
Targeting Adrenergic Receptors in Metabolic Therapies for Heart Failure


Abstract
1. Introduction
2. Cardiac ARs
3. ARs in Heart Failure
4. Metabolic Regulation of Normal vs. Failing Heart
4.1. Glucose Utilization
4.2. Fatty Acid Metabolism
4.3. General Metabolic Therapies for HF
5. ARs in Cardiac Metabolism and as Potential Therapeutics
5.1. α1-ARs
5.2. α1A-AR Therapeutics
5.3. β2-ARs
5.4. β1-AR Therapeutics
6. Conclusions
Funding
Conflicts of Interest
References
- Perez, D.M.; Doze, V.A. Cardiac and neuroprotection regulated by α1-adrenergic receptor subtypes. J. Recept. Signal. Transduct. Res. 2011, 31, 98–110. [Google Scholar] [CrossRef]
- Stadel, J.M.; Namb, I.P.; Shorr, R.G.; Sawyer, D.F.; Caron, M.G.; Lefkowitz, R.J. Catecholamine-induced desensitization of turkey erythrocyte adenylate cyclase is associated with phosphorylation of the β-adrenergic receptor. Proc. Natl. Acad. Sci. USA 1983, 80, 3173–3177. [Google Scholar] [CrossRef]
- Hausdorff, W.P.; Caron, M.G.; Lefkowitz, R.J. Turning off the signal: Desensitization of b-adrenergic receptor function. FASEB J. 1990, 4, 2881–2889. [Google Scholar] [CrossRef] [PubMed]
- Bristow, M.R.; Ginsburg, R.; Umans, V.; Fowler, M.; Minobe, W.; Rasmussen, R.; Zera, P.; Menlove, R.; Shah, P.; Jamieson, S.; et al. β1- and β2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: Coupling of both receptor subtypes to muscle contraction and selective β1-receptor down-regulation in heart failure. Circ. Res. 1986, 59, 297–309. [Google Scholar] [CrossRef]
- Brodde, O.E. β1- and β2-adrenoceptors in the human heart: Properties, function, and alterations in chronic heart failure. Pharmacol. Rev. 1991, 43, 203–242. [Google Scholar] [PubMed]
- Sulakhe, P.V.; Vo, X.T. Regulation of phospholamban and troponin-I phosphorylation in the intact rat cardiomyocytes by adrenergic and cholinergic stimuli: Roles of cyclic nucleotides, calcium, protein kinases and phosphatases and depolarization. Mol. Cell Biochem. 1995, 149–150, 103–126. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Kobilka, B.K. Myocyte adrenoceptor signaling pathways. Science 2003, 300, 1530–1532. [Google Scholar] [CrossRef]
- Xiao, R.P.; Zhu, W.; Zheng, M.; Cao, C.; Zhang, Y.; Lakatta, E.G.; Han, Q. Subtype-specific α1- and β-adrenoceptor signaling in the heart. Trends Pharmacol. Sci. 2006, 27, 330–337. [Google Scholar] [CrossRef]
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef]
- Communal, C.; Singh, K.; Sawyer, D.B.; Colucci, W.S. Opposing effects of β1- and β2-adrenergic receptors on cardiac myocyte apoptosis: Role of a pertussis toxin-sensitive G protein. Circulation 1999, 100, 2210–2212. [Google Scholar] [CrossRef] [PubMed]
- Chesley, A.; Lundberg, M.S.; Asai, T.; Xiao, R.P.; Ohtani, S.; Lakatta, E.G.; Crow, M.T. The β2-adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through G(i)-dependent coupling to phosphatidylinositol 3′-kinase. Circ. Res. 2000, 87, 1172–1179. [Google Scholar] [CrossRef]
- Zhu, W.Z.; Zheng, M.; Koch, W.J.; Lefkowitz, R.J.; Kobilka, B.K.; Xiao, R.P. Dual modulation of cell survival and cell death by β2-adrenergic signaling in adult mouse cardiac myocytes. Proc. Natl. Acad. Sci. USA 2001, 98, 1607–1612. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, C.; Tavernier, G.; Charpentier, F.; Langin, D.; Le Marec, H. Functional β3-adrenoceptor in the human heart. J. Clin. Investig. 1996, 98, 556–562. [Google Scholar] [CrossRef]
- Gauthier, C.; Leblais, V.; Kobzik, L.; Trochu, J.N.; Khandoudi, N.; Bril, A.; Balligand, J.L.; Le Marec, H. The negative inotropic effect of β3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J. Clin. Investig. 1998, 102, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Varghese, P.; Harrison, R.W.; Lofthouse, R.A.; Georgakopoulos, D.; Berkowitz, D.E.; Hare, J.M. β3-adrenoceptor deficiency blocks nitric oxide-dependent inhibition of myocardial contractility. J. Clin. Investig. 2000, 106, 697–703. [Google Scholar] [CrossRef]
- Tavernier, G.; Toumaniantz, G.; Erfanian, M.; Heymann, M.F.; Laurent, K.; Langin, D.; Gauthier, C. β3-Adrenergic stimulation produces a decrease of cardiac contractility ex vivo in mice overexpressing the human β3-adrenergic receptor. Cardiovasc. Res. 2003, 2, 288–296. [Google Scholar] [CrossRef]
- Steinfath, M.; Chen, Y.Y.; Lavicky, J.; Magnussen, O.; Nose, M.; Rosswag, S.; Schmitz, W.; Scholz, H. Cardiac α1-adrenoceptor densities in different mammalian species. Br. J. Pharmacol. 1992, 107, 185–188. [Google Scholar] [CrossRef]
- Michel, M.C.; Hanft, G.; Gross, G. Radioligand binding studies of α1-adrenoceptor subtypes in rat heart. Br. J. Pharmacol. 1994, 111, 533–538. [Google Scholar] [CrossRef]
- Scofield, M.A.; Liu, F.; Abel, P.W.; Jeffries, W.B. Quantification of steady state expression of mRNA for α1-adrenergic receptor subtypes using reverse transcription and a competitive polymerase chain reaction. J. Pharmacol. Exp. Ther. 1995, 275, 1035–1042. [Google Scholar]
- Turnbull, L.; McCloskey, D.T.; O’Connell, T.D.; Simpson, P.C.; Baker, A.J. α1-adrenergic receptor responses in α1AB-AR knockout mouse hearts suggest the presence of α1D-AR. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1104–H1109. [Google Scholar] [CrossRef]
- Jensen, B.C.; Swigart, P.M.; De Marco, T.; Hoopes, C.; Simpson, P.C. α1-Adrenergic receptor subtypes in nonfailing and failing human myocardium. Circ. Heart Fail. 2009, 2, 654–663. [Google Scholar] [CrossRef]
- Methven, L.; Simpson, P.C.; McGrath, J.C. α1A/B-knockout mice explain the native α1D-adrenoceptor’s role in vasoconstriction and show that its location is independent of the other α1-subtypes. Br. J. Pharmacol. 2009, 158, 1663–1675. [Google Scholar] [CrossRef]
- Rokosh, D.G.; Simpson, P.C. Knockout of the α1A/C-adrenergic receptor subtype: The α1A/C is expressed in resistance arteries and is required to maintain arterial blood pressure. Proc. Natl. Acad. Sci. USA 2002, 99, 9474–9479. [Google Scholar] [CrossRef] [PubMed]
- Piascik, M.T.; Perez, D.M. α1-adrenergic receptors: New insights and directions. J. Pharmacol. Exp. Ther. 2001, 298, 403–410. [Google Scholar] [PubMed]
- Otani, H.; Otani, H.; Das, D.K. α1-adrenoceptor-mediated phosphoinositide breakdown and inotropic response in rat left ventricular papillary muscles. Circ. Res. 1988, 62, 8–17. [Google Scholar] [CrossRef]
- Pucéat, M.; Terzic, A.; Clément, O.; Scamps, F.; Vogel, S.M.; Vassort, G. Cardiac α1-adrenoceptors mediate positive inotropy via myofibrillar sensitization. Trends Pharmacol. Sci. 1992, 13, 263–265. [Google Scholar] [CrossRef]
- Endoh, M. Cardiac α1-Adrenoceptors and Inotropy: Myofilament Ca2+ Sensitivity, Intracellular Ca2+ Mobilization, Signaling Pathway, and Pathophysiological Relevance. Circ. Res. 2016, 119, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Venema, R.C.; Raynor, R.L.; Noland, T.A., Jr.; Kuo, J.F. Role of protein kinase C in the phosphorylation of cardiac myosin light chain. Biochem. J. 1993, 294, 401–406. [Google Scholar] [CrossRef]
- Snabaitis, A.K.; Yokoyama, H.; Avkiran, M. Roles of mitogen-activated protein kinases and protein kinase C in α1A-adrenoceptor-mediated stimulation of the sarcolemmal Na+-H+ exchanger. Circ. Res. 2000, 86, 214–220. [Google Scholar] [CrossRef]
- Yu, Z.Y.; Tan, J.C.; McMahon, A.C.; Iismaa, S.E.; Xiao, X.H.; Kesteven, S.H.; Reichelt, M.E.; Mohl, M.C.; Smith, N.J.; Fatkin, D.; et al. RhoA/ROCK signaling and pleiotropic α1A-adrenergic receptor regulation of cardiac contractility. PLoS ONE 2014, 9, e99024. [Google Scholar] [CrossRef]
- Taniguchi, M.; Okamoto, R.; Ito, M.; Goto, I.; Fujita, S.; Konishi, K.; Mizutani, H.; Dohi, K.; Hartshorne, D.J.; Itoh, T. New Isoform of Cardiac Myosin Light Chain Kinase and the Role of Cardiac Myosin Phosphorylation in α1-Adrenoceptor Mediated Inotropic Response. PLoS ONE 2015, 10, e0141130. [Google Scholar] [CrossRef]
