (↓) Heme: cofactor for the mitochondrial electron transport chain (complexes II, III, IV) | [106,107] [118] |
| PINK1/DJ1; LRRK2; SNCA; PARK2 | No direct experimental data are yet available. Further, devoted studies are needed. | |
| INFLAMMATION | BV (↓) BLVRA, UCB, CO (↓) HMOX1 (↓) | [111] [96,103,111,125,126,127,128,129,130,131,132] [134,135] |
| Microglia and astrocyte activation | No direct experimental data are yet available. Further, devoted studies are needed. | |
| α-synuclein | ||
| iNOS and COX | BLVRA, UCB, CO (↓) | [96,103,125,126,127,128,129,130,131,132] |
| TNFα | BLVRA, UCB, CO (↓) | [96,103,125,126,127,128,129,130,131,132] |
| IL6 | BLVRA, UCB, CO (↓) | [96,103,125,126,127,128,129,130,131,132] |
| IL1β; IFNγ ; Il2; IL10; CXCLY2 | No direct experimental data are yet available. Further, devoted studies are needed. | |
| CD8+ and CD4+ T cells | BLVRA (↓) BLVRA, UCB, CO (↓) | [14,105] [96,103,125,126,127,128,129,130,131,132] |
| LRRK2; SNCA | No direct experimental data are yet available. Further, devoted studies are needed. | |
| PROTEIN DEGRADATION | ||
| UPS | No direct experimental data are yet available. Further, devoted studies are needed. | |
| Autophagy | BLVRA (↓) | [14,105] |
| LRKK2; GBA; SMPD1; SNCA; PARK2; PINK1/DJ1; SCARB2 | No direct experimental data are yet available. Further, devoted studies are needed. | |
| GLUTAMATE TOXICITY | UCB (↓) BV (↓) | [106] [110] |
3.1.3. The YPs in Parkinson’s Disease (PD)
| YPs | Modulation | Ref. |
|---|---|---|
| HMOX1 | (↑) In DOPAn, microglia, and astroglia of the SN. (↑) In neurons of the neo-cortex with Lewy bodies. (↑) In in vitro model of PD. Genetic variants of HMOX1 (leading to a reduced transcription and induction of the gene) are more frequent in PD subjects and correlate with an early onset of the disease. | [136,144,145,146] [136] [134,135] [136] |
| HMOX2 | Genetic variants of the neuronal constitutive HMOX2 (leading to a reduced transcription) are more frequent in PD subjects. | [145] |
| TSB | (↑) In early clinical stages of PD. (↑) In PD patients with less severe symptoms. (↓) In late/more severe clinical stages of PD. | [140,150] [140] [137,150] |
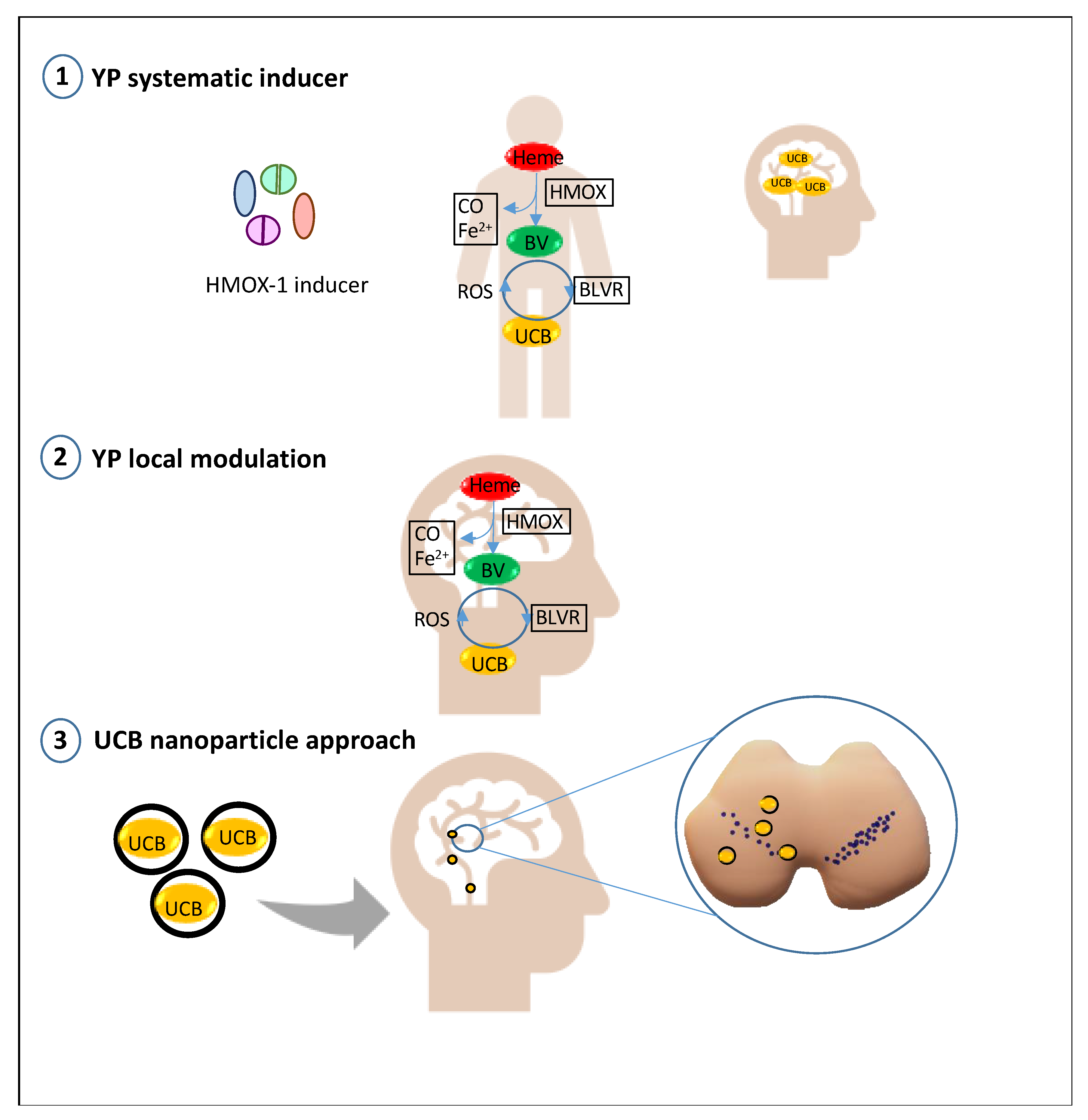
4. Future Prospective: Bilirubin as a Treatment in PD and Its Modulatory/Delivery System
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Feigin, V.L.; Forouzanfar, M.H.; Afshin, A.; Alexander, L.T.; Anderson, H.R.; Bhutta, Z.A.; Biryukov, S.; Brauer, M.; Burnett, R.; Cercy, K.; et al. Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Bloem, B.R. The Parkinson Pandemic—A Call to Action. JAMA Neurol. 2018, 75, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hamilton, J.L.; Kopil, C.; Beck, J.C.; Tanner, C.M.; Albin, R.L.; Dorsey, E.R.; Dahodwala, N.; Cintina, I.; Hogan, P.; et al. Current and projected future economic burden of Parkinson’s disease in the U.S. NPJ Parkinson’s Dis. 2020, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Gilbert, R.M. Epidemiology of Parkinson Disease. Neurol. Clin. 2016, 34, 955–965. [Google Scholar] [CrossRef]
- De Virgilio, A.; Greco, A.; Fabbrini, G.; Inghilleri, M.; Rizzo, M.I.; Gallo, A.; Conte, M.; Rosato, C.; Appiani, M.C.; de Vincentiis, M. Parkinson’s disease: Autoimmunity and neuroinflammation. Autoimmun. Rev. 2016, 15, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Qin, W.-Z.; Zhang, A.; Ye, N. Recent advances in dopaminergic strategies for the treatment of Parkinson’s disease. Acta Pharmacol. Sin. 2020, 41, 471–482. [Google Scholar] [CrossRef]
- Belvisi, D.; Pellicciari, R.; Fabbrini, G.; Tinazzi, M.; Berardelli, A.; Defazio, G. Modifiable risk and protective factors in disease development, progression and clinical subtypes of Parkinson’s disease: What do prospective studies suggest? Neurobiol. Dis. 2020, 134, 104671. [Google Scholar] [CrossRef]
- Liu, Z.; Cheung, H.-H. Stem Cell-Based Therapies for Parkinson Disease. Int. J. Mol. Sci. 2020, 21, 8060. [Google Scholar] [CrossRef] [PubMed]
- Eblesa, J.; Etrigo-Damas, I.; Equiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef]
- Calabrese, V.; Santoro, A.; Monti, D.; Crupi, R.; Di Paola, R.; Latteri, S.; Cuzzocrea, S.; Zappia, M.; Giordano, J.; Calabrese, E.J.; et al. Aging and Parkinson’s Disease: Inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic. Biol. Med. 2018, 115, 80–91. [Google Scholar] [CrossRef]
- Surmeier, D.J. Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J. 2018, 285, 3657–3668. [Google Scholar] [CrossRef]
- Cheong, S.L.; Federico, S.; Spalluto, G.; Klotz, K.-N.; Pastorin, G. The current status of pharmacotherapy for the treatment of Parkinson’s disease: Transition from single-target to multitarget therapy. Drug Discov. Today 2019, 24, 1769–1783. [Google Scholar] [CrossRef] [PubMed]
