
MYC-Induced Replicative Stress: A Double-Edged Sword for Cancer Development and Treatment

Abstract
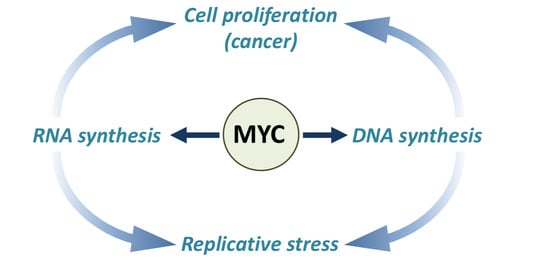
{kind=link}
{kind=link}
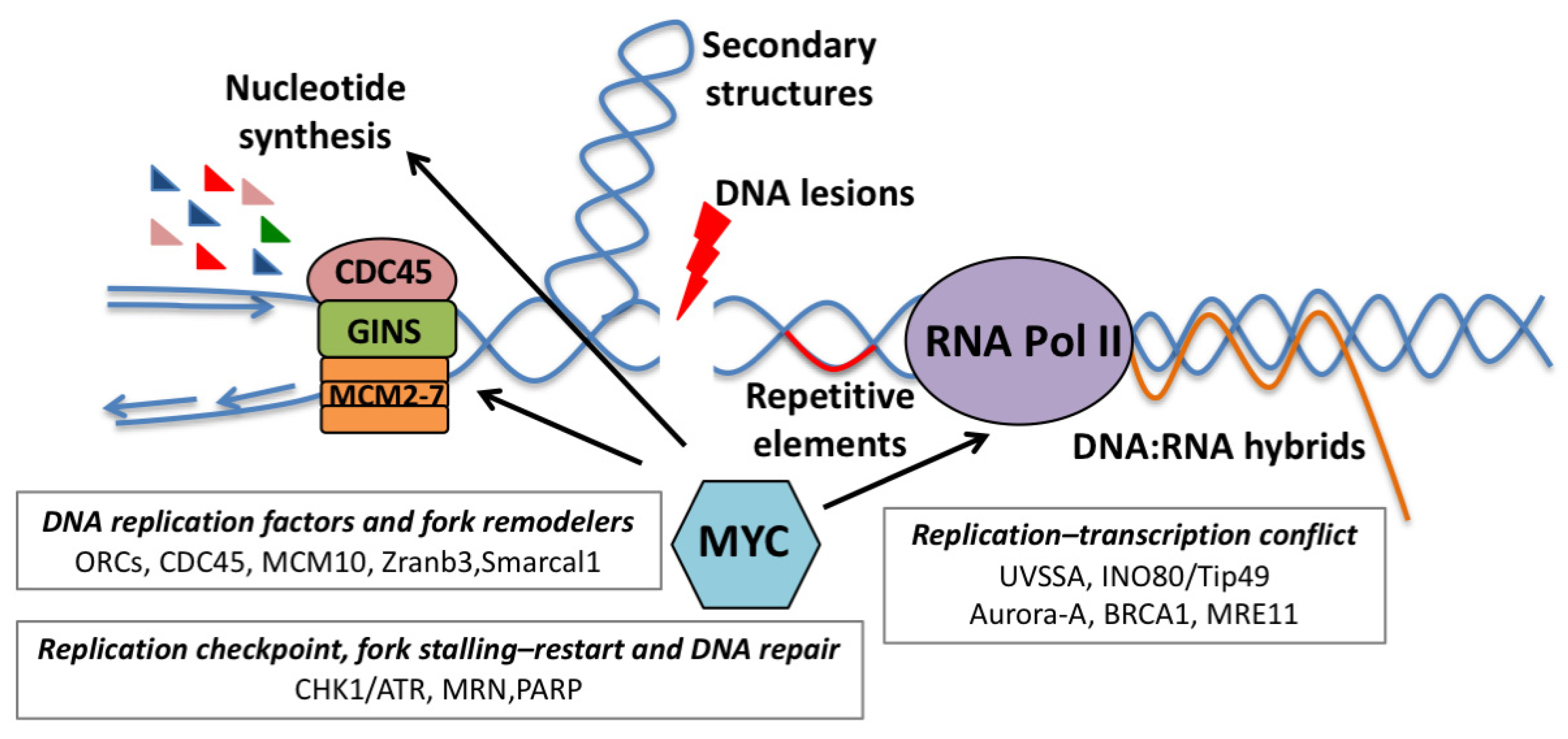{kind=link}
{kind=link}
1. MYC Is a Transcription Factor and Oncogene
2. The Landscape of MYC-Dependent Transcription
3. Structure and Function Insights
4. Activation of Transcription by MYC
5. Transcriptional Repression by MYC
6. MYC Controls DNA Replication and Replicative Stress
7. Linking MYC-Dependent Transcription to Replicative Stress
8. Replicative Stress Is a Therapeutic Target for MYC-Driven Cancers
9. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sheiness, D.; Bishop, J.M. DNA and RNA from uninfected vertebrate cells contain nucleotide sequences related to the putative transforming gene of avian myelocytomatosis virus. J. Virol. 1979, 31, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Duesberg, P.H.; Vogt, P.K. Avian acute leukemia viruses MC29 and MH2 share specific RNA sequences: Evidence for a second class of transforming genes. Proc. Natl. Acad Sci. USA 1979, 76, 1633–1637. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Kress, T.R.; Sabo, A.; Amati, B. MYC: Connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 2015, 15, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Carroll, P.A.; Freie, B.W.; Mathsyaraja, H.; Eisenman, R.N. The MYC transcription factor network: Balancing metabolism, proliferation and oncogenesis. Front. Med. 2018, 12, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Felsher, D.W. MYC Inactivation Elicits Oncogene Addiction through Both Tumor Cell-Intrinsic and Host-Dependent Mechanisms. Genes Cancer 2010, 1, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, J.R.; Beaulieu, M.E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10. [Google Scholar] [CrossRef]
- Wolf, E.; Lin, C.Y.; Eilers, M.; Levens, D.L. Taming of the beast: Shaping Myc-dependent amplification. Trends Cell Biol. 2015, 25, 241–248. [Google Scholar] [CrossRef]
- Guccione, E.; Martinato, F.; Finocchiaro, G.; Luzi, L.; Tizzoni, L.; Dall’ Olio, V.; Zardo, G.; Nervi, C.; Bernard, L.; Amati, B. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat. Cell Biol. 2006, 8, 764–770. [Google Scholar] [CrossRef]
- Lorenzin, F.; Benary, U.; Baluapuri, A.; Walz, S.; Jung, L.A.; von Eyss, B.; Kisker, C.; Wolf, J.; Eilers, M.; Wolf, E. Different promoter affinities account for specificity in MYC-dependent gene regulation. eLife 2016, 5. [Google Scholar] [CrossRef]
- Sabò, A.; Kress, T.R.; Pelizzola, M.; de Pretis, S.; Gorski, M.M.; Tesi, A.; Morelli, M.J.; Bora, P.; Doni, M.; Verrecchia, A.; et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014, 511, 488–492. [Google Scholar] [CrossRef]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Walz, S.; Lorenzin, F.; Morton, J.; Wiese, K.E.; von Eyss, B.; Herold, S.; Rycak, L.; Dumay-Odelot, H.; Karim, S.; Bartkuhn, M.; et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014, 511, 483–487. [Google Scholar] [CrossRef]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef]
- See, Y.X.; Chen, K.; Fullwood, M.J. MYC overexpression leads to increased chromatin interactions at superenhancers and c-Myc binding sites. BioRxiv 2021. [Google Scholar] [CrossRef]
- Beaulieu, M.E.; Castillo, F.; Soucek, L. Structural and Biophysical Insights into the Function of the Intrinsically Disordered Myc Oncoprotein. Cells 2020, 9, 1038. [Google Scholar] [CrossRef] [PubMed]
- Pellanda, P.; Dalsass, M.; Filipuzzi, M.; Loffreda, A.; Verrecchia, A.; Castillo Cano, V.; Thabussot, H.; Doni, M.; Morelli, M.J.; Soucek, L.; et al. Integrated requirement of non-specific and sequence-specific DNA binding in Myc-driven transcription. EMBO J. 2021. [Google Scholar] [CrossRef]