- Endoh, M.; Hiramoto, T.; Ishihata, A.; Takanashi, M.; Inui, J. Myocardial α1-adrenoceptors mediate positive inotropic effect and changes in phosphatidylinositol metabolism. Species differences in receptor distribution and the intracellular coupling process in mammalian ventricular myocardium. Circ. Res. 1991, 68, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Owens, W.A.; Chen, S.; Stevens, M.E.; Kesteven, S.; Arthur, J.F.; Woodcock, E.A.; Feneley, M.P.; Graham, R.M. Targeted α1A-adrenergic receptor overexpression induces enhanced cardiac contractility but not hypertrophy. Circ. Res. 2001, 89, 343–350. [Google Scholar] [CrossRef]
- Janssen, P.M.L.; Canan, B.D.; Kilic, A.; Whitson, B.A.; Baker, A.J. Human Myocardium Has a Robust α1A-Subtype Adrenergic Receptor Inotropic Response. J. Cardiovasc. Pharmacol. 2018, 72, 136–142. [Google Scholar] [CrossRef]
- Ross, S.A.; Rorabaugh, B.R.; Chalothorn, D.; Yun, J.; Gonzalez-Cabrera, P.J.; McCune, D.F.; Piascik, M.T.; Perez, D.M. The α1B-adrenergic receptor decreases the inotropic response in the mouse Langendorff heart model. Cardiovasc. Res. 2003, 60, 598–607. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Uchi, J.; Sasaki, H.; Morimoto, S.; Kusakari, Y.; Shinji, H.; Obata, T.; Hongo, K.; Komukai, K.; Kurihara, S. Interaction of α1-adrenoceptor subtypes with different G proteins induces opposite effects on cardiac L-type Ca2+ channel. Circ. Res. 2008, 102, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- Cowley, P.M.; Wang, G.; Chang, A.N.; Makwana, O.; Swigart, P.M.; Lovett, D.H.; Stull, J.T.; Simpsom, P.C.; Baker, A.J. The α1A-adrenergic receptor subtype mediates increased contraction of failing right ventricular myocardium. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H888–H896. [Google Scholar] [CrossRef] [PubMed]
- Cowley, P.M.; Wang, G.; Joshi, S.; Swigart, P.M.; Lovett, D.H.; Simpson, P.C.; Baker, A.J. α1A-Subtype adrenergic agonist therapy for the failing right ventricle. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1109–H1118. [Google Scholar] [CrossRef] [PubMed]
- Cowley, P.M.; Wang, G.; Swigart, P.M.; Raghunathan, A.; Reddy, N.; Dulam, P.; Lovett, D.H.; Simpson, P.C.; Baker, A.J. Reversal of right ventricular failure by chronic α1A-subtype adrenergic agonist therapy. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H224–H232. [Google Scholar] [CrossRef] [PubMed]
- Myagmar, B.E.; Flynn, J.M.; Cowley, P.M.; Swigart, P.M.; Montgomery, M.D.; Thai, K.; Nair, D.; Gupta, R.; Deng, D.X.; Hosoda, C.; et al. Adrenergic Receptors in Individual Ventricular Myocytes: The β1 and α1B Are in All Cells, the α1A Is in a Subpopulation, and the β2 and β3 Are Mostly Absent. Circ. Res. 2017, 120, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Philipp, M.; Hein, L. Adrenergic receptor knockout mice: Distinct functions of 9 receptor subtypes. Pharmacol. Ther. 2004, 101, 65–74. [Google Scholar] [CrossRef]
- Hein, L.; Altman, J.D.; Kobilka, B.K. Two functionally distinct α2-adrenergic receptors regulate sympathetic neurotransmission. Nature. 1999, 402, 181–184. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenal adrenoceptors in heart failure: Fine-tuning cardiac stimulation. Trends Mol. Med. 2007, 13, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Brede, M.; Wiesmann, F.; Jahns, R.; Hadamek, K.; Arnolt, C.; Neubauer, S.; Lohse, M.J.; Hein, L. Feedback inhibition of catecholamine release by two different α2-adrenoceptor subtypes prevent progression of heart failure. Circulation 2002, 106, 2491–2496. [Google Scholar] [CrossRef] [PubMed]
- Bristow, M.R. Mechanistic and clinical rationales for using beta-blockers in heart failure. J. Card. Fail. 2000, 6, 8–14. [Google Scholar] [PubMed]
- Todd, G.L.; Baroldi, G.; Pieper, G.M.; Clayton, F.C.; Eliot, R.S. Experimental catecholamine-induced myocardial necrosis. I. Morphology, quantification and regional distribution of acute contraction band lesions. J. Mol. Cell. Cardiol. 1985, 17, 317–338. [Google Scholar] [CrossRef]
- Mann, D.L.; Kent, R.L.; Parsons, B.; Cooper, G., 4th. Adrenergic effects on the biology of the adult mammalian cardiocyte. Circulation 1992, 85, 790–804. [Google Scholar] [CrossRef]
- Communal, C.; Singh, K.; Pimentel, D.R.; Colucci, W.S. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the β-adrenergic pathway. Circulation 1998, 98, 1329–1334. [Google Scholar] [CrossRef] [PubMed]
- Felker, G.M.; O’Connor, C.M. Inotropic therapy for heart failure: An evidence-based approach. Am. Heart J. 2001, 142, 393–401. [Google Scholar] [CrossRef]
- Zaugg, M.; Xu, W.; Lucchinetti, E.; Shafiq, S.A.; Jamali, N.Z.; Siddiqui, M.A. β-adrenergic receptor subtypes differentially affect apoptosis in adult rat ventricular myocytes. Circulation 2000, 102, 344–350. [Google Scholar] [CrossRef]
- Ahmet, I.; Krawczyk, M.; Heller, P.; Moon, C.; Lakatta, E.G.; Talan, M.I. Beneficial effects of chronic pharmacological manipulation of β-adrenoreceptor subtype signaling in rodent dilated ischemic cardiomyopathy. Circulation 2004, 110, 1083–1090. [Google Scholar] [CrossRef]
- Ahmet, I.; Krawczyk, M.; Zhu, W.; Woo, A.Y.; Morrell, C.; Poosala, S.; Xiao, R.P.; Lakatta, E.G.; Talan, M.I. Cardioprotective and survival benefits of long-term combined therapy with β2 adrenoreceptor (AR) agonist and β1 AR blocker in dilated cardiomyopathy postmyocardial infarction. J. Pharmacol. Exp. Ther. 2008, 325, 491–499. [Google Scholar] [CrossRef]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. β2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Bristow, M.R.; Cohn, J.N. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. carvedilol heart failure study group. N. Engl. J. Med. 1996, 334, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Antonopoulos, G.V.; Berlin, J.A.; Chittams, J.; Konstam, M.A.; Udelson, J.E. Comparative effects of carvedilol and metoprolol on left ventricular ejection fraction in heart failure: Results of a meta-analysis. Am. Heart J. 2001, 141, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, E.; Lechat, P.; Verkenne, P.; Wiemann, H. Results from post-hoc analyses of the CIBIS II trial: Effect of bisoprolol in high-risk patient groups with chronic heart failure. Eur. J. Heart Fail. 2001, 3, 469–479. [Google Scholar]
- Leineweber, K.; Rohe, P.; Beilfuss, A.; Wolf, C.; Sporkmann, H.; Bruck, H.; Jakob, H.G.; Heusch, G.; Philipp, T.; Brodde, O.E. G-protein-coupled receptor kinase activity in human heart failure: Effects of β-adrenoceptor blockade. Cardiovasc. Res. 2005, 66, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Prieto, J.; Garcia-Ruiz, J.M.; Sanz-Rosa, D.; Pun, A.; Garcia-Alvarez, A.; Davidson, S.M.; Fernández-Friera, L.; Nuno-Ayala, M.; Fernández-Jiménez, R.; Bernal, J.A.; et al. β3-adrenergic receptor selective stimulation during ischemia/ reperfusion improves cardiac function in translational models through inhibition of mPTP opening in cardiomyocytes. Basic Res. Cardiol. 2014, 109, 422. [Google Scholar] [CrossRef]
- Cannavo, A.; Koch, W.J. Targeting β3-Adrenergic Receptors in the Heart: Selective Agonism and β-Blockade. J. Cardiovasc. Pharmacol. 2017, 69, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Bristow, M.R.; Ginsburg, R.; Minobe, W.; Cubicciotti, R.S.; Sageman, W.S.; Lurie, K.; Billingham, M.E.; Harrison, D.C.; Stinson, E.B. Decreased catecholamine sensitivity and β-adrenergic-receptor density in failing human hearts. N. Engl. J. Med. 1982, 307, 205–211. [Google Scholar] [CrossRef]
- Hwang, K.C.; Gray, C.D.; Sweet, W.E.; Moravec, C.S.; Im, M.J. α1-adrenergic receptor coupling with Gh in the failing human heart. Circulation 1996, 94, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Vago, T.; Bevilacqua, M.; Norbiato, G.; Baldi, G.; Chebat, E.; Bertora, P.; Baroldi, R.; Accinni, R. Identification of α1-adrenergic receptors on sarcolemma from normal subjects and patients with idiopathic dilated cardiomyopathy: Characteristics and linkage to GTP-binding protein. Circ. Res. 1989, 64, 474–481. [Google Scholar] [CrossRef]