- Rane, P.; Sarmah, D.; Bhute, S.; Kaur, H.; Goswami, A.; Kalia, K.; Borah, A.; Dave, K.R.; Sharma, N.; Bhattacharya, P. Novel Targets for Parkinson’s Disease: Addressing Different Therapeutic Paradigms and Conundrums. ACS Chem. Neurosci. 2018, 10, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Gazzin, S.; Vitek, L.; Watchko, J.; Shapiro, S.M.; Tiribelli, C. A Novel Perspective on the Biology of Bilirubin in Health and Disease. Trends Mol. Med. 2016, 22, 758–768. [Google Scholar] [CrossRef]
- Schipper, H.M.; Song, W.; Tavitian, A.; Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Prog. Neurobiol. 2019, 172, 40–70. [Google Scholar] [CrossRef] [PubMed]
- Vítek, L. Bilirubin as a signaling molecule. Med. Res. Rev. 2020, 40, 1335–1351. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Tarakad, A. Diagnosis and Management of Parkinson’s Disease. Semin. Neurol. 2017, 37, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Balado, J.; Riba-Llena, I.; Garde, E.; Valor, M.; Gutiérrez, B.; Pujadas, F.; Delgado, P. Prevalence of hippocampal enlarged perivascular spaces in a sample of patients with hypertension and their relation with vascular risk factors and cognitive function. J. Neurol. Neurosurg. Psychiatry 2018, 89, 651–656. [Google Scholar] [CrossRef]
- Stocchi, F.; Torti, M.; Fossati, C. Advances in dopamine receptor agonists for the treatment of Parkinson’s disease. Expert Opin. Pharmacother. 2016, 17, 1889–1902. [Google Scholar] [CrossRef]
- Borovac, J.A. Side effects of a dopamine agonist therapy for Parkinson’s disease: A mini-review of clinical pharmacology. Yale J. Boil. Med. 2016, 89, 37–47. [Google Scholar]
- Connolly, B.S.; Lang, A.E. Pharmacological Treatment of Parkinson Disease. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef]
- Robottom, B. Efficacy, safety, and patient preference of monoamine oxidase B inhibitors in the treatment of Parkinson’s disease. Patient Prefer. Adherence 2011, 5, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Wawruch, M.; Macugova, A.; Kostkova, L.; Luha, J.; Dukat, A.; Murin, J.; Drobna, V.; Wilton, L.; Kuzelova, M. The use of medications with anticholinergic properties and risk factors for their use in hospitalised elderly patients. Pharmacoepidemiol. Drug Saf. 2011, 21, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Zhang, Z.-W.; Liang, L.-W.; Shen, Q.; Wang, X.-D.; Ren, S.-M.; Ma, H.-J.; Jiao, S.-J.; Liu, P. Treatment strategies for Parkinson’s disease. Neurosci. Bull. 2010, 26, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Cui, Y.; Li, S.; Le, W. Current Pharmaceutical Treatments and Alternative Therapies of Parkinson’s Disease. Curr. Neuropharmacol. 2016, 14, 339–355. [Google Scholar] [CrossRef]
- Lees, A. Alternatives to Levodopa in the Initial Treatment of Early Parkinson’s Disease. Drugs Aging 2005, 22, 731–740. [Google Scholar] [CrossRef]
- Kogan, M.; McGuire, M.; Riley, J. Deep Brain Stimulation for Parkinson Disease. Neurosurg. Clin. N. Am. 2019, 30, 137–146. [Google Scholar] [CrossRef]
- Hartmann, C.J.; Fliegen, S.; Groiss, S.J.; Wojtecki, L.; Schnitzler, A. An update on best practice of deep brain stimulation in Parkinson’s disease. Ther. Adv. Neurol. Disord. 2019, 12, 1756286419838096. [Google Scholar] [CrossRef] [PubMed]
- Arenas, E. Towards stem cell replacement therapies for Parkinson’s disease. Biochem. Biophys. Res. Commun. 2010, 396, 152–156. [Google Scholar] [CrossRef]
- Kim, T.W.; Koo, S.Y.; Studer, L. Pluripotent Stem Cell Therapies for Parkinson Disease: Present Challenges and Future Opportunities. Front. Cell Dev. Biol. 2020, 8, 729. [Google Scholar] [CrossRef]
- Stoddard-Bennett, T.; Pera, R.R. Treatment of Parkinson’s Disease through Personalized Medicine and Induced Pluripotent Stem Cells. Cells 2019, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.M.; Wilson-Delfosse, A.L.; Mieyal, J.J. Dysregulation of Glutathione Homeostasis in Neurodegenerative Diseases. Nutrients 2012, 4, 1399–1440. [Google Scholar] [CrossRef]
- Puspita, L.; Chung, S.Y.; Shim, J.-W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 1–12. [Google Scholar] [CrossRef]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53, S26–S38. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Huang, Y.; Przedborski, S. Oxidative Stress in Parkinson’s Disease. Ann. N. Y. Acad. Sci. 2008, 1147, 93–104. [Google Scholar] [CrossRef]
- Wei, Z.; Li, X.; Li, X.; Liu, Q.; Cheng, Y. Oxidative Stress in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Mol. Neurosci. 2018, 11, 236. [Google Scholar] [CrossRef]
- Moon, H.E.; Paek, S.H. Mitochondrial Dysfunction in Parkinson’s Disease. Exp. Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-H.; Chen, C.-M. The Role of Oxidative Stress in Parkinson’s Disease. Antioxidants 2020, 9, 597. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson’s Disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, Paraquat, and Parkinson’s Disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef]