- Kalkat, M.; Resetca, D.; Lourenco, C.; Chan, P.K.; Wei, Y.; Shiah, Y.J.; Vitkin, N.; Tong, Y.; Sunnerhagen, M.; Done, S.J.; et al. MYC Protein Interactome Profiling Reveals Functionally Distinct Regions that Cooperate to Drive Tumorigenesis. Mol. Cell 2018, 72, 836–848.e7. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.B.; Wood, M.A.; Cole, M.D. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol. Cell Biol. 2000, 20, 556–562. [Google Scholar] [CrossRef]
- McMahon, S.B.; Van Buskirk, H.A.; Dugan, K.A.; Copeland, T.D.; Cole, M.D. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell 1998, 94, 363–374. [Google Scholar] [CrossRef]
- Wood, M.A.; McMahon, S.B.; Cole, M.D. An ATPase/helicase complex is an essential cofactor for oncogenic transformation by c-Myc. Mol. Cell 2000, 5, 321–330. [Google Scholar] [CrossRef]
- Thomas, L.R.; Adams, C.M.; Fesik, S.W.; Eischen, C.M.; Tansey, W.P. Targeting MYC through WDR5. Mol. Cell Oncol. 2020, 7, 1709388. [Google Scholar] [CrossRef]
- Thomas, L.R.; Adams, C.M.; Wang, J.; Weissmiller, A.M.; Creighton, J.; Lorey, S.L.; Liu, Q.; Fesik, S.W.; Eischen, C.M.; Tansey, W.P. Interaction of the oncoprotein transcription factor MYC with its chromatin cofactor WDR5 is essential for tumor maintenance. Proc. Natl. Acad Sci. USA 2019, 116, 25260–25268. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Resetca, D.; Li, Z.; Johansson-Akhe, I.; Ahlner, A.; Helander, S.; Wallenhammar, A.; Morad, V.; Raught, B.; Wallner, B.; et al. Multiple direct interactions of TBP with the MYC oncoprotein. Nat. Struct. Mol. Biol. 2019, 26, 1035–1043. [Google Scholar] [CrossRef]
- Baluapuri, A.; Hofstetter, J.; Dudvarski Stankovic, N.; Endres, T.; Bhandare, P.; Vos, S.M.; Adhikari, B.; Schwarz, J.D.; Narain, A.; Vogt, M.; et al. MYC Recruits SPT5 to RNA Polymerase II to Promote Processive Transcription Elongation. Mol. Cell 2019, 74, 674–687.e11. [Google Scholar] [CrossRef] [PubMed]
- Endres, T.; Solvie, D.; Heidelberger, J.B.; Andrioletti, V.; Baluapuri, A.; Ade, C.P.; Muhar, M.; Eilers, U.; Vos, S.M.; Cramer, P.; et al. Ubiquitylation of MYC couples transcription elongation with double-strand break repair at active promoters. Mol. Cell 2021, 81, 830–844.e13. [Google Scholar] [CrossRef] [PubMed]
- Zippo, A.; Serafini, R.; Rocchigiani, M.; Pennacchini, S.; Krepelova, A.; Oliviero, S. Histone crosstalk between H3S10ph and H4K16ac generates a histone code that mediates transcription elongation. Cell 2009, 138, 1122–1136. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ma, Q.; Wong, K.; Li, W.; Ohgi, K.; Zhang, J.; Aggarwal, A.; Rosenfeld, M.G. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell 2013, 155, 1581–1595. [Google Scholar] [CrossRef] [PubMed]
- Rahl, P.B.; Lin, C.Y.; Seila, A.C.; Flynn, R.A.; McCuine, S.; Burge, C.B.; Sharp, P.A.; Young, R.A. c-Myc regulates transcriptional pause release. Cell 2010, 141, 432–445. [Google Scholar] [CrossRef]
- Gargano, B.; Amente, S.; Majello, B.; Lania, L. P-TEFb is a crucial co-factor for Myc transactivation. Cell Cycle 2007, 6, 2031–2037. [Google Scholar] [CrossRef]
- Donato, E.; Croci, O.; Sabo, A.; Muller, H.; Morelli, M.J.; Pelizzola, M.; Campaner, S. Compensatory RNA polymerase 2 loading determines the efficacy and transcriptional selectivity of JQ1 in Myc-driven tumors. Leukemia 2016. [Google Scholar] [CrossRef] [PubMed]
- Leon, J.; Ferrandiz, N.; Acosta, J.C.; Delgado, M.D. Inhibition of cell differentiation: A critical mechanism for MYC-mediated carcinogenesis? Cell Cycle 2009, 8, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- de Pretis, S.; Kress, T.R.; Morelli, M.J.; Sabo, A.; Locarno, C.; Verrecchia, A.; Doni, M.; Campaner, S.; Amati, B.; Pelizzola, M. Integrative analysis of RNA polymerase II and transcriptional dynamics upon MYC activation. Genome Res. 2017, 27, 1658–1664. [Google Scholar] [CrossRef]