- Grigore, A.; Poindexter, B.; Vaughn, W.K.; Nussmeier, N.; Frazier, O.H.; Cooper, J.R.; Gregoric, I.D.; Buja, L.M.; Bick, R.J. Alterations in α-adrenoreceptor density and localization after mechanical left ventricular unloading with the Jarvik flowmaker left ventricular assist device. J. Heart Lung Transpl. 2005, 24, 609–613. [Google Scholar] [CrossRef]
- Zhao, M.; Hagler, H.K.; Muntz, K.H. Regulation of α1-, β1-, and β2-adrenergic receptors in rat heart by norepinephrine. Am. J. Physiol. 1996, 271, H1762–H1768. [Google Scholar] [PubMed]
- Limas, C.J.; Limas, C.; Goldenberg, I.F. Intracellular distribution of adrenoceptors in the failing human myocardium. Am. Heart J. 1989, 117, 1310–1316. [Google Scholar] [CrossRef]
- Fischer, V.; Gabauer, I.; Tillinger, A.; Novakova, M.; Pechan, I.; Krizanova, O.; Myslivecek, J. Heart adrenoceptor gene expression and binding sites in the human failing heart. Ann. N. Y. Acad. Sci. 2008, 1148, 400–408. [Google Scholar] [CrossRef]
- Shi, T.; Moravec, C.S.; Perez, D.M. Novel proteins associated with human dilated cardiomyopathy: Selective reduction in α1A-adrenergic receptors and increased desensitization proteins. J. Recept. Signal. Transduct. Res. 2013, 33, 96–106. [Google Scholar] [CrossRef][Green Version]
- Corr, P.B.; Shayman, J.A.; Kramer, J.B.; Kipnis, R.J. Increased α-adrenergic receptors in ischemic cat myocardium: A potential mediator of electrophysiological derangements. J. Clin. Investig. 1981, 67, 1232–1236. [Google Scholar] [CrossRef]
- Maisel, A.S.; Motulsky, H.J.; Ziegler, M.G.; Insel, P.A. Ischemia- and agonist-induced changes in α- and β-adrenergic receptor traffic in guinea pig hearts. Am. J. Physiol. 1987, 253, H1159–H1166. [Google Scholar] [CrossRef]
- Butterfield, M.C.; Chess-Williams, R. Enhanced α-adrenoceptor responsiveness and receptor number during global ischaemia in the Langendorff perfused rat heart. Br. J. Pharmacol. 1990, 100, 641–645. [Google Scholar] [CrossRef]
- Itaya, T.; Hashimoto, H.; Satoh, R.; Uematsu, T.; Nakashima, M. Increases in α- but not β-adrenoceptors in hypertrophied non-infarcted cardiac muscles from rats with chronic myocardial infarction. Jpn. J. Pharmacol. 1990, 53, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Kurz, T.; Yamada, K.A.; DaTorre, S.D.; Corr, P.B. α1-adrenergic system and arrhythmias in ischaemic heart disease. Eur. Heart J. 1991, 12, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Eckhart, A.D.; Zhu, Z.; Arendshorst, W.J.; Faber, J.E. Oxygen modulates α1B-adrenergic receptor gene expression by arterial but not venous vascular smooth muscle. Am. J. Physiol. 1996, 271, H1599–H1608. [Google Scholar] [PubMed]
- Böhm, M.; Diet, F.; Feiler, G.; Kemkes, B.; Erdmann, E. α-adrenoceptors and α-adrenoceptor-mediated positive inotropic effects in failing human myocardium. J. Cardiovasc. Pharmacol. 1988, 12, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Skomedal, T.; Borthne, K.; Aass, H.; Geiran, O.; Osnes, J.B. Comparison between α1- adrenoceptor-mediated and β-adrenoceptor-mediated inotropic components elicited by norepinephrine in failing human ventricular muscle. J. Pharmacol. Exp. Ther. 1997, 280, 721–729. [Google Scholar]
- Sjaastad, I.; Schiander, I.; Sjetnan, A.; Qvigstad, E.; Bøkenes, J.; Sandnes, D.; Osnes, J.-B.; Sejersted, O.M.; Skomedal, T. Increased contribution of α1- vs. β-adrenoceptor-mediated inotropic response in rats with congestive heart failure. Acta Physiol. Scand. 2003, 177, 449–458. [Google Scholar] [CrossRef]
- Du, X.J.; Fang, L.; Gao, X.M.; Kiriazis, H.; Feng, X.; Hotchkin, E.; Finch, A.M.; Chaulet, H.; Graham, R.M. Genetic enhancement of ventricular contractility protects against pressure-overload-induced cardiac dysfunction. J. Mol. Cell Cardiol. 2004, 37, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Du, X.J.; Gao, X.M.; Kiriazis, H.; Moore, X.L.; Ming, Z.; Su, Y.; Finch, A.M.; Hannan, R.A.; Dart, A.M.; Graham, R.M. Transgenic α1A-adrenergic activation limits post-infarct ventricular remodeling and dysfunction and improves survival. Cardiovasc. Res. 2006, 71, 735–743. [Google Scholar] [CrossRef]
- Beak, J.; Huang, W.; Parker, J.S.; Hicks, S.T.; Patterson, C.; Simpson, P.C.; Ma, A.; Jin, J.; Jensen, B.C. An Oral Selective α1A-Adrenergic Receptor Agonist Prevents Doxorubicin Cardiotoxicity. JACC Basic Transl. Sci. 2017, 2, 39–53. [Google Scholar] [CrossRef]
- Montgomery, M.D.; Chan, T.; Swigart, P.M.; Myagmar, B.E.; Dash, R.; Simpson, P.C. An α1A-Adrenergic Receptor Agonist Prevents Acute Doxorubicin Cardiomyopathy in Male Mice. PLoS ONE 2017, 12, e0168409. [Google Scholar] [CrossRef]
- Akhter, S.A.; Milano, C.A.; Shotwell, K.F.; Cho, M.C.; Rockman, H.A.; Lefkowitz, R.J.; Koch, W.J. Transgenic mice with cardiac overexpression of α1B-adrenergic receptors. In vivo α1-adrenergic receptor-mediated regulation of β-adrenergic signaling. J. Biol. Chem. 1997, 272, 21253–21259. [Google Scholar] [CrossRef]
- Grupp, I.L.; Lorenz, J.N.; Walsh, R.A.; Boivin, G.P.; Rindt, H. Overexpression of α1B-adrenergic receptor induces left ventricular dysfunction in the absence of hypertrophy. Am. J. Physiol. 1998, 275, H1338–H1350. [Google Scholar] [CrossRef]
- Lemire, I.; Ducharme, A.; Tardif, J.C.; Poulin, F.; Jones, L.R.; Allen, B.G.; Hebert, T.E.; Rindt, H. Cardiac-directed overexpression of wild-type α1B-adrenergic receptor induces dilated cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H931–H938. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.H.; Du, X.J.; Autelitano, D.J.; Milano, C.A.; Woodcock, E.A. Adverse effects of constitutively active α1B-adrenergic receptors after pressure overload in mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H1079–H1086. [Google Scholar] [CrossRef]
- Elia, M. Organ and tissue contribution to metabolic rate. In Energy Metabolism: Tissue Determinants and Cellular Corollaries; Kinney, J.M., Tucker, H.N., Eds.; Raven Press: New York, NY, USA, 1992; pp. 61–80. [Google Scholar]
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef]
- Olson, R.E.; Schwartz, W.B. Myocardial metabolism in congestive heart failure. Medicine 1951, 30, 21–41. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.L.; Wang, C.H.; Shiao, M.S.; Liu, M.H.; Huang, Y.Y.; Huang, C.Y.; Mao, C.-T.; Lin, J.-F.; Ho, H.-Y.; Yang, N.-I. Metabolic disturbances identified in plasma are associated with outcomes in patients with heart failure: Diagnostic and prognostic value of metabolomics. J. Am. Coll. Cardiol. 2015, 65, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Peoples, J.; Maxmillian, T.; Le, Q.; Nadtochiy, S.M.; Brookes, P.S.; Porter, G.A., Jr.; Davidson, V.L.; Ebert, S.N. Metabolomics reveals critical adrenergic regulatory checkpoints in glycolysis and pentose-phosphate pathways in embryonic heart. J. Biol. Chem. 2018, 293, 6925–6941. [Google Scholar] [CrossRef]
- Becker, C.; Sevilla, L.; Tomas, E.; Palacin, M.; Zorzano, A.; Fischer, Y. The endosomal compartment is an insulin-sensitive recruitment site for GLUT4 and GLUT1 glucose transporters in cardiac myocytes. Endocrinology 2001, 142, 5267–5276. [Google Scholar] [CrossRef]
- Depré, C.; Rider, M.H.; Hue, L. Mechanisms of control of heart glycolysis. Eur. J. Biochem. 1998, 258, 277–290. [Google Scholar] [CrossRef]
- Stanton, R.C. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. Iubmb Life 2012, 64, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Brenner, D.A.; Cui, L.; Lim, C.C.; Wang, B.; Pimentel, D.R.; Koh, S.; Sawyer, D.B.; Leopold, J.A.; Handy, D.E.; et al. Glucose-6-phosphate dehydrogenase modulates cytosolic redox status and contractile phenotype in adult cardiomyocytes. Circ. Res. 2003, 93, e9–e16. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- Turrens, J.F. Superoxide production by the mitochondrial respiratory chain. Biosci. Rep. 1997, 17, 3–8. [Google Scholar] [CrossRef]