- Pissadaki, E.K.; Bolam, J.P. The energy cost of action potential propagation in dopamine neurons: Clues to susceptibility in Parkinson’s disease. Front. Comput. Neurosci. 2013, 7, 13. [Google Scholar] [CrossRef]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef]
- Kannarkat, G.T.; Boss, J.M.; Tansey, M.G. The Role of Innate and Adaptive Immunity in Parkinson’s Disease. J. Parkinson’s Dis. 2013, 3, 493–514. [Google Scholar] [CrossRef]
- Jayanti, S.; Moretti, R.; Tiribelli, C.; Gazzin, S. Bilirubin and inflammation in neurodegenerative and other neurological diseases. Neuroimmunol. Neuroinflamm. 2020, 7, 92–108. [Google Scholar] [CrossRef]
- Ben, M.D.; Bongiovanni, R.; Tuniz, S.; Fioriti, E.; Tiribelli, C.; Moretti, R.; Gazzin, S. Earliest Mechanisms of Dopaminergic Neurons Sufferance in a Novel Slow Progressing Ex Vivo Model of Parkinson Disease in Rat Organotypic Cultures of Substantia Nigra. Int. J. Mol. Sci. 2019, 20, 2224. [Google Scholar] [CrossRef]
- Ferreira, S.A.; Romero-Ramos, M. Microglia Response During Parkinson’s Disease: Alpha-Synuclein Intervention. Front. Cell. Neurosci. 2018, 12, 247. [Google Scholar] [CrossRef] [PubMed]
- Novellino, F.; Saccà, V.; Donato, A.; Zaffino, P.; Spadea, M.F.; Vismara, M.; Arcidiacono, B.; Malara, N.; Presta, I.; Donato, G. Innate Immunity: A Common Denominator between Neurodegenerative and Neuropsychiatric Diseases. Int. J. Mol. Sci. 2020, 21, 1115. [Google Scholar] [CrossRef] [PubMed]
- Kouli, A.; Torsney, K.M.; Kuan, W.-L.; Stoker, T.B.; Greenland, J.C. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018. [Google Scholar]
- Tansey, M.G.; Romero-Ramos, M. Immune system responses in Parkinson’s disease: Early and dynamic. Eur. J. Neurosci. 2018, 49, 364–383. [Google Scholar] [CrossRef]
- Su, X.; Maguire-Zeiss, K.A.; Giuliano, R.; Prifti, L.; Venkatesh, K.; Federoff, H.J. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol. Aging 2008, 29, 1690–1701. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Standaert, D.G. Ten Unsolved Questions About Neuroinflammation in Parkinson’s Disease. Mov. Disord. 2021, 36, 16–24. [Google Scholar] [CrossRef]
- Ojha, S.; Javed, H.; Azimullah, S.; Haque, M.E. β-Caryophyllene, a phytocannabinoid attenuates oxidative stress, neuroinflammation, glial activation, and salvages dopaminergic neurons in a rat model of Parkinson disease. Mol. Cell. Biochem. 2016, 418, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.-H.; Zhang, Y.-N.; Zhang, J.-N.; Gu, L.; Yang, H.-M.; Huang, Y.-Y.; Xia, N.; Zhang, H. Urate inhibits microglia activation to protect neurons in an LPS-induced model of Parkinson’s disease. J. Neuroinflamm. 2018, 15, 131. [Google Scholar] [CrossRef] [PubMed]
- Hassanzadeh, K.; Rahimmi, A. Oxidative stress and neuroinflammation in the story of Parkinson’s disease: Could targeting these pathways write a good ending? J. Cell. Physiol. 2019, 234, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Lieu, C.A.; DeMaria, M.; Laberge, R.-M.; Campisi, J.; Andersen, J.K. Environmental stress, ageing and glial cell senescence: A novel mechanistic link to Parkinson’s disease? J. Intern. Med. 2013, 273, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Barres, B.A. Astrocyte heterogeneity: An underappreciated topic in neurobiology. Curr. Opin. Neurobiol. 2010, 20, 588–594. [Google Scholar] [CrossRef]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.-M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2008, 119, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.P.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Williams-Gray, C.H.; Wijeyekoon, R.; Yarnall, A.; Lawson, R.A.; Breen, D.P.; Evans, J.R.; Cummins, G.A.; Duncan, G.W.; Khoo, T.K.; Burn, D.; et al. Serum immune markers and disease progression in an incident Parkinson’s disease cohort (ICICLE-PD). Mov. Disord. 2016, 31, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef]
- Deleidi, M.; Maetzler, W. Protein Clearance Mechanisms of Alpha-Synuclein and Amyloid-Beta in Lewy Body Disorders. Int. J. Alzheimer’s Dis. 2012, 2012, 1–9. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; Wahlster, L.; McLean, P.J. Protein degradation pathways in Parkinson’s disease: Curse or blessing. Acta Neuropathol. 2012, 124, 153–172. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, Š.; Sonninen, T.-M.; Wojciechowski, S.; Goldsteins, G.; Koistinaho, J. Dysfunction of Cellular Proteostasis in Parkinson’s Disease. Front. Neurosci. 2019, 13, 457. [Google Scholar] [CrossRef]