- Dominguez-Sola, D.; Gautier, J. MYC and the control of DNA replication. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef]
- Bretones, G.; Delgado, M.D.; Leon, J. Myc and cell cycle control. Biochim. Biophys. Acta 2015, 1849, 506–516. [Google Scholar] [CrossRef]
- Perna, D.; Faga, G.; Verrecchia, A.; Gorski, M.M.; Barozzi, I.; Narang, V.; Khng, J.; Lim, K.C.; Sung, W.K.; Sanges, R.; et al. Genome-wide mapping of Myc binding and gene regulation in serum-stimulated fibroblasts. Oncogene 2012, 31, 1695–1709. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Li, F.; Handler, J.; Huang, C.R.; Xiang, Y.; Neretti, N.; Sedivy, J.M.; Zeller, K.I.; Dang, C.V. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS ONE 2008, 3, e2722. [Google Scholar] [CrossRef]
- Kaczmarek, L.; Hyland, J.K.; Watt, R.; Rosenberg, M.; Baserga, R. Microinjected c-myc as a competence factor. Science 1985, 228, 1313–1315. [Google Scholar] [CrossRef]
- Henriksson, M.; Classon, M.; Hammarskjold, M.L.; Klein, G.; Sumegi, J. The replication activity of SV40 DNA correlates with the level of c-myc expression in human tumor cell lines. Curr. Top. Microbiol. Immunol. 1988, 141, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Classon, M.; Henriksson, M.; Klein, G.; Sumegi, J. The effect of myc proteins on SV40 replication in human lymphoid cells. Oncogene 1990, 5, 1371–1376. [Google Scholar]
- Classon, M.; Henriksson, M.; Sumegi, J.; Klein, G.; Hammarskjold, M.L. Elevated c-myc expression facilitates the replication of SV40 DNA in human lymphoma cells. Nature 1987, 330, 272–274. [Google Scholar] [CrossRef]
- Dominguez-Sola, D.; Ying, C.Y.; Grandori, C.; Ruggiero, L.; Chen, B.; Li, M.; Galloway, D.A.; Gu, W.; Gautier, J.; Dalla-Favera, R. Non-transcriptional control of DNA replication by c-Myc. Nature 2007, 448, 445–451. [Google Scholar] [CrossRef]
- Srinivasan, S.V.; Dominguez-Sola, D.; Wang, L.C.; Hyrien, O.; Gautier, J. Cdc45 is a critical effector of myc-dependent DNA replication stress. Cell Rep. 2013, 3, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Nepon-Sixt, B.S.; Bryant, V.L.; Alexandrow, M.G. Myc-driven chromatin accessibility regulates Cdc45 assembly into CMG helicases. Commun. Biol. 2019, 2, 110. [Google Scholar] [CrossRef] [PubMed]
- Swarnalatha, M.; Singh, A.K.; Kumar, V. The epigenetic control of E-box and Myc-dependent chromatin modifications regulate the licensing of lamin B2 origin during cell cycle. Nucleic. Acids. Res. 2012, 40, 9021–9035. [Google Scholar] [CrossRef]
- Primo, L.M.F.; Teixeira, L.K. DNA replication stress: Oncogenes in the spotlight. Genet. Mol. Biol. 2019, 43, e20190138. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, M.C.; Noguchi, E. Regulation of DNA Replication through Natural Impediments in the Eukaryotic Genome. Genes 2017, 8, 98. [Google Scholar] [CrossRef]
- Voutsinos, V.; Munk, S.H.N.; Oestergaard, V.H. Common Chromosomal Fragile Sites-Conserved Failure Stories. Genes 2018, 9, 580. [Google Scholar] [CrossRef]
- Barra, V.; Fachinetti, D. The dark side of centromeres: Types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat. Commun. 2018, 9, 4340. [Google Scholar] [CrossRef]
- Gomez-Gonzalez, B.; Aguilera, A. Transcription-mediated replication hindrance: A major driver of genome instability. Genes Dev. 2019, 33, 1008–1026. [Google Scholar] [CrossRef]
- Kotsantis, P.; Petermann, E.; Boulton, S.J. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov. 2018, 8, 537–555. [Google Scholar] [CrossRef] [PubMed]