- Viola, H.M.; Hool, L.C. Qo site of mitochondrial complex III is the source of increased superoxide after transient exposure to hydrogen peroxide. J. Mol. Cell. Cardiol. 2010, 49, 875–885. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Belch, J.J.; Bridges, A.B.; Scott, N.; Chopra, M. Oxygen free radicals and congestive heart failure. Br. Heart J. 1991, 65, 245–248. [Google Scholar] [CrossRef]
- Hill, M.F.; Singal, P.K. Right and left myocardial antioxidant responses during heart failure subsequent to myocardial infarction. Circulation 1997, 96, 2414–2420. [Google Scholar] [CrossRef]
- Mallat, Z.; Philip, I.; Lebret, M.; Chatel, D.; Maclouf, J.; Tedgui, A. Elevated levels of 8-iso-prostaglandin F2alpha in pericardial fluid of patients with heart failure: A potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation 1998, 97, 1536–1539. [Google Scholar] [CrossRef]
- Nakamura, K.; Kusano, K.; Nakamura, Y.; Kakishita, M.; Ohta, K.; Nagase, S.; Yamamoto, M.; Miyaji, K.; Saito, H.; Morita, H.; et al. Carvedilol decreases elevated oxidative stress in human failing myocardium. Circulation 2002, 105, 2867–2871. [Google Scholar] [CrossRef]
- Sam, F.; Kerstetter, D.L.; Pimental, D.R.; Mulukutla, S.; Tabaee, A.; Bristow, M.R.; Colucci, W.S.; Sawyer, D.B. Increased reactive oxygen species production and functional alterations in antioxidant enzymes in human failing myocardium. J. Card. Fail. 2005, 11, 473–480. [Google Scholar] [CrossRef]
- Sheeran, F.L.; Pepe, S. Posttranslational modifications and dysfunction of mitochondrial enzymes in human heart failure. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E449–E460. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Cui, L.; Brenner, D.A.; Wang, B.; Handy, D.E.; Leopold, J.A.; Loscalzo, J.; Apstein, C.S.; Liao, R. Increased myocardial dysfunction after ischemia-reperfusion in mice lacking glucose-6-phosphate dehydrogenase. Circulation 2004, 109, 898–903. [Google Scholar] [CrossRef]
- Long, W.K.; Wilson, S.W.; Frenkel, E.P. Associations between red cell glucose-6-phosphate dehydrogenase variants and vascular diseases. Am. J. Hum. Genet. 1967, 19, 35–53. [Google Scholar] [PubMed]
- Hecker, P.A.; Lionetti, V.; Ribeiro, R.F., Jr.; Rastogi, S.; Brown, B.H.; O’Connell, K.A.; Cox, J.W.; Shekar, K.C.; Gamble, D.M.; Sabbah, H.N.; et al. Glucose 6-phosphate dehydrogenase deficiency increases redox stress and moderately accelerates the development of heart failure. Circ. Heart Fail. 2013, 6, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Badolia, R.; Ramadurai, D.K.A.; Abel, E.D.; Ferrin, P.; Taleb, I.; Shankar, T.S.; Krokidi, A.T.; Navankasattusas, S.; McKellar, S.H.; Yin, M.; et al. The Role of Nonglycolytic Glucose Metabolism in Myocardial Recovery Upon Mechanical Unloading and Circulatory Support in Chronic Heart Failure. Circulation 2020, 142, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Diakos, N.A.; Navankasattusas, S.; Abel, E.D.; Rutter, J.; McCreath, L.; Ferrin, P.; McKellar, S.H.; Miller, D.V.; Park, S.Y.; Richardson, R.S.; et al. Evidence of Glycolysis Up-Regulation and Pyruvate Mitochondrial Oxidation Mismatch During Mechanical Unloading of the Failing Human Heart: Implications for Cardiac Reloading and Conditioning. JACC Basic Transl. Sci. 2016, 1, 432–444. [Google Scholar] [CrossRef]
- Lei, B.; Lionetti, V.; Young, M.E.; Chandler, M.P.; d’Agostino, C.; Kang, E.; Altarejos, M.; Matsuo, K.; Hintze, T.H.; Stanley, W.C.; et al. Paradoxical downregulation of the glucose oxidation pathway despite enhanced flux in severe heart failure. J. Mol. Cell. Cardiol. 2004, 36, 567–576. [Google Scholar] [CrossRef]
- Razeghi, P.; Young, M.E.; Alcorn, J.L.; Moravec, C.S.; Frazier, O.H.; Taegtmeyer, H. Metabolic gene expression in fetal and failing human heart. Circulation 2001, 104, 2923–2931. [Google Scholar] [CrossRef]
- Gupte, S.A.; Levine, R.J.; Gupte, R.S.; Young, M.E.; Lionetti, V.; Labinskyy, V.; Floyd, B.C.; Ojaimi, C.; Bellomo, M.; Wolin, M.S.; et al. Glucose-6-phosphate dehydrogenase-derived NADPH fuels superoxide production in the failing heart. J. Mol. Cell. Cardiol. 2006, 41, 340–349. [Google Scholar] [CrossRef]
- Gupte, R.S.; Vijay, V.; Marks, B.; Levine, R.J.; Sabbah, H.N.; Wolin, M.S.; Recchia, F.A.; Gupte, S.A. Upregulation of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase activity increases oxidative stress in failing human heart. J. Card. Fail. 2007, 13, 497–506. [Google Scholar] [CrossRef]
- Serpillon, S.; Floyd, B.C.; Gupte, R.S.; George, S.; Kozicky, M.; Neito, V.; Recchia, F.; Stanley, W.; Wolin, M.S.; Gupte, S.A. Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose-6-phosphate dehydrogenase-derived NADPH. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H153–H162. [Google Scholar] [CrossRef]
- Vimercati, C.; Qanud, K.; Mitacchione, G.; Sosnowska, D.; Ungvari, Z.; Sarnari, R.; Mania, D.; Patel, N.; Hintze, T.H.; Gupte, S.; et al. Beneficial effects of acute inhibition of the oxidative pentose phosphate pathway in the failing heart. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H709–H717. [Google Scholar] [CrossRef]
- Cocco, P.; Todde, P.; Fornera, S.; Manca, M.B.; Manca, P.; Sias, A.R. Mortality in a cohort of men expressing the glucose-6-phosphate dehydrogenase deficiency. Blood 1998, 91, 706–709. [Google Scholar] [CrossRef]
- Meloni, L.; Manca, M.R.; Loddo, I.; Cioglia, G.; Cocco, P.; Schwartz, A.; Muntoni, S.; Muntoni, S. Glucose-6-phosphate dehydrogenase deficiency protects against coronary heart disease. J. Inherit. Metab. Dis. 2008, 31, 412–417. [Google Scholar] [CrossRef]
- Sansbury, B.E.; DeMartino, A.M.; Xie, Z.; Brooks, A.C.; Brainard, R.E.; Watson, L.J.; DeFilippis, A.P.; Cummins, T.D.; Harbeson, M.A.; Brittian, K.R.; et al. Metabolomic analysis of pressure-overloaded and infarcted mouse hearts. Circ. Heart Fail. 2014, 7, 634–642. [Google Scholar] [CrossRef]
- Contaifer, D., Jr.; Buckley, L.F.; Wohlford, G.; Kumar, N.G.; Morriss, J.M.; Ranasinghe, A.D.; Carbone, S.; Canada, J.M.; Trankle, C.; Abbate, A.; et al. Metabolic modulation predicts heart failure tests performance. PLoS ONE 2019, 14, e0218153. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization Working Group. Glucose-6-phosphate dehydrogenase deficiency. WHO Working Group. Bull. World Health Organ. 1989, 67, 601–611. [Google Scholar]
- Cappellini, M.D.; Fiorelli, G. Glucose-6-phosphate dehydrogenase deficiency. Lancet 2008, 371, 64–74. [Google Scholar] [CrossRef]
- Luzzatto, L. Glucose-6-phosphate dehydrogenase deficiency. Advanced Medicine-Twelve. In Proceedings of the Conference Held at the Royal College of Physicians of London, Churchill Livingstone, UK, 11–14 February 1986. [Google Scholar]
- Opie, L.H. Metabolism of the heart in health and disease. I. Am. Heart J. 1968, 76, 685–698. [Google Scholar] [CrossRef]
- Opie, L.H. Metabolism of the heart in health and disease. II. Am. Heart J. 1969, 77, 100–122. [Google Scholar] [CrossRef]
- Neely, J.R.; Morgan, H.E. Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Annu. Rev. Physiol. 1974, 36, 413–459. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef] [PubMed]
- Koonen, D.P.; Glatz, J.F.; Bonen, A.; Luiken, J.J. Long-chain fatty acid uptake and FAT/CD36 translocation in heart and skeletal muscle. Biochim. Biophys. Acta 2005, 3, 163–180. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Brown, N.F. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem. 1997, 244, 1–14. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Gandour, R.D.; van der Leij, F.R. Molecular enzymology of carnitine transfer and transport. Biochim. Biophys. Acta 2001, 1546, 21–43. [Google Scholar] [CrossRef]
- Conway, M.A.; Allis, J.; Ouwerkerk, R.; Niioka, T.; Rajagopalan, B.; Radda, G.K. Detection of low phosphocreatine to ATP ratio in failing hypertrophied human myocardium by 31P magnetic resonance spectroscopy. Lancet 1991, 338, 973–976. [Google Scholar] [CrossRef]