- Bi, M.; Du, X.; Jiao, Q.; Chen, X.; Jiang, H. Expanding the role of proteasome homeostasis in Parkinson’s disease: Beyond protein breakdown. Cell Death Dis. 2021, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Vigouroux, S.; Wong, H.; Guttman, M.; Rajput, A.H.; Ang, L.; Kish, S.J.; Briand, Y.; Buldanlioglu, U. Brain proteasomal function in sporadic Parkinson’s disease and related disorders. Ann. Neurol. 2002, 51, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Bentea, E.; Verbruggen, L.; Massie, A. The Proteasome Inhibition Model of Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 31–63. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, C.; De Snoo, M.L.; Gondard, E.; Neudorfer, C.; Chau, H.; Ngana, S.G.; O’Hara, D.M.; Brotchie, J.M.; Koprich, J.B.; Lozano, A.M.; et al. Early-onset impairment of the ubiquitin-proteasome system in dopaminergic neurons caused by α-synuclein. Acta Neuropathol. Commun. 2020, 8, 1–16. [Google Scholar] [CrossRef]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef]
- Marras, C.; Lang, A.; Van De Warrenburg, B.P.; Sue, C.M.; Tabrizi, S.J.; Bertram, L.; Mercimek-Mahmutoglu, S.; Ebrahimi-Fakhari, D.; Warner, T.T.; Durr, A.; et al. Nomenclature of genetic movement disorders: Recommendations of the international Parkinson and movement disorder society task force. Mov. Disord. 2016, 31, 436–457. [Google Scholar] [CrossRef] [PubMed]
- Bi, M.; Kang, S.; Du, X.; Jiao, Q.; Jiang, H. Association between SNCA rs356220 polymorphism and Parkinson’s disease: A meta-analysis. Neurosci. Lett. 2020, 717, 134703. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Dion, P.A.; Rouleau, G.A. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 2015, 11, 1443–1457. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Lang, A.E.; Postuma, R.B.; Maetzler, W.; Deuschl, G.; Gasser, T.; Siderowf, A.; Schapira, A.H.; Oertel, W.; Obeso, J.A.; et al. Changing the research criteria for the diagnosis of Parkinson’s disease: Obstacles and opportunities. Lancet Neurol. 2013, 12, 514–524. [Google Scholar] [CrossRef]
- Hayes, M.T. Parkinson’s Disease and Parkinsonism. Am. J. Med. 2019, 132, 802–807. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Garcia-Ruiz, P.J.; Castrillo, J.C.M.; Alonso-Canovas, A.; Barcenas, A.H.; Vela, L.; Alonso, P.S.; Mata, M.; Gonzalez, N.O.; Fernandez, I.M. Impulse control disorder in patients with Parkinson’s disease under dopamine agonist therapy: A multicentre study. J. Neurol. Neurosurg. Psychiatry 2014, 85, 840–844. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Miocinovic, S.; Somayajula, S.; Chitnis, S.; Vitek, J.L. History, Applications, and Mechanisms of Deep Brain Stimulation. JAMA Neurol. 2013, 70, 163–171. [Google Scholar] [CrossRef]
- Kuusimäki, T.; Korpela, J.; Pekkonen, E.; Martikainen, M.H.; Antonini, A.; Kaasinen, V. Deep brain stimulation for monogenic Parkinson’s disease: A systematic review. J. Neurol. 2020, 267, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.-Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson’s Disease. N. Engl. J. Med. 2020, 382, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- McTague, A.; Rossignoli, G.; Ferrini, A.; Barral, S.; Kurian, M.A. Genome Editing in iPSC-Based Neural Systems: From Disease Models to Future Therapeutic Strategies. Front. Genome Ed. 2021, 3. [Google Scholar] [CrossRef]
- Konnova, E.A.; Swanberg, M. Animal Models of Parkinson’s Disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018. [Google Scholar]
- Gazzin, S.; Masutti, F.; Vítek, L.; Tiribelli, C. The molecular basis of jaundice: An old symptom revisited. Liver Int. 2016, 37, 1094–1102. [Google Scholar] [CrossRef]
- Chen, J.; Tu, Y.; Moon, C.; Nagata, E.; Ronnett, G.V. Heme oxygenase-1 and heme oxygenase-2 have distinct roles in the proliferation and survival of olfactory receptor neurons mediated by cGMP and bilirubin, respectively. J. Neurochem. 2003, 85, 1247–1261. [Google Scholar] [CrossRef]
- Park, J.-S.; Nam, E.; Lee, H.-K.; Lim, M.H.; Rhee, H.-W. In Cellulo Mapping of Subcellular Localized Bilirubin. ACS Chem. Biol. 2016, 11, 2177–2185. [Google Scholar] [CrossRef]
- Takeda, T.-A.; Mu, A.; Tai, T.T.; Kitajima, S.; Taketani, S. Continuous de novo biosynthesis of haem and its rapid turnover to bilirubin are necessary for cytoprotection against cell damage. Sci. Rep. 2015, 5, 10488. [Google Scholar] [CrossRef]
- Abraham, N.G.; Kappas, A. Pharmacological and Clinical Aspects of Heme Oxygenase. Pharmacol. Rev. 2008, 60, 79–127. [Google Scholar] [CrossRef] [PubMed]
- Gozzelino, R. The Pathophysiology of Heme in the Brain. Curr. Alzheimer Res. 2016, 13, 174–184. Available online: https://www.eurekaselect.com/135089/article (accessed on 27 July 2020). [CrossRef] [PubMed]
- Maines, M.D. New Insights into Biliverdin Reductase Functions: Linking Heme Metabolism to Cell Signaling. Physiology 2005, 20, 382–389. [Google Scholar] [CrossRef]
- Nitti, M.; Piras, S.; Brondolo, L.; Marinari, U.M.; Pronzato, M.A.; Furfaro, A.L. Heme Oxygenase 1 in the Nervous System: Does It Favor Neuronal Cell Survival or Induce Neurodegeneration? Int. J. Mol. Sci. 2018, 19, 2260. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M.K. Heme Oxygenase-1/Carbon Monoxide: From Basic Science to Therapeutic Applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Wagner, K.-H.; Wallner, M.; Mölzer, C.; Gazzin, S.; Bulmer, A.C.; Tiribelli, C.; Vitek, L. Looking to the horizon: The role of bilirubin in the development and prevention of age-related chronic diseases. Clin. Sci. 2015, 129, 1–25. [Google Scholar] [CrossRef]