- Ekholm-Reed, S.; Mendez, J.; Tedesco, D.; Zetterberg, A.; Stillman, B.; Reed, S.I. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J. Cell Biol. 2004, 165, 789–800. [Google Scholar] [CrossRef]
- Frum, R.A.; Singh, S.; Vaughan, C.; Mukhopadhyay, N.D.; Grossman, S.R.; Windle, B.; Deb, S.; Deb, S.P. The human oncoprotein MDM2 induces replication stress eliciting early intra-S-phase checkpoint response and inhibition of DNA replication origin firing. Nucleic. Acids. Res. 2014, 42, 926–940. [Google Scholar] [CrossRef]
- Liontos, M.; Koutsami, M.; Sideridou, M.; Evangelou, K.; Kletsas, D.; Levy, B.; Kotsinas, A.; Nahum, O.; Zoumpourlis, V.; Kouloukoussa, M.; et al. Deregulated overexpression of hCdt1 and hCdc6 promotes malignant behavior. Cancer Res. 2007, 67, 10899–10909. [Google Scholar] [CrossRef] [PubMed]
- Sarni, D.; Kerem, B. Oncogene-Induced Replication Stress Drives Genome Instability and Tumorigenesis. Int. J. Mol. Sci. 2017, 18, 1339. [Google Scholar] [CrossRef]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.M.; Gagos, S.; Tzetis, M.; et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef]
- Lamm, N.; Maoz, K.; Bester, A.C.; Im, M.M.; Shewach, D.S.; Karni, R.; Kerem, B. Folate levels modulate oncogene-induced replication stress and tumorigenicity. EMBO Mol. Med. 2015, 7, 1138–1152. [Google Scholar] [CrossRef]
- Aird, K.M.; Zhang, G.; Li, H.; Tu, Z.; Bitler, B.G.; Garipov, A.; Wu, H.; Wei, Z.; Wagner, S.N.; Herlyn, M.; et al. Suppression of Nucleotide Metabolism Underlies the Establishment and Maintenance of Oncogene-Induced Senescence. Cell Rep. 2013, 3, 1252–1265. [Google Scholar] [CrossRef]
- Xie, M.; Yen, Y.; Owonikoko, T.K.; Ramalingam, S.S.; Khuri, F.R.; Curran, W.J.; Doetsch, P.W.; Deng, X. Bcl2 induces DNA replication stress by inhibiting ribonucleotide reductase. Cancer Res. 2014, 74, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Dang, C.V. c-Myc overexpression uncouples DNA replication from mitosis. Mol. Cell Biol. 1999, 19, 5339–5351. [Google Scholar] [CrossRef]
- Gandarillas, A.; Davies, D.; Blanchard, J.M. Normal and c-Myc-promoted human keratinocyte differentiation both occur via a novel cell cycle involving cellular growth and endoreplication. Oncogene 2000, 19, 3278–3289. [Google Scholar] [CrossRef]
- Pierce, S.B.; Yost, C.; Britton, J.S.; Loo, L.W.; Flynn, E.M.; Edgar, B.A.; Eisenman, R.N. dMyc is required for larval growth and endoreplication in Drosophila. Development 2004, 131, 2317–2327. [Google Scholar] [CrossRef] [PubMed]
- Gatti, G.; Maresca, G.; Natoli, M.; Florenzano, F.; Nicolin, A.; Felsani, A.; D’Agnano, I. MYC prevents apoptosis and enhances endoreduplication induced by paclitaxel. PLoS ONE 2009, 4, e5442. [Google Scholar] [CrossRef] [PubMed]
- Sheen, J.H.; Dickson, R.B. Overexpression of c-Myc alters G(1)/S arrest following ionizing radiation. Mol. Cell Biol. 2002, 22, 1819–1833. [Google Scholar] [CrossRef]
- Albajar, M.; Gomez-Casares, M.T.; Llorca, J.; Mauleon, I.; Vaque, J.P.; Acosta, J.C.; Bermudez, A.; Donato, N.; Delgado, M.D.; Leon, J. MYC in chronic myeloid leukemia: Induction of aberrant DNA synthesis and association with poor response to imatinib. Mol. Cancer Res. 2011, 9, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Cowling, V.H.; Chandriani, S.; Whitfield, M.L.; Cole, M.D. A conserved Myc protein domain, MBIV, regulates DNA binding, apoptosis, transformation, and G2 arrest. Mol. Cell Biol. 2006, 26, 4226–4239. [Google Scholar] [CrossRef]
- Felsher, D.W.; Zetterberg, A.; Zhu, J.; Tlsty, T.; Bishop, J.M. Overexpression of MYC causes p53-dependent G2 arrest of normal fibroblasts. Proc. Natl. Acad Sci. USA 2000, 97, 10544–10548. [Google Scholar] [CrossRef] [PubMed]