- Beer, M.; Seyfarth, T.; Sandstede, J.; Landschütz, W.; Lipke, C.; Köstler, H.; von Kienlin, M.; Harre, K.; Hahn, D.; Neubauer, S. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J. Am. Coll. Cardiol. 2002, 40, 1267–1274. [Google Scholar] [CrossRef]
- Tian, R.; Nascimben, L.; Kaddurah-Daouk, R.; Ingwall, J.S. Depletion of energy reserve via the creatine kinase reaction during the evolution of heart failure in cardiomyopathic hamsters. J. Mol. Cell. Cardiol. 1996, 28, 755–765. [Google Scholar] [CrossRef]
- Piacentino, V., III; Weber, C.R.; Chen, X.; Weisser-Thomas, J.; Margulies, K.B.; Bers, D.M.; Houser, S.R. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ. Res. 2003, 92, 651–658. [Google Scholar] [CrossRef]
- Noland, R.C.; Koves, T.R.; Seiler, S.E.; Lum, H.; Lust, R.M.; Ilkayeva, O.; Stevens, R.D.; Hegardt, F.G.; Muoio, D.M. Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. J. Biol. Chem. 2009, 284, 22840–22852. [Google Scholar] [CrossRef]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.; Newgard, C.B.; et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell. Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Akhmedov, A.T.; Rybin, V.; Marin-Garcia, J. Mitochondrial oxidative metabolism and uncoupling proteins in the failing heart. Heart Fail. Rev. 2015, 20, 227–249. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, B.A.; Zhao, X.; Heidenreich, P.A.; Peterson, E.D.; Bhatt, D.L.; Cannon, C.P.; Hernandez, A.F.; Fonarow, G.C.; Get With the Guidelines Scientific Advisory Committee and Investigators. Trends in patients hospitalized with heart failure and preserved left ventricular ejection fraction: Prevalence, therapies, and outcomes. Circulation 2012, 126, 65–75. [Google Scholar] [CrossRef]
- Li, X. SIRT1 and energy metabolism. Acta Biochim. Biophys. Sin. 2013, 45, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W.; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef]
- Lu, T.M.; Tsai, J.Y.; Chen, Y.C.; Huang, C.Y.; Hsu, H.L.; Weng, C.F.; Shih, C.C.; Hsu, C.P. Downregulation of Sirt1 as aging change in advanced heart failure. J. Biomed. Sci. 2014, 21, 57. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Nakashima, R.; Yuan, Q.; Marks, A.R. Intracellular calcium release channels: An update. J. Physiol. 2017, 595, 3041–3051. [Google Scholar] [CrossRef]
- Bugger, H.; Schwarzer, M.; Chen, D.; Schrepper, A.; Amorim, P.A.; Schoepe, M.; Nguyen, T.D.; Mohr, F.W.; Khalimonchuk, O.; Weimer, B.C.; et al. Proteomic remodelling of mitochondrial oxidative pathways in pressure overload-induced heart failure. Cardiovasc. Res. 2010, 85, 376–384. [Google Scholar] [CrossRef]
- Doenst, T.; Pytel, G.; Schrepper, A.; Amorim, P.; Farber, G.; Shingu, Y.; Mohr, F.W.; Schwarzer, M. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc. Res. 2010, 86, 461–470. [Google Scholar] [CrossRef]
- Osorio, J.C.; Stanley, W.C.; Linke, A.; Castellari, M.; Diep, Q.N.; Panchal, A.R.; Hintze, T.H.; Lopaschuk, G.D.; Recchia, F.A. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor-alpha in pacing-induced heart failure. Circulation 2002, 106, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Qanud, K.; Mamdani, M.; Pepe, M.; Khairallah, R.J.; Gravel, J.; Lei, B.; Gupte, S.A.; Sharov, V.G.; Sabbah, H.N.; Stanley, W.C.; et al. Reverse changes in cardiac substrate oxidation in dogs recovering from heart failure. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2098–H2105. [Google Scholar] [CrossRef][Green Version]
- Neglia, D.; De Caterina, A.; Marraccini, P.; Natali, A.; Ciardetti, M.; Vecoli, C.; Gastaldelli, A.; Ciociaro, D.; Pellegrini, P.; Testa, R.; et al. Impaired myocardial metabolic reserve and substrate selection flexibility during stress in patients with idiopathic dilated cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3270–H3278. [Google Scholar] [CrossRef]
- Ingwall, J.S. On substrate selection for ATP synthesis in the failing human myocardium. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3225–H3226. [Google Scholar] [CrossRef] [PubMed]
- Zordoky, B.N.; Sung, M.M.; Ezekowitz, J.; Mandal, R.; Han, B.; Bjorndahl, T.C.; Bouatra, S.; Anderson, T.; Oudit, G.Y.; Wishart, D.S.; et al. Metabolomic fingerprint of heart failure with preserved ejection fraction. PLoS ONE 2015, 10, e0124844. [Google Scholar]
- Hunter, W.G.; Kelly, J.P.; McGarrah, R.W., III; Khouri, M.G.; Craig, D.; Haynes, C.; Ilkayeva, O.; Stevens, R.D.; Bain, J.R.; Muehlbauer, M.J.; et al. Metabolomic Profiling Identifies Novel Circulating Biomarkers of Mitochondrial Dysfunction Differentially Elevated in Heart Failure with Preserved Versus Reduced Ejection Fraction: Evidence for Shared Metabolic Impairments in Clinical Heart Failure. J. Am. Heart Assoc. 2016, 5, e003190. [Google Scholar] [CrossRef] [PubMed]
- De Jong, K.A.; Lopaschuk, G.D. Complex Energy Metabolic Changes in Heart Failure with Preserved Ejection Fraction and Heart Failure with Reduced Ejection Fraction. Can. J. Cardiol. 2017, 33, 860–871. [Google Scholar] [CrossRef]
- Senni, M.; Redfield, M.M. Heart failure with preserved systolic function: A different natural history? J. Am. Coll. Cardiol. 2001, 38, 1277–1282. [Google Scholar] [CrossRef]
- Hogg, K.; Swedberg, K.; McMurray, J. Heart failure with preserved left ventricular systolic function; epidemiology, clinical characteristics, and prognosis. J. Am. Coll. Cardiol. 2004, 43, 317–327. [Google Scholar] [CrossRef]
- Owan, T.E.; Hodge, D.O.; Herges, R.M.; Jacobsen, S.J.; Roger, V.L.; Redfield, M.M. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N. Engl. J. Med. 2006, 355, 251–259. [Google Scholar] [CrossRef]
- Davila-Roman, V.G.; Vedala, G.; Herrero, P.; de las Fuentes, L.; Rogers, J.G.; Kelly, D.P.; Gropler, R.J. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 2002, 40, 271–277. [Google Scholar] [CrossRef]
- Tuunanen, H.; Engblom, E.; Naum, A.; Någren, K.; Hesse, B.; Airaksinen, K.E.; Nuutila, P.; Iozzo, P.; Ukkonen, H.; Opie, L.H.; et al. Free fatty acid depletion acutely decreases cardiac work and efficiency in cardiomyopathic heart failure. Circulation 2006, 114, 2130–2137. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Niizuma, S.; Inuzuka, Y.; Kawashima, T.; Okuda, J.; Tamaki, Y.; Iwanaga, Y.; Narazaki, M.; Matsuda, T.; Soga, T.; et al. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ. Heart Fail. 2010, 3, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Christe, M.E.; Rodgers, R.L. Cardiac glucose and fatty acid oxidation in the streptozotocin-induced diabetic spontaneously hypertensive rat. Hypertension 1995, 25, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.D.; Kaulbach, H.C.; Tian, R.; Hopkins, J.C.; Duffy, J.; Doetschman, T.; Minnemann, T.; Boers, M.E.; Hadro, E.; Oberste-Berghaus, C.; et al. Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J. Clin. Investig. 1999, 104, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Folmes, C.D.; Stanley, W.C. Cardiac energy metabolism in obesity. Circ. Res. 2007, 101, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Mori, J.; Basu, R.; McLean, B.A.; Das, S.K.; Zhang, L.; Patel, V.B.; Wagg, C.S.; Kassiri, Z.; Lopaschuk, G.D.; Oudit, G.Y. Agonist-induced hypertrophy and diastolic dysfunction are associated with selective reduction in glucose oxidation. A metabolic contribution to heart failure with normal ejection fraction. Circ. Heart Fail. 2012, 5, 493–503. [Google Scholar] [CrossRef]
- Mori, J.; Alrob, O.A.; Wagg, C.S.; Harris, R.A.; Lopaschuk, G.D.; Oudit, G.Y. ANG II causes insulin resistance and induces cardiac metabolic switch and inefficiency: A critical role of Pdkam. J. Physiol. Heart Circ. Physiol. 2013, 304, H1103–H1113. [Google Scholar] [CrossRef]