- Vitek, L. Bilirubin Chemistry and Metabolism; Harmful and Protective Aspects. Curr. Pharm. Des. 2009, 15, 2869–2883. [Google Scholar] [CrossRef] [PubMed]
- Diamond, I.; Schmid, R. Experimental bilirubin encephalopathy. The mode of entry of bilirubin-14C into the central nervous system. J. Clin. Investig. 1966, 45, 678–689. [Google Scholar] [CrossRef]
- Wennberg, R.P.; Ahlfors, C.E.; Bhutani, V.K.; Johnson, L.H.; Shapiro, S.M. Toward Understanding Kernicterus: A Challenge to Improve the Management of Jaundiced Newborns. Pediatrics 2006, 117, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef]
- Baranano, D.E.; Rao, M.; Ferris, C.D.; Snyder, S.H. Biliverdin reductase: A major physiologic cytoprotectant. Proc. Natl. Acad. Sci. USA 2002, 99, 16093–16098. [Google Scholar] [CrossRef] [PubMed]
- Sedlak, T.W.; Snyder, S.H. Bilirubin Benefits: Cellular Protection by a Biliverdin Reductase Antioxidant Cycle. Pediatrics 2004, 113, 1776–1782. [Google Scholar] [CrossRef]
- Sedlak, T.W.; Saleh, M.; Higginson, D.S.; Paul, B.D.; Juluri, K.R.; Snyder, S.H. Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc. Natl. Acad. Sci. USA 2009, 106, 5171–5176. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, B.; Wang, X.; Luo, L.; Li, P.; Paty, D.W.; Cynader, M.S. Bilirubin as a potent antioxidant suppresses experimental autoimmune encephalomyelitis: Implications for the role of oxidative stress in the development of multiple sclerosis. J. Neuroimmunol. 2003, 139, 27–35. [Google Scholar] [CrossRef]
- Kaur, H.; Hughes, M.N.; Green, C.J.; Naughton, P.; Foresti, R.; Motterlini, R. Interaction of bilirubin and biliverdin with reactive nitrogen species. FEBS Lett. 2003, 543, 113–119. [Google Scholar] [CrossRef]
- Mancuso, C.; Barone, E.; Guido, P.; Miceli, F.; Di Domenico, F.; Perluigi, M.; Santangelo, R.; Preziosi, P. Inhibition of lipid peroxidation and protein oxidation by endogenous and exogenous antioxidants in rat brain microsomes in vitro. Neurosci. Lett. 2012, 518, 101–105. [Google Scholar] [CrossRef]
- Datla, S.R.; Dusting, G.J.; Mori, T.A.; Taylor, C.J.; Croft, K.D.; Jiang, F. Induction of Heme Oxygenase-1 In Vivo Suppresses NADPH Oxidase–Derived Oxidative Stress. Hypertension 2007, 50, 636–642. [Google Scholar] [CrossRef]
- Qaisiya, M.; Zabetta, C.D.C.; Bellarosa, C.; Tiribelli, C. Bilirubin mediated oxidative stress involves antioxidant response activation via Nrf2 pathway. Cell. Signal. 2014, 26, 512–520. [Google Scholar] [CrossRef]
- Dang, T.N.; Robinson, S.R.; Dringen, R.; Bishop, G.M. Uptake, metabolism and toxicity of hemin in cultured neurons. Neurochem. Int. 2011, 58, 804–811. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, Y.; Tang, Z.; Shan, Y.; Wu, X.; Liu, H. Hemin treatment protects neonatal rats from sevoflurane-induced neurotoxicity via the phosphoinositide 3-kinase/Akt pathway. Life Sci. 2020, 242, 117151. [Google Scholar] [CrossRef] [PubMed]
- Vasavda, C.; Kothari, R.; Malla, A.P.; Tokhunts, R.; Lin, A.; Ji, M.; Ricco, C.; Xu, R.; Saavedra, H.; Sbodio, J.I.; et al. Bilirubin Links Heme Metabolism to Neuroprotection by Scavenging Superoxide. Cell Chem. Biol. 2019, 26, 1450–1460.e7. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, P.; Lu, J.; Xiong, W.; Oger, J.; Tetzlaff, W.; Cynader, M. Bilirubin Possesses Powerful Immunomodulatory Activity and Suppresses Experimental Autoimmune Encephalomyelitis. J. Immunol. 2008, 181, 1887–1897. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.C.; Shapiro, S.M. Biliverdin-induced brainstem auditory evoked potential abnormalities in the jaundiced Gunn rat. Brain Res. 2006, 1107, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.M.P.; Solá, S.; Brito, M.A.; Brites, D.; Moura, J.J. Bilirubin directly disrupts membrane lipid polarity and fluidity, protein order, and redox status in rat mitochondria. J. Hepatol. 2002, 36, 335–341. [Google Scholar] [CrossRef]
- Rodrigues, C.M.P.; Solá, S.; Brites, D. Bilirubin induces apoptosis via the mitochondrial pathway in developing rat brain neurons. Hepatology 2002, 35, 1186–1195. [Google Scholar] [CrossRef]
- Rawat, V.; Bortolussi, G.; Gazzin, S.; Tiribelli, C.; Muro, A.F. Bilirubin-Induced Oxidative Stress Leads to DNA Damage in the Cerebellum of Hyperbilirubinemic Neonatal Mice and Activates DNA Double-Strand Break Repair Pathways in Human Cells. Oxidative Med. Cell. Longev. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bulters, D.; Gaastra, B.; Zolnourian, A.; Alexander, S.; Ren, D.; Blackburn, S.L.; Borsody, M.; Dore, S.; Galea, J.; Iihara, K.; et al. Haemoglobin scavenging in intracranial bleeding: Biology and clinical implications. Nat. Rev. Neurol. 2018, 14, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Righy, C.; Bozza, M.T.; Oliveira, M.F.; Bozza, F.A. Molecular, Cellular and Clinical Aspects of Intracerebral Hemorrhage: Are the Enemies Within? Curr. Neuropharmacol. 2016, 14, 392–402. [Google Scholar] [CrossRef]