- Kuschak, T.I.; Kuschak, B.C.; Taylor, C.L.; Wright, J.A.; Wiener, F.; Mai, S. c-Myc initiates illegitimate replication of the ribonucleotide reductase R2 gene. Oncogene 2002, 21, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Maya-Mendoza, A.; Ostrakova, J.; Kosar, M.; Hall, A.; Duskova, P.; Mistrik, M.; Merchut-Maya, J.M.; Hodny, Z.; Bartkova, J.; Christensen, C.; et al. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Mol. Oncol. 2015, 9, 601–616. [Google Scholar] [CrossRef]
- Macheret, M.; Halazonetis, T.D. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 2018, 555, 112–116. [Google Scholar] [CrossRef]
- Stork, C.T.; Bocek, M.; Crossley, M.P.; Sollier, J.; Sanz, L.A.; Chedin, F.; Swigut, T.; Cimprich, K.A. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. eLife 2016, 5. [Google Scholar] [CrossRef]
- Gorthi, A.; Romero, J.C.; Loranc, E.; Cao, L.; Lawrence, L.A.; Goodale, E.; Iniguez, A.B.; Bernard, X.; Masamsetti, V.P.; Roston, S.; et al. EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature 2018, 555, 387–391. [Google Scholar] [CrossRef]
- Lavado, A.; Park, J.Y.; Pare, J.; Finkelstein, D.; Pan, H.; Xu, B.; Fan, Y.; Kumar, R.P.; Neale, G.; Kwak, Y.D.; et al. The Hippo Pathway Prevents YAP/TAZ-Driven Hypertranscription and Controls Neural Progenitor Number. Dev. Cell 2018, 47, 576–591.e8. [Google Scholar] [CrossRef]
- Poli, J.; Gasser, S.M.; Papamichos-Chronakis, M. The INO80 remodeller in transcription, replication and repair. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef]
- Hristova, R.H.; Stoynov, S.S.; Tsaneva, I.R.; Gospodinov, A.G. Deregulated levels of RUVBL1 induce transcription-dependent replication stress. Int. J. Biochem. Cell Biol. 2020, 128, 105839. [Google Scholar] [CrossRef]
- Sato, M.; Liebau, A.W.; Liu, Z.; Liu, L.; Rabadan, R.; Gautier, J. The UVSSA complex alleviates MYC-driven transcription stress. J. Cell Biol. 2021, 220. [Google Scholar] [CrossRef]
- Steurer, B.; Marteijn, J.A. Traveling Rocky Roads: The Consequences of Transcription-Blocking DNA Lesions on RNA Polymerase II. J. Mol. Biol. 2017, 429, 3146–3155. [Google Scholar] [CrossRef]
- Tufegdžić Vidaković, A.; Mitter, R.; Kelly, G.P.; Neumann, M.; Harreman, M.; Rodríguez-Martínez, M.; Herlihy, A.; Weems, J.C.; Boeing, S.; Encheva, V.; et al. Regulation of the RNAPII Pool Is Integral to the DNA Damage Response. Cell 2020, 180, 1245–1261.e21. [Google Scholar] [CrossRef]
- Nakazawa, Y.; Hara, Y.; Oka, Y.; Komine, O.; van den Heuvel, D.; Guo, C.; Daigaku, Y.; Isono, M.; He, Y.; Shimada, M.; et al. Ubiquitination of DNA Damage-Stalled RNAPII Promotes Transcription-Coupled Repair. Cell 2020, 180, 1228–1244.e24. [Google Scholar] [CrossRef]
- van der Weegen, Y.; Golan-Berman, H.; Mevissen, T.E.T.; Apelt, K.; González-Prieto, R.; Goedhart, J.; Heilbrun, E.E.; Vertegaal, A.C.O.; van den Heuvel, D.; Walter, J.C.; et al. The cooperative action of CSB, CSA, and UVSSA target TFIIH to DNA damage-stalled RNA polymerase II. Nat. Commun. 2020, 11, 2104. [Google Scholar] [CrossRef]
- Roeschert, I.; Poon, E.; Henssen, A.G.; Dorado Garcia, H.; Gatti, M.; Giansanti, C.; Jamin, Y.; Ade, C.P.; Gallant, P.; Schülein-Völk, C.; et al. Combined inhibition of Aurora-A and ATR kinases results in regression of MYCN-amplified neuroblastoma. Nat. Cancer 2021, 2, 312–326. [Google Scholar] [CrossRef]
- Herold, S.; Kalb, J.; Büchel, G.; Ade, C.P.; Baluapuri, A.; Xu, J.; Koster, J.; Solvie, D.; Carstensen, A.; Klotz, C.; et al. Recruitment of BRCA1 limits MYCN-driven accumulation of stalled RNA polymerase. Nature 2019, 567, 545–549. [Google Scholar] [CrossRef]