- Zhang, L.; Jaswal, J.S.; Ussher, J.R.; Sankaralingam, S.; Wagg, L.; Zaugg, M.; Lopaschuk, G.D. Cardiac insulin-resistance and decreased mitochondrial energy production precede the development of systolic heart failure after pressure-overload hypertrophy. Circ. Heart Fail. 2013, 6, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Sankaralingam, S.; Abo Alrob, O.; Zhang, L.; Jaswal, J.S.; Wagg, C.S.; Fukushima, A.; Padwal, R.S.; Johnstone, D.E.; Sharma, A.M.; Lopaschuk, G.D. Lowering body weight in obese mice with diastolic heart failure improves cardiac insulin sensitivity and function: Implications for the obesity paradox. Diabetes 2015, 64, 1643–1657. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, V.; Stanley, W.C.; Recchia, F.A. Modulating fatty acid oxidation in heart failure. Cardiovasc. Res. 2011, 90, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, N.; Mori, J.; Lopaschuk, G.D. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 2014, 171, 2080–2090. [Google Scholar] [CrossRef] [PubMed]
- Sorokina, N.; O’Donnell, J.M.; McKinney, R.D.; Pound, K.M.; Woldegiorgis, G.; LaNoue, K.F.; Ballal, K.; Taegtmeyer, H.; Buttrick, P.M.; Lewandowski, E.D. Recruitment of compensatory pathways to sustain oxidative flux with reduced carnitine palmitoyltransferase I activity characterizes inefficiency in energy metabolism in hypertrophied hearts. Circulation 2007, 115, 2033–2041. [Google Scholar] [CrossRef]
- Jarreta, D.; Orus, J.; Barrientos, A.; Miro, O.; Roig, E.; Heras, M.; Moraes, C.T.; Cardellach, F.; Casademont, J. Mitochondrial function in heart muscle from patients with idiopathic dilated cardiomyopathy. Cardiovasc. Res. 2000, 45, 860–865. [Google Scholar] [CrossRef]
- Quigley, A.F.; Kapsa, R.M.; Esmore, D.; Hale, G.; Byrne, E. Mitochondrial respiratory chain activity in idiopathic dilated cardiomyopathy. J. Card. Fail. 2000, 6, 47–55. [Google Scholar] [CrossRef]
- Sheeran, F.L.; Pepe, S. Energy deficiency in the failing heart: Linking increased reactive oxygen species and disruption of oxidative phosphorylation rate. Biochim. Biophys. Acta 2006, 1757, 543–552. [Google Scholar] [CrossRef]
- Scheubel, R.J.; Tostlebe, M.; Simm, A.; Rohrbach, S.; Prondzinsky, R.; Gellerich, F.N.; Silber, R.E.; Holtz, J. Dysfunction of mitochondrial respiratory chain complex I in human failing myocardium is not due to disturbed mitochondrial gene expression. J. Am. Coll. Cardiol. 2002, 40, 2174–2181. [Google Scholar] [CrossRef]
- Kjekshus, J.K.; Mjos, O.D. Effect WI inhibition of lipolysis on myocardial oxygen consumption in the presence of isoproterenol. J. Clin. Investig. 1972, 51, 1767–1776. [Google Scholar] [CrossRef]
- Mjos, O.D. Effect of inhibition of lipolysis on myocardial oxygen consumption in the presence of isoproterenol. J. Clin. Investig. 1971, 50, 1869–1873. [Google Scholar] [CrossRef]
- Liu, B.; Clanachan, A.S.; Schulz, R.; Lopaschuk, G.D. Cardiac efficiency is improved after ischemia by altering both the source and fate of protons. Circ. Res. 1996, 79, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Docherty, J.C.; Rendell, J.C.T.; Clanachan, A.S.; Lopaschuk, G.D. High levels of fatty acids delay the recovery of intracellular pH and cardiac efficiency in post-ischemic hearts by inhibiting glucose oxidation. J. Am. Coll. Cardiol. 2002, 39, 718–725. [Google Scholar] [CrossRef]
- Folmes, C.D.; Clanachan, A.S.; Lopaschuk, G.D. Fatty acids attenuate insulin regulation of 5′-AMP-activated protein kinase and insulin cardioprotection after ischemia. Circ. Res. 2006, 99, 61–68. [Google Scholar] [CrossRef]
- Vogel, S.; Sperelakis, N. Blockade of myocardial slow inward current at low pH. Am. J. Phys. 1977, 233, C99–C103. [Google Scholar] [CrossRef]
- Steenbergen, C.; Deleeuw, G.; Rich, T.; Williamson, J.R. Effects of acidosis and ischemia on contractility and intracellular pH of rat heart. Circ. Res. 1977, 41, 849–858. [Google Scholar] [CrossRef]
- Beanlands, R.S.B.; Armstrong, W.F.; Hicks, R.J.; Nicklas, J.; Moore, C.; Hutchins, G.D.; Wolpers, H.G.; Schwaiger, M. The effects of afterload reduction on myocardial carbon 11-labeled acetate kinetics and noninvasively estimated mechanical efficiency in patients with dilated cardiomyopathy. J. Nucl. Cardiol. 1994, 1, 3–16. [Google Scholar] [CrossRef]
- Masoud, W.G.; Ussher, J.R.; Wang, W.; Jaswal, J.S.; Wagg, C.S.; Dyck, J.R.; Lygate, C.A.; Neubauer, S.; Clanachan, A.S.; Lopaschuk, G.D. Failing mouse hearts utilize energy inefficiently and benefit from improved coupling of glycolysis and glucose oxidation. Cardiovasc. Res. 2014, 101, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Barr, R.; Thomas, P.D.; Dyck, J.R. Beneficial effects of trimetazidine in ex vivo working ischemic hearts are due to a stimulation of glucose oxidation secondary to inhibition of long-chain 3-ketoacyl coenzyme a thiolase. Circ. Res. 2003, 93, e33–e37. [Google Scholar] [CrossRef] [PubMed]
- Dyck, J.R.; Cheng, J.F.; Stanley, W.C.; Barr, R.; Chandler, M.P.; Brown, S.; Wallace, D.; Arrhenius, T.; Harmon, C.; Yang, G.; et al. Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ. Res. 2004, 94, e78–e84. [Google Scholar] [CrossRef] [PubMed]
- Dyck, J.R.; Hopkins, T.A.; Bonnet, S.; Michelakis, E.D.; Young, M.E.; Watanabe, M.; Kawase, Y.; Jishage, K.; Lopaschuk, G.D. Absence of malonyl coenzyme A decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation 2006, 114, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Ussher, J.R.; Wang, W.; Gandhi, M.; Keung, W.; Samokhvalov, V.; Oka, T.; Wagg, C.S.; Jaswal, J.S.; Harris, R.A.; Clanachan, A.S.; et al. Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovasc. Res. 2012, 94, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, A.; Milner, K.; Gupta, A.; Lopaschuk, G.D. Myocardial Energy Substrate Metabolism in Heart Failure: From Pathways to Therapeutic Targets. Curr. Pharm. Des. 2015, 21, 3654–3664. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, N.; Levasseur, J.L.; Fukushima, A.; Wagg, C.S.; Wang, W.; Dyck, J.R.B.; Lopaschuk, G.D. Uncoupling of glycolysis from glucose oxidation accompanies the development of heart failure with preserved ejection fraction. Mol. Med. 2018, 24, 3. [Google Scholar] [CrossRef]
- Li, T.; Xu, J.; Qin, X.; Hou, Z.; Guo, Y.; Liu, Z.; Wu, J.; Zheng, H.; Zhang, X.; Gao, F. Glucose oxidation positively regulates glucose uptake and improves cardiac function recovery after myocardial reperfusion. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E577–E585. [Google Scholar] [CrossRef]
- Wargovich, T.J.; MacDonald, R.G.; Hill, J.A.; Feldman, R.L.; Stacpoole, P.W.; Pepine, C.J. Myocardial metabolic and hemodynamic effects of dichloroacetate in coronary artery disease. Am. J. Cardiol. 1988, 61, 65–70. [Google Scholar] [CrossRef]
- McGarry, J.D.; Takabayashi, Y.; Foster, D.W. The role of malonyl-CoA in the coordination of fatty acid synthesis and oxidation in isolated rat hepatocytes. J. Biol. Chem. 1978, 253, 8294–8300. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Wal, l.S.R.; Olley, P.M.; Davies, N.J. Etomoxir, a carnitine palmitoyltransferase I inhibitor, protects hearts from fatty acid-induced ischemic injury independent of changes in long chain acylcarnitine. Circ. Res. 1988, 63, 1036–1043. [Google Scholar] [CrossRef]
- Wall, S.R.; Lopaschuk, G.D. Glucose oxidation rates in fatty acid-perfused isolated working hearts from diabetic rats. Biochim. Biophys. Acta 1989, 1006, 97–103. [Google Scholar] [CrossRef]
- Schmidt-Schweda, S.; Holubarsch, C. First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin. Sci. 2000, 99, 27–35. [Google Scholar] [CrossRef]
- Lee, L.; Campbell, R.; Scheuermann-Freestone, M.; Taylor, R.; Gunaruwan, P.; Williams, L.; Ashrafian, H.; Horowitz, J.; Fraser, A.G.; Clarke, K.; et al. Metabolic modulation with perhexiline in chronic heart failure: A randomized, controlled trial of short-term use of a novel treatment. Circulation 2005, 112, 3280–3288. [Google Scholar] [CrossRef] [PubMed]
- Holubarsch, C.J.; Rohrbach, M.; Karrasch, M.; Boehm, E.; Polonski, L.; Ponikowski, P.; Rhein, S. A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: The ERGO (Etomoxir for the Recovery of Glucose Oxidation) study. Clin. Sci. 2007, 113, 205–212. [Google Scholar] [CrossRef]