- Chiabrando, D.; Fiorito, V.; Petrillo, S.; Tolosano, E. Unraveling the Role of Heme in Neurodegeneration. Front. Neurosci. 2018, 12, 712. [Google Scholar] [CrossRef]
- Schipper, H.M. Heme oxygenase-1: Role in brain aging and neurodegeneration. Exp. Gerontol. 2000, 35, 821–830. [Google Scholar] [CrossRef]
- Schipper, H.M. Brain iron deposition and the free radical-mitochondrial theory of ageing. Ageing Res. Rev. 2004, 3, 265–301. [Google Scholar] [CrossRef]
- Andrade, V.M.; Aschner, M.; Dos Santos, A.P.M. Neurotoxicity of Metal Mixtures. In Neurotoxicity of Metals; Aschner, M., Costa, L.G., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 227–265. [Google Scholar]
- Qaisiya, M.; Brischetto, C.; Jasprova, J.; Vítek, L.; Tiribelli, C.; Bellarosa, C. Bilirubin-induced ER stress contributes to the inflammatory response and apoptosis in neuronal cells. Arch. Toxicol. 2017, 91, 1847–1858. [Google Scholar] [CrossRef]
- Fernandes, A.; Falcão, A.S.; Silva, R.F.M.; Gordo, A.C.; Gama, M.J.; Brito, M.A.; Brites, D. Inflammatory signalling pathways involved in astroglial activation by unconjugated bilirubin. J. Neurochem. 2006, 96, 1667–1679. [Google Scholar] [CrossRef]
- Fernandes, A.; Falcão, A.S.; Silva, R.F.M.; Brito, M.A.; Brites, D. MAPKs are key players in mediating cytokine release and cell death induced by unconjugated bilirubin in cultured rat cortical astrocytes. Eur. J. Neurosci. 2007, 25, 1058–1068. [Google Scholar] [CrossRef]
- Zou, Z.; Liu, J.; Chang, C.; Li, J.; Luo, J.; Jin, Y.; Ma, Z.; Wang, T.; Shao, J. Biliverdin administration regulates the microRNA-mRNA expressional network associated with neuroprotection in cerebral ischemia reperfusion injury in rats. Int. J. Mol. Med. 2019, 43, 1356–1372. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Murase, N.; Ho, C.; Toyokawa, H.; Billiar, T.R.; Kanno, S. Biliverdin Administration Prevents the Formation of Intimal Hyperplasia Induced by Vascular Injury. Circulation 2005, 112, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Wegiel, B.; Tampe, J.; Neubauer, O.; Wagner, K.-H.; Otterbein, L.E.; Bulmer, A.C. Biliverdin modulates the expression of C5aR in response to endotoxin in part via mTOR signaling. Biochem. Biophys. Res. Commun. 2014, 449, 94–99. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wegiel, B.; Gallo, D.; Csizmadia, E.; Roger, T.; Kaczmarek, E.; Harris, C.; Zuckerbraun, B.S.; Otterbein, L.E. Biliverdin inhibits Toll-like receptor-4 (TLR4) expression through nitric oxide-dependent nuclear translocation of biliverdin reductase. Proc. Natl. Acad. Sci. USA 2011, 108, 18849–18854. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. Heme oxygenase in neuroprotection: From mechanisms to therapeutic implications. Rev. Neurosci. 2014, 25, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A. Heme Oxygenase-1 as a Therapeutic Target in Neurodegenerative Diseases and Brain Infections. Curr. Pharm. Des. 2008, 14, 429–442. [Google Scholar] [CrossRef]
- Peng, F.; Deng, X.; Yu, Y.; Chen, X.; Shen, L.; Zhong, X.; Qiu, W.; Jiang, Y.; Zhang, J.; Hu, X. Serum bilirubin concentrations and multiple sclerosis. J. Clin. Neurosci. 2011, 18, 1355–1359. [Google Scholar] [CrossRef]
- Lee, H.; Choi, Y.K. Regenerative Effects of Heme Oxygenase Metabolites on Neuroinflammatory Diseases. Int. J. Mol. Sci. 2018, 20, 78. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Shan, Y.; Tang, Z.; Wu, X.; Bi, C.; Zhang, Y.; Gao, Y.; Liu, H. The Neuroprotective Effect of Hemin and the Related Mechanism in Sevoflurane Exposed Neonatal Rats. Front. Neurosci. 2019, 13, 537. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.-Y.; Liou, H.-C.; Kang, K.-H.; Wu, R.-M.; Wen, C.-C.; Fu, W.-M. Overexpression of Heme Oxygenase-1 Protects Dopaminergic Neurons against 1-Methyl-4-Phenylpyridinium-Induced Neurotoxicity. Mol. Pharmacol. 2008, 74, 1564–1575. [Google Scholar] [CrossRef]
- Yoo, M.S.; Chun, H.S.; Son, J.J.; A DeGiorgio, L.; Kim, D.J.; Peng, C.; Son, J.H. Oxidative stress regulated genes in nigral dopaminergic neuronal cells: Correlation with the known pathology in Parkinson’s disease. Mol. Brain Res. 2003, 110, 76–84. [Google Scholar] [CrossRef]
- Ayuso, P.; Martínez, C.; Pastor, P.; Lorenzo-Betancor, O.; Luengo, A.; Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; Agúndez, J.A.G.; García-Martín, E. An association study between Heme oxygenase-1 genetic variants and Parkinson’s disease. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef]