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015, 10, 425–448. [Google Scholar] [CrossRef]
- Mannava, S.; Grachtchouk, V.; Wheeler, L.J.; Im, M.; Zhuang, D.; Slavina, E.G.; Mathews, C.K.; Shewach, D.S.; Nikiforov, M.A. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 2008, 7, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Luoto, K.R.; Meng, A.X.; Wasylishen, A.R.; Zhao, H.; Coackley, C.L.; Penn, L.Z.; Bristow, R.G. Tumor Cell Kill by c-MYC Depletion: Role of MYC-Regulated Genes that Control DNA Double-Strand Break Repair. Cancer Res. 2010. [Google Scholar] [CrossRef]
- Cui, F.; Fan, R.; Chen, Q.; He, Y.; Song, M.; Shang, Z.; Zhang, S.; Zhu, W.; Cao, J.; Guan, H.; et al. The involvement of c-Myc in the DNA double-strand break repair via regulating radiation-induced phosphorylation of ATM and DNA-PKcs activity. Mol. Cell. Biochem. 2015, 406, 43–51. [Google Scholar] [CrossRef]
- Petroni, M.; Sardina, F.; Heil, C.; Sahún-Roncero, M.; Colicchia, V.; Veschi, V.; Albini, S.; Fruci, D.; Ricci, B.; Soriani, A.; et al. The MRN complex is transcriptionally regulated by MYCN during neural cell proliferation to control replication stress. Cell Death Differ. 2016, 23, 197–206. [Google Scholar] [CrossRef]
- Falck, J.; Forment, J.V.; Coates, J.; Mistrik, M.; Lukas, J.; Bartek, J.; Jackson, S.P. CDK targeting of NBS1 promotes DNA-end resection, replication restart and homologous recombination. EMBO Rep. 2012, 13, 561–568. [Google Scholar] [CrossRef]
- Chang, E.Y.; Tsai, S.; Aristizabal, M.J.; Wells, J.P.; Coulombe, Y.; Busatto, F.F.; Chan, Y.A.; Kumar, A.; Dan Zhu, Y.; Wang, A.Y.; et al. MRE11-RAD50-NBS1 promotes Fanconi Anemia R-loop suppression at transcription-replication conflicts. Nat. Commun. 2019, 10, 4265. [Google Scholar] [CrossRef]
- Grandori, C.; Wu, K.J.; Fernandez, P.; Ngouenet, C.; Grim, J.; Clurman, B.E.; Moser, M.J.; Oshima, J.; Russell, D.W.; Swisshelm, K.; et al. Werner syndrome protein limits MYC-induced cellular senescence. Genes Dev. 2003, 17, 1569–1574. [Google Scholar] [CrossRef]
- Robinson, K.; Asawachaicharn, N.; Galloway, D.A.; Grandori, C. c-Myc accelerates S-phase and requires WRN to avoid replication stress. PLoS ONE 2009, 4, e5951. [Google Scholar] [CrossRef] [PubMed]
- Puccetti, M.V.; Adams, C.M.; Kushinsky, S.; Eischen, C.M. Smarcal1 and Zranb3 Protect Replication Forks from Myc-Induced DNA Replication Stress. Cancer Res. 2019, 79, 1612–1623. [Google Scholar] [CrossRef] [PubMed]
- Kondratick, C.M.; Washington, M.T.; Spies, M. Making Choices: DNA Replication Fork Recovery Mechanisms. Semin. Cell Dev. Biol. 2021, 113, 27–37. [Google Scholar] [CrossRef]
- Kurashima, K.; Sekimoto, T.; Oda, T.; Kawabata, T.; Hanaoka, F.; Yamashita, T. Poleta, a Y-family translesion synthesis polymerase, promotes cellular tolerance of Myc-induced replication stress. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Murayama, T.; Takeuchi, Y.; Yamawaki, K.; Natsume, T.; Li, M.; Marcela, R.-C.N.; Nishimura, T.; Kogure, Y.; Nakata, A.; Tominaga, K.; et al. MCM10 compensates for Myc-induced DNA replication stress in breast cancer stem-like cells. Cancer Sci. 2021, 112, 1209–1224. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Xie, W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019, 20, 535–550. [Google Scholar] [CrossRef]