- Abozguia, K.; Elliott, P.; McKenna, W.; Phan, T.T.; Nallur-Shivu, G.; Ahmed, I.; Maher, A.R.; Kaur, K.; Taylor, J.; Henning, A.; et al. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation 2010, 122, 1562–1569. [Google Scholar] [CrossRef]
- Fragasso, G.; Palloshi, A.; Puccetti, P.; Silipigni, C.; Rossodivita, A.; Pala, M.; Calori, G.; Alfieri, O.; Margonato, A. A randomized clinical trial of trimetazidine, a partial free fatty acid oxidation inhibitor, in patients with heart failure. J. Am. Coll. Cardiol. 2006, 48, 992–998. [Google Scholar] [CrossRef]
- Tuunanen, H.; Engblom, E.; Naum, A.; Nagren, K.; Scheinin, M.; Hesse, B.; Juhani Airaksinen, K.E.; Nuutila, P.; Iozzo, P.; Ukkonen, H.; et al. Trimetazidine, a metabolic modulator, has cardiac and extracardiac benefits in idiopathic dilated cardiomyopathy. Circulation 2008, 118, 1250–1258. [Google Scholar] [CrossRef]
- Gao, D.; Ning, N.; Niu, X.; Hao, G.; Meng, Z. Trimetazidine: A meta-analysis of randomized controlled trials in heart failure. Heart 2011, 97, 278–286. [Google Scholar] [CrossRef] [PubMed]
- El Alaoui-Talibi, Z.; Landormy, S.; Loireau, A.; Moravec, J. Fatty acid oxidation and mechanical performance of volume-overloaded rat hearts. Am. J. Physiol. 1992, 262, H1068–H1074. [Google Scholar] [CrossRef] [PubMed]
- El Alaoui-Talibi, Z.; Moravec, J. Carnitine transport and exogenous palmitate oxidation in chronically volume-overloaded rat hearts. Biochim. Biophys. Acta 1989, 1003, 109–114. [Google Scholar] [CrossRef]
- Ruiz, M.; Labarthe, F.; Fortier, A.; Bouchard, B.; Legault Thompson, J.; Bolduc, V.; Rigal, O.; Chen, J.; Ducharme, A.; Crawford, P.A.; et al. Circulating acylcarnitine profile in human heart failure: A surrogate of fatty acid metabolic dysregulation in mitochondria and beyond. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H768–H781. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.Y.; Quaife, C.J.; Palmiter, R.D. Targeted disruption of the tyrosine hydroxylase gene reveals that catecholamines are required for mouse fetal development. Nature 1995, 374, 640–643. [Google Scholar] [CrossRef]
- Baker, C.N.; Gidus, S.A.; Price, G.F.; Peoples, J.N.; Ebert, S.N. Impaired cardiac energy metabolism in embryos lacking adrenergic stimulation. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E402–E413. [Google Scholar] [CrossRef]
- Bao, X.; Lu, C.M.; Liu, F.; Gu, Y.; Dalton, N.D.; Zhu, B.Q.; Foster, E.; Chen, J.; Karliner, J.S.; Ross, J.; et al. Epinephrine is required for normal cardiovascular responses to stress in the phenylethanolamine N-methyltransferase knockout mouse. Circulation 2007, 116, 1024–1031. [Google Scholar] [CrossRef]
- Ebert, S.N.; Rong, Q.; Boe, S.; Pfeifer, K. Catecholamine-synthesizing cells in the embryonic mouse heart. Ann. N. Y. Acad. Sci. 2008, 1148, 317–324. [Google Scholar] [CrossRef]
- Goutis, A.; Felts, J.M. Effects of epinephrine, norepinephrine, glucose and insulin on extraction and oxidation of free fatty acid by myocardium. Circulation 1963, 28, 729. [Google Scholar]
- Gold, M.; Atlas, H.J.; Scott, J.C.; Spitzen, J.J. Effect of norepinephrine on myocardial free fatty acid uptake and oxidation. Proc. Sot. Exp. Biol. Med. 1965, 118, 876–879. [Google Scholar] [CrossRef]
- Crass, M.F., III; Shipp, J.C.; Pieper, G.M. Effects of catecholamines on myocardial endogenous substrates and contractility. Am. J. Physiol. 1975, 228, 618–627. [Google Scholar] [CrossRef]
- Murthy, V.K.; Bauman, M.D.; Shipp, J.C. Effects of epinephrine and perfusion pressure on the peak aortic pressure development and glucose transport in the isolated perfused heart of normal and diabetic rats. Basic Res. Cardiol. 1983, 78, 281–288. [Google Scholar] [CrossRef]
- Collins-Nakai, R.L.; Noseworthy, D.; Lopaschuk, G.D. Epinephrine increases ATP production in hearts by preferentially increasing glucose metabolism. Am. J. Physiol. Heart Circ. Physiol. 1994, 267, H1862–H1871. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P.; Porte, D., Jr. Adrenergic modulation of basal insulin secretion in man. Diabetes 1973, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lerner, R.L.; Porte, D., Jr. Epinephrine: Selective inhibition of the acute insulin response to glucose. J. Clin. Investig. 1971, 50, 2453–2457. [Google Scholar] [CrossRef] [PubMed]
- Christensen, N.J.; Videbaek, J. Plasma catecholamines and carbohydrate metabolism in patients with acute myocardial infarction. J. Clin. Investig. 1974, 54, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Hue, L.; Feliu, J.E.; Hers, H.G. Control of gluconeogenesis and of enzymes of glycogen metabolism in isolated rat hepatocytes. A parallel study of the effect of phenylephrine and of glucagon. Biochem. J. 1978, 176, 791–797. [Google Scholar] [CrossRef] [PubMed]
- García-Sáinz, J.A.; Hernández-Sotomayor, S.M. Adrenergic regulation of gluconeogenesis: Possible involvement of two mechanisms of signal transduction in α1-adrenergic action. PNAS 1985, 82, 6727–6730. [Google Scholar] [CrossRef] [PubMed]
- Stark, B.; Keller, U. a1-adrenergic stimulation of ketogenesis and fatty acid oxidation is associate with inhibition of lipogenesis in rat hepatocytes. Experientia 1987, 43, 1104–1106. [Google Scholar] [CrossRef]
- Chan, T.M.; Exton, J.H. Studies on α-adrenergic activation of hepatic glucose output. Studies on α-adrenergic inhibition of hepatic pyruvate kinase and activation of gluconeogenesis. J. Biol. Chem. 1978, 253, 6393–6400. [Google Scholar] [CrossRef]
- De Oliveira, A.L.; de Paula, M.N.; Comar, J.F.; Vilela, V.R.; Peralta, R.M.; Bracht, A. Adrenergic metabolic and hemodynamic effects of octopamine in the liver. Int. J. Mol. Sci. 2013, 14, 21858–21872. [Google Scholar] [CrossRef] [PubMed]
- Dileepan, K.N.; Khawaja, A.M.; Wagle, S.R. Studies on the mechanism of action of somatostatin on renal gluconeogenesis: Evidence for the involvement of α1- adrenergic stimuli. Arch. Biochem. Biophys. 1982, 213, 169–176. [Google Scholar] [CrossRef]
- Dileepan, K.N.; Wagle, S.R. Somatostatin: A metabolic regulator. Life Sci. 1985, 37, 2335–2343. [Google Scholar] [CrossRef]
- Hutson, N.J.; Brumley, F.T.; Assimacopoulos, F.D.; Harper, S.C.; Exton, J.H. Studies on the α-adrenergic activation of hepatic glucose output. I. Studies on the α-adrenergic activation of phosphorylase and gluconeogenesis and inactivation of glycogen synthase in isolated rat liver parenchymal cells. J. Biol. Chem. 1976, 251, 5200–5208. [Google Scholar] [CrossRef]
- Assimacopoulos-Jeannet, F.D.; Blackmore, P.F.; Exton, J.H. Studies on α-adrenergic activation of hepatic glucose output. Studies on role of calcium in α-adrenergic activation of phosphorylase. J. Biol. Chem. 1977, 252, 2662–2669. [Google Scholar] [CrossRef]
- Packer, M. Lessons learned from the DAPA-HF trial concerning the mechanisms of benefit of SGLT2 inhibitors on heart failure events in the context of other large-scale trials nearing completion. Cardiovasc. Diabetol. 2019, 18, 129. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Docherty, K.F.; Jhund, P.S. The dapagliflozin and prevention of adverse outcomes in heart failure trial (DAPA-HF) in context. Eur. Heart J. 2020, ii, ehz916. [Google Scholar] [CrossRef]
- Swe, M.T.; Thongnak, L.; Jaikumkao, K.; Pongchaidecha, A.; Chatsudthipong, V.; Lungkaphin, A. Dapagliflozin not only improves hepatic injury and pancreatic endoplasmic reticulum stress, but also induces hepatic gluconeogenic enzymes expression in obese rats. Clin. Sci. 2019, 133, 2415–2430. [Google Scholar] [CrossRef]
- Basu, R.; Shah, P.; Basu, A.; Norby, B.; Dicke, B.; Chandramouli, V.; Cohen, O.; Landau, B.R.; Rizza, R.A. Comparison of the effects of pioglitazone and metformin on hepatic and extra-hepatic insulin action in people with type 2 diabetes. Diabetes 2008, 57, 24–31. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef]
- Cahill, G.F., Jr.; Veech, R.L. Ketoacids? Good medicine? Trans. Am. Clin. Climatol. Assoc. 2003, 114, 149–163. [Google Scholar]