- Macías-García, D.; Barrio, C.M.-D.; Jesús, S.; Labrador, M.A.; Adarmes-Gómez, A.; Vargas-González, L.; Carrillo, F.; Gómez-Garre, P.; Mir, P. Increased bilirubin levels in Parkinson’s disease. Parkinsonism Relat. Disord. 2019, 63, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Hatano, T.; Saiki, S.; Okuzumi, A.; Mohney, R.P.; Hattori, N. Identification of novel biomarkers for Parkinson’s disease by metabolomic technologies. J. Neurol. Neurosurg. Psychiatry 2015, 87, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Luan, H.; Liu, L.-F.; Tang, Z.; Mok, V.C.; Li, M.; Cai, Z. Elevated excretion of biopyrrin as a new marker for idiopathic Parkinson’s disease. Parkinsonism Relat. Disord. 2015, 21, 1371–1372. [Google Scholar] [CrossRef] [PubMed]
- Moccia, M.; Picillo, M.; Erro, R.; Longo, K.; Amboni, M.; Santangelo, G.; Palladino, R.; Allocca, R.; Caporale, O.; Triassi, M.; et al. Increased bilirubin levels inde novoParkinson’s disease. Eur. J. Neurol. 2015, 22, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Scigliano, G.; Girotti, F.; Soliveri, P.; Musicco, M.; Radice, D.; Caraceni, T. Increased plasma bilirubin in Parkinson patients on L-dopa: Evidence against the free radical hypothesis? Neurol. Sci. 1997, 18, 69–72. [Google Scholar] [CrossRef]
- Ayuso, P.; Martínez, C.; Lorenzo-Betancor, O.; Pastor, P.; Luengo, A.; Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; Villalba, M.T.; Agundez, J.; García-Martín, E. A polymorphism located at an ATG transcription start site of the heme oxygenase-2 gene is associated with classical Parkinson’s disease. Pharmacogenet. Genom. 2011, 21, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Kothari, V.; Velly, A.M.; Cressatti, M.; Liberman, A.; Gornitsky, M.; Schipper, H.M. Evaluation of salivary heme oxygenase-1 as a potential biomarker of early Parkinson’s disease. Mov. Disord. 2018, 33, 583–591. [Google Scholar] [CrossRef]
- Castellani, R.; Smith, M.; Richey, G.; Perry, G. Glycoxidation and oxidative stress in Parkinson disease and diffuse Lewy body disease. Brain Res. 1996, 737, 195–200. [Google Scholar] [CrossRef]
- Schipperab, H.M.; Liberman, A.; Stopa, E. Neural Heme Oxygenase-1 Expression in Idiopathic Parkinson’s Disease. Exp. Neurol. 1998, 150, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Song, W.; Zukor, H.; Hascalovici, J.R.; Zeligman, D. Heme oxygenase-1 and neurodegeneration: Expanding frontiers of engagement. J. Neurochem. 2009, 110, 469–485. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M. Is glial heme oxygenase-1 suppression in neurodegenerative disorders permissive for neural repair? Neural Regen. Res. 2015, 10, 208–210. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Song, W.; Schipper, H.M. Astroglia overexpressing heme oxygenase-1 predispose co-cultured PC12 cells to oxidative injury. J. Neurosci. Res. 2007, 85, 2186–2195. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di Domenico, F.; Mancuso, C.; Butterfield, D.A. The Janus face of the heme oxygenase/biliverdin reductase system in Alzheimer disease: It’s time for reconciliation. Neurobiol. Dis. 2014, 62, 144–159. [Google Scholar] [CrossRef]
- Lee, D.Y.; Oh, M.; Kim, S.-J.; Oh, J.S.; Chung, S.J.; Kim, J.S. Bilirubin-Related Differential Striatal [18F]FP-CIT Uptake in Parkinson Disease. Clin. Nucl. Med. 2019, 44, 855–859. [Google Scholar] [CrossRef]
- Li, C.; Hossieny, P.; Wu, B.J.; Qawasmeh, A.; Beck, K.; Stocker, R. Pharmacologic Induction of Heme Oxygenase-1. Antioxid. Redox Signal. 2007, 9, 2227–2240. [Google Scholar] [CrossRef]
- Strasky, Z.; Zemankova, L.; Nemeckova, I.; Rathouska, J.; Wong, R.J.; Muchova, L.; Subhanova, I.; Vanikova, J.; Vanova, K.; Vitek, L.; et al. Spirulina platensis and phycocyanobilin activate atheroprotective heme oxygenase-1: A possible implication for atherogenesis. Food Funct. 2013, 4, 1586–1594. [Google Scholar] [CrossRef]
- Vitek, L.; Bellarosa, C.; Tiribelli, C. Induction of Mild Hyperbilirubinemia: Hype or Real Therapeutic Opportunity? Clin. Pharmacol. Ther. 2019, 106, 568–575. [Google Scholar] [CrossRef]
- Kim, J.S.; Yoon, T.-J.; Yu, K.N.; Kim, B.G.; Park, S.J.; Kim, H.W.; Lee, K.H.; Park, S.B.; Lee, J.-K.; Cho, M.H. Toxicity and Tissue Distribution of Magnetic Nanoparticles in Mice. Toxicol. Sci. 2005, 89, 338–347. [Google Scholar] [CrossRef]
- Petters, C.; Irrsack, E.; Koch, M.; Dringen, R. Uptake and Metabolism of Iron Oxide Nanoparticles in Brain Cells. Neurochem. Res. 2014, 39, 1648–1660. [Google Scholar] [CrossRef]
- Sim, T.M.; Tarini, D.; Dheen, S.T.; Bay, B.H.; Srinivasan, D.K. Nanoparticle-Based Technology Approaches to the Management of Neurological Disorders. Int. J. Mol. Sci. 2020, 21, 6070. [Google Scholar] [CrossRef]
- Teleanu, D.M.; Chircov, C.; Grumezescu, A.M.; Volceanov, A.; Teleanu, R.I. Blood-Brain Delivery Methods Using Nanotechnology. Pharmaceutics 2018, 10, 269. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, L.B.; Thomsen, M.S.; Moos, T. Targeted drug delivery to the brain using magnetic nanoparticles. Ther. Deliv. 2015, 6, 1145–1155. [Google Scholar] [CrossRef]