- Arnould, C.; Rocher, V.; Finoux, A.-L.; Clouaire, T.; Li, K.; Zhou, F.; Caron, P.; Mangeot, P.E.; Ricci, E.P.; Mourad, R.; et al. Loop extrusion as a mechanism for formation of DNA damage repair foci. Nature 2021, 590, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Villa-Hernandez, S.; Bermejo, R. Cohesin dynamic association to chromatin and interfacing with replication forks in genome integrity maintenance. Curr. Genet. 2018, 64, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Busslinger, G.A.; Stocsits, R.R.; van der Lelij, P.; Axelsson, E.; Tedeschi, A.; Galjart, N.; Peters, J.-M. Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl. Nature 2017, 544, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Rohban, S.; Cerutti, A.; Morelli, M.J.; d’Adda di Fagagna, F.; Campaner, S. The cohesin complex prevents Myc-induced replication stress. Cell Death Dis. 2017, 8, e2956. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Murga, M.; Fernandez-Capetillo, O. Targeting ATR and Chk1 kinases for cancer treatment: A new model for new (and old) drugs. Mol. Oncol. 2011, 5, 368–373. [Google Scholar] [CrossRef]
- Gilad, O.; Nabet, B.Y.; Ragland, R.L.; Schoppy, D.W.; Smith, K.D.; Durham, A.C.; Brown, E.J. Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Res. 2010, 70, 9693–9702. [Google Scholar] [CrossRef]
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montana, M.F.; D’Artista, L.; Schleker, T.; Guerra, C.; Garcia, E.; et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [Google Scholar] [CrossRef]
- Schoppy, D.W.; Ragland, R.L.; Gilad, O.; Shastri, N.; Peters, A.A.; Murga, M.; Fernandez-Capetillo, O.; Diehl, J.A.; Brown, E.J. Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J. Clin. Investig. 2012, 122, 241–252. [Google Scholar] [CrossRef]
- Rohban, S.; Campaner, S. Myc induced replicative stress response: How to cope with it and exploit it. Biochim. Biophys Acta 2015, 1849, 517–524. [Google Scholar] [CrossRef]
- Kruger, K.; Geist, K.; Stuhldreier, F.; Schumacher, L.; Blumel, L.; Remke, M.; Wesselborg, S.; Stork, B.; Klocker, N.; Bormann, S.; et al. Multiple DNA damage-dependent and DNA damage-independent stress responses define the outcome of ATR/Chk1 targeting in medulloblastoma cells. Cancer Lett. 2018, 430, 34–46. [Google Scholar] [CrossRef]
- Qiu, Z.; Fa, P.; Liu, T.; Prasad, C.B.; Ma, S.; Hong, Z.; Chan, E.R.; Wang, H.; Li, Z.; He, K.; et al. A Genome-Wide Pooled shRNA Screen Identifies PPP2R2A as a Predictive Biomarker for the Response to ATR and CHK1 Inhibitors. Cancer Res. 2020, 80, 3305–3318. [Google Scholar] [CrossRef]
- Cottini, F.; Hideshima, T.; Suzuki, R.; Tai, Y.T.; Bianchini, G.; Richardson, P.G.; Anderson, K.C.; Tonon, G. Synthetic Lethal Approaches Exploiting DNA Damage in Aggressive Myeloma. Cancer Discov. 2015, 5, 972–987. [Google Scholar] [CrossRef]
- Young, L.A.; O’Connor, L.O.; de Renty, C.; Veldman-Jones, M.H.; Dorval, T.; Wilson, Z.; Jones, D.R.; Lawson, D.; Odedra, R.; Maya-Mendoza, A.; et al. Differential Activity of ATR and WEE1 Inhibitors in a Highly Sensitive Subpopulation of DLBCL Linked to Replication Stress. Cancer Res. 2019, 79, 3762–3775. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.C.; Venkataraman, S.; Subramanian, A.; Desisto, J.; Balakrishnan, I.; Prince, E.; Pierce, A.; Griesinger, A.; Green, A.; Eberhardt, C.G.; et al. Targeting MYC-driven replication stress in medulloblastoma with AZD1775 and gemcitabine. J. Neuro Oncol. 2020, 147, 531–545. [Google Scholar] [CrossRef]
- Song, X.; Wang, L.; Wang, T.; Hu, J.; Wang, J.; Tu, R.; Su, H.; Jiang, J.; Qing, G.; Liu, H. Synergistic targeting of CHK1 and mTOR in MYC-driven tumors. Carcinogenesis 2021, 42, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, D.; Scionti, F.; Juli, G.; Altomare, E.; Golino, G.; Todoerti, K.; Grillone, K.; Riillo, C.; Arbitrio, M.; Iannone, M.; et al. Exploiting MYC-induced PARPness to target genomic instability in multiple myeloma. Haematologica 2021, 106, 185–195. [Google Scholar] [CrossRef]
- Maifrede, S.; Martin, K.; Podszywalow-Bartnicka, P.; Sullivan-Reed, K.; Langer, S.K.; Nejati, R.; Dasgupta, Y.; Hulse, M.; Gritsyuk, D.; Nieborowska-Skorska, M.; et al. IGH/MYC Translocation Associates with BRCA2 Deficiency and Synthetic Lethality to PARP1 Inhibitors. Mol. Cancer Res. 2017, 15, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Bosch, N.; Iglesias, M.; Munne-Collado, J.; Martinez-Caceres, C.; Moreno, M.; Guerra, C.; Yelamos, J.; Navarro, P. Parp-1 genetic ablation in Ela-myc mice unveils novel roles for Parp-1 in pancreatic cancer. J. Pathol. 2014, 234, 214–227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, B.; Wu, W.; Li, L.; Broom, B.M.; Basourakos, S.P.; Korentzelos, D.; Luan, Y.; Wang, J.; Yang, G.; et al. Targeting the MYCN-PARP-DNA Damage Response Pathway in Neuroendocrine Prostate Cancer. Clin. Cancer Res. 2018, 24, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Colicchia, V.; Petroni, M.; Guarguaglini, G.; Sardina, F.; Sahun-Roncero, M.; Carbonari, M.; Ricci, B.; Heil, C.; Capalbo, C.; Belardinilli, F.; et al. PARP inhibitors enhance replication stress and cause mitotic catastrophe in MYCN-dependent neuroblastoma. Oncogene 2017, 36, 4682–4691. [Google Scholar] [CrossRef]
- King, D.; Li, X.D.; Almeida, G.S.; Kwok, C.; Gravells, P.; Harrison, D.; Burke, S.; Hallsworth, A.; Jamin, Y.; George, S.; et al. MYCN expression induces replication stress and sensitivity to PARP inhibition in neuroblastoma. Oncotarget 2020, 11, 2141–2159. [Google Scholar] [CrossRef] [PubMed]
- Barone, G.; Staples, C.J.; Ganesh, A.; Patterson, K.W.; Bryne, D.P.; Myers, K.N.; Patil, A.A.; Eyers, C.E.; Maslen, S.; Skehel, J.M.; et al. Human CDK18 promotes replication stress signaling and genome stability. Nucleic Acids Res. 2016, 44, 8772–8785. [Google Scholar] [CrossRef] [PubMed]
- Spehalski, E.; Capper, K.M.; Smith, C.J.; Morgan, M.J.; Dinkelmann, M.; Buis, J.; Sekiguchi, J.M.; Ferguson, D.O. MRE11 Promotes Tumorigenesis by Facilitating Resistance to Oncogene-Induced Replication Stress. Cancer Res. 2017, 77, 5327–5338. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, M.; Teo, S.X.; Muller, J.; Mok, W.C.; Sahu, S.K.; Vardy, L.A.; Bonday, Z.Q.; Guccione, E. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 2013, 27, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.Y.; Simon, L.M.; Neill, N.J.; Marcotte, R.; Sayad, A.; Bland, C.S.; Echeverria, G.V.; Sun, T.; Kurley, S.J.; Tyagi, S.; et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 2015, 525, 384–388. [Google Scholar] [CrossRef]
- Cossa, G.; Roeschert, I.; Prinz, F.; Baluapuri, A.; Silveira Vidal, R.; Schulein-Volk, C.; Chang, Y.C.; Ade, C.P.; Mastrobuoni, G.; Girard, C.; et al. Localized Inhibition of Protein Phosphatase 1 by NUAK1 Promotes Spliceosome Activity and Reveals a MYC-Sensitive Feedback Control of Transcription. Mol. Cell 2020, 77, 1322–1339.e11. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curti, L.; Campaner, S. MYC-Induced Replicative Stress: A Double-Edged Sword for Cancer Development and Treatment. Int. J. Mol. Sci. 2021, 22, 6168. https://doi.org/10.3390/ijms22126168
Curti L, Campaner S. MYC-Induced Replicative Stress: A Double-Edged Sword for Cancer Development and Treatment. International Journal of Molecular Sciences. 2021; 22(12):6168. https://doi.org/10.3390/ijms22126168
Chicago/Turabian StyleCurti, Laura, and Stefano Campaner. 2021. "MYC-Induced Replicative Stress: A Double-Edged Sword for Cancer Development and Treatment" International Journal of Molecular Sciences 22, no. 12: 6168. https://doi.org/10.3390/ijms22126168
APA StyleCurti, L., & Campaner, S. (2021). MYC-Induced Replicative Stress: A Double-Edged Sword for Cancer Development and Treatment. International Journal of Molecular Sciences, 22(12), 6168. https://doi.org/10.3390/ijms22126168