- Israel, L.L.; Galstyan, A.; Holler, E.; Ljubimova, J.Y. Magnetic iron oxide nanoparticles for imaging, targeting and treatment of primary and metastatic tumors of the brain. J. Control. Release 2020, 320, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, S.; Panghal, A.; Flora, S.J.S. Nanotechnology: A Promising Approach for Delivery of Neuroprotective Drugs. Front. Neurosci. 2020, 14, 494. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Men, P.; Perry, G.; Smith, M.A. Nanoparticle and Iron Chelators as a Potential Novel Alzheimer Therapy. Methods Mol. Biol. 2010, 610, 123–144. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, S.; Lee, D.Y.; Yu, B.; Miao, W.; Jon, S. Multistimuli-Responsive Bilirubin Nanoparticles for Anticancer Therapy. Angew. Chem. Int. Ed. 2016, 55, 10676–10680. [Google Scholar] [CrossRef]



| Treatment | Targets | Clinical Effect | Side Effects/Limitations | References |
|---|---|---|---|---|
| Pharmacological Treatment | ||||
| L-DOPA | Dopamine precursor | Improving motor symptoms | Dyskinesia, nausea, hypotension, muscular rigidity, wearing off effects | [17,18] |
| Decarboxylase inhibitor (carbidopa, benserazide) | Paired with levodopa to inhibit its peripheral conversion to dopamine | Reducing peripheral L-DOPA side effects: vomiting, nausea, arrhythmia, and postural hypertension | [12] | |
| Dopamine agonists Ergoline-derived agonist (bromocriptine, cabergoline, pergolide, lisuride) Non-ergoline-derived agonist (pramipexole, ropinirole, rotigotine, apomorphine) | Mimicking the endogenous dopamine and stimulating dopamine receptors Binding to dopamine receptor (D1, D2), 5-HT, and adrenergic receptor Specifically binding to dopamine receptor (mainly D2, D3) | Ameliorating motor fluctuations and delaying levodopa administration | Spesific risks of peritoneal, pulmonary, and cardiac/valvular fibrosis Hypotension, impulse control disorder, psychosis, hallucination | [19,20] |
| Catechol-O-methyl transferase inhibitors (tolcapone, entacapone) | Inhibiting catechol-O-methyltransferase to prevent dopamine degradation | Reducing wearing-off-type motor fluctuations | Nausea, diarrhea, orthostatic hypotension, dyskinesia, risk for hepatotoxicity | [21] |
| Monoamine oxidase type B (MAO-B) inhibitors (rasagiline, selegiline) | Inhibiting MAO-B to prevent dopamine metabolism | Improving mild symptoms and “off” period | Sleep disturbances, anxiety, nausea, stomatitis, orthostatic hypotension, hallucinations | [20,22] |
| Anticholinergics (trihexyphenidyl, benztropine) | Antagonism of muscarinic acetylcholine receptor helps to maintain the balance of dopamine and acetylcholine | Mitigating the mild symptoms of tremor and rigidity | Immobilization, urinary dysfunction, gastroduodenal ulcer, depression, epilepsy | [23,24,25] |
| N-Methyl-D- Aspartate glutamate receptor antagonist (amantadine, memantine) | Enhancing dopamine release and blocks dopamine reuptake | Useful in the control of dyskinesia | Livedo reticularis, ankle edema, confusion, nightmares, withdrawal encephalopathy, and mild peripheral antimuscarinic effects | [12,26] |
| Non-pharmacological treatment | ||||
| Deep brain stimulation | Stereotactic surgery ablations of either the globus pallidus internus or subthalamic nucleus | Improving appendicular motor symptoms (brady/akinesia, rigidity, and tremor), lowering the L-DOPA dose needed, alleviating hyperdopaminergic behaviors, neuropsychiatric fluctuations | Aggravate visuomotor, depressive symptoms, dementia, and surgical complications (intracranial hemorrhage, infections, microlesion) | [27,28] |
| Cell replacement therapy | Transplantation of hESCs or iPSCs to replace the dopaminergic neuron loss | Under monitoring (ongoing clinical trial phase) | Poor survival of DA neurons, risk of neural tissue overgrowth and neuroepithelial tumors, and could carry mutations | [29,30,31] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jayanti, S.; Moretti, R.; Tiribelli, C.; Gazzin, S. Bilirubin: A Promising Therapy for Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 6223. https://doi.org/10.3390/ijms22126223
Jayanti S, Moretti R, Tiribelli C, Gazzin S. Bilirubin: A Promising Therapy for Parkinson’s Disease. International Journal of Molecular Sciences. 2021; 22(12):6223. https://doi.org/10.3390/ijms22126223
Chicago/Turabian StyleJayanti, Sri, Rita Moretti, Claudio Tiribelli, and Silvia Gazzin. 2021. "Bilirubin: A Promising Therapy for Parkinson’s Disease" International Journal of Molecular Sciences 22, no. 12: 6223. https://doi.org/10.3390/ijms22126223
APA StyleJayanti, S., Moretti, R., Tiribelli, C., & Gazzin, S. (2021). Bilirubin: A Promising Therapy for Parkinson’s Disease. International Journal of Molecular Sciences, 22(12), 6223. https://doi.org/10.3390/ijms22126223