- Abel, E.D.; O’Shea, K.M.; Ramasamy, R. Insulin resistance: Metabolic mechanisms and consequences in the heart. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2068–2076. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell. Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, S.G.; Kim, S.H.; Kim, J.H.; Choi, E.; Cho, W.; Rim, J.H.; Hwang, I.; Lee, C.J.; Lee, M.; et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 2020, 11, 2127. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Egert, S.; Nguyen, N.; Schwaiger, M. Contribution of α-adrenergic and β-adrenergic stimulation to ischemia-induced glucose transporter (GLUT) 4 and GLUT1 translocation in the isolated perfused rat heart. Circ. Res. 1999, 84, 1407–1415. [Google Scholar] [CrossRef]
- Doenst, T.; Taegtmeyer, H. α-Adrenergic Stimulation Mediates Glucose Uptake Through Phosphatidylinositol 3-Kinase in Rat Heart. Circ. Res. 1999, 84, 467–474. [Google Scholar] [CrossRef]
- Shi, T.; Papay, R.S.; Perez, D.M. The role of α1-adrenergic receptors in regulating metabolism: Increased glucose tolerance, leptin secretion and lipid oxidation. J. Recept. Signal. Transduct. Res. 2017, 37, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Evans, B.A.; Sandström, A.L.; Chia, L.Y.; Mukaida, S.; Thai, B.S.; Nguyen, A.; Lim, L.; Tan, C.; Baltos, J.A.; et al. α1A-Adrenoceptors activate mTOR signalling and glucose uptake in cardiomyocytes. Biochem. Pharmacol. 2018, 148, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Papay, R.S.; Perez, D.M. α1-Adrenergic receptors increase glucose oxidation under normal and ischemic conditions in adult mouse cardiomyocytes. J. Recept. Signal. Transduct. Res. 2021, 41, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Liu, I.M.; Tsai, C.C.; Lai, T.Y.; Cheng, J.T. Stimulatory effect of isoferulic acid on α1A-adrenoceptor to increase glucose uptake into cultured myoblast C2C12 cells of mice. Auton. Neurosci. 2001, 88, 175–180. [Google Scholar] [CrossRef]
- Hutchinson, D.S.; Bengtsson, T. α1A-adrenoceptors activate glucose Uptake in L6 muscle cells through a phospholipase C-, phosphatidylinositol-3 kinase-, andatypical protein kinase C-dependent pathway. Endocrinology 2005, 146, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, D.S.; Bengtsson, T. AMP-activated protein kinase activation by adrenoceptors in L6 skeletal muscle cells: Mediation by α1-adrenoceptors causing glucose uptake. Diabetes 2006, 55, 682–690. [Google Scholar] [CrossRef]
- Faintrenie, G.; Géloën, A. α1-adrenergic stimulation of glucose uptake in rat white adipocytes. J. Pharmacol. Exp. Ther. 1998, 1286, 607–610. [Google Scholar]
- Cheng, J.-T.; Liu, I.-M.; Yen, S.-T.; Chen, P.-C. Role of α1A-adrenoceptor in the regulation of glucose uptake into white adipocyte of rats in vitro. Auton. Neurosci. 2000, 84, 140–146. [Google Scholar] [CrossRef]
- Boschmann, M.; Krupp, G.; Luft, F.C.; Klaus, S.; Jordan, J. In vivo response to α1-adrenoreceptor stimulation in human white adipose tissue. Obes. Res. 2002, 10, 555–558. [Google Scholar] [CrossRef]
- Flechtner-Mors, M.; Jenkinson, C.P.; Alt, A.; Adler, G.; Ditschuneit, H.H. In vivo α1-adrenergic lipolytic activity in subcutaneous adipose tissue of obese subjects. J. Pharmacol. Exp. Ther. 2002, 301, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Flechtner-Mors, M.; Jenkinson, C.P.; Alt, A.; Biesalski, H.K.; Adler, G.; Ditschuneit, H.H. Sympathetic regulation of glucose uptake by the α1-adrenoceptor in human obesity. Obes. Res. 2004, 12, 612–620. [Google Scholar] [CrossRef]
- Shi, T.; Papay, R.S.; Perez, D.M. α1A-Adrenergic receptor prevents cardiac ischemic damage through PKCδ/GLUT1/4-mediated glucose uptake. J. Recept. Signal. Transduct. Res. 2016, 36, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Lattion, A.L.; Hummler, E.; Nenniger, M.; Pedrazzini, T.; Aubert, J.F.; Michel, M.C.; Yang, M.; Lembo, G.; Vecchione, C.; et al. Decreased blood pressure response in mice deficient of the α1b-adrenergic receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 11589–115894. [Google Scholar] [CrossRef]
- Zuscik, M.J.; Sand, S.; Ross, S.A.; Waugh, D.J.J.; Gaivin, R.J.; Morilak, D.; Perez, D.M. Overexpression of the α1b-Adrenergic receptor causes apoptotic neurodegeneration: A multiple system atrophy. Nat. Med. 2000, 6, 1388–1394. [Google Scholar] [CrossRef]
- Zuscik, M.J.; Chalothorn, D.; Hellard, D.; Deighan, C.; McGee, A.; Daly, C.; Waugh, D.J.; Ross, S.A.; Gaivin, R.J.; Morehead, A.J.; et al. Hypotension, autonomic failure and cardiac hypertrophy in transgenic mice over-expressing the α1b-adrenergic receptor. J. Biol. Chem. 2001, 276, 13738–13743. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, C.; Maraldi, T.; Ghelli, A.; Rugolo, M.; Leoncini, E.; Hakim, G.; Hrelia, S. Green tea modulates α1-adrenergic stimulated glucose transport in cultured rat cardiomyocytes. J. Agric. Food Chem. 2007, 55, 7553–7558. [Google Scholar] [CrossRef] [PubMed]
- Rorabaugh, B.R.; Gaivin, R.J.; Papay, R.S.; Shi, T.; Simpson, P.C.; Perez, D.M. Both α1A- and α1B-Adrenergic Receptors Cross-talk to Downregulate β1-ARs in Mouse Heart: Coupling to Differential PTX-Sensitive Pathways. J. Mol. Cell. Cardiol. 2005, 39, 777–784. [Google Scholar] [CrossRef]
- Nishino, Y.; Miura, T.; Miki, T.; Sakamoto, J.; Nakamura, Y.; Ikeda, Y.; Kobayashi, H.; Shimamoto, K. Ischemic preconditioning activates AMPK in a PKC-dependent manner and induces GLUT4 up-regulation in the late phase of cardioprotection. Cardiovasc. Res. 2004, 61, 610–619. [Google Scholar] [CrossRef]
- Gundewar, S.; Calvert, J.W.; Jha, S.; Toedt-Pingel, I.; Ji, S.Y.; Nunez, D.; Ramachandran, A.; Anaya-Cisneros, M.; Tian, R.; Lefer, D.J. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ. Res. 2009, 104, 403–411. [Google Scholar] [CrossRef]
- Turrell, H.E.; Rodrigo, G.C.; Norman, R.I.; Dickens, M.; Standen, N.B. Phenylephrine preconditioning involves modulation of cardiac sarcolemmal K(ATP) current by PKC delta, AMPK and p38 MAPK. J. Mol. Cell. Cardiol. 2011, 51, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Zarrinpashneh, E.; Beauloye, C.; Ginion, A.; Pouleur, A.C.; Havaux, X.; Hue, L.; Viollet, B.; Vanoverschelde, J.L.; Bertrand, L. AMPKα2 counteracts the development of cardiac hypertrophy induced by isoproterenol. Biochem. Biophys. Res. Commun. 2008, 376, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Ma, X.; Feng, W.; Fu, Y.; Lu, Z.; Xu, M.; Shen, Q.; Zhu, Y.; Zhang, Y. Metformin attenuates cardiac fibrosis by inhibiting the TGFb1-Smad3 signalling pathway. Cardiovasc. Res. 2010, 87, 504–513. [Google Scholar] [CrossRef]
- Gaskin, F.S.; Kamada, K.; Zuidema, M.Y.; Jones, A.W.; Rubin, L.J.; Korthuis, R.J. Isoform-selective 5’-AMP-activated protein kinase-dependent preconditioning mechanisms to prevent postischemic leukocyte-endothelial cell adhesive interactions. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1352–H1360. [Google Scholar] [CrossRef]
- Cieslik, K.A.; Taffet, G.E.; Crawford, J.R.; Trial, J.; Mejia Osuna, P.; Entman, M.L. AICAR-dependent AMPK activation improves scar formation in the aged heart in a murine model of reperfused myocardial infarction. J. Mol. Cell. Cardiol. 2013, 63, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Fu, Y.; Xiao, H.; Song, Y.; Chen, R.; Shen, J.; An, X.; Shen, Q.; Li, Z.; Zhang, Y. Cardiac Fibrosis Alleviated by Exercise Training Is AMPK-Dependent. PLoS ONE 2015, 10, e0129971. [Google Scholar] [CrossRef]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell. 2016, 15, 416–427. [Google Scholar] [CrossRef]
- Garg, G.; Singh, S.; Singh, A.K.; Rizvi, S.I. Metformin Alleviates Altered Erythrocyte Redox Status During Aging in Rats. Rejuvenation Res. 2017, 20, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Na, H.J.; Park, J.S.; Pyo, J.H.; Jeon, H.J.; Kim, Y.S.; Arking, R.; Yoo, M.-A. Metformin inhibits age-related centrosome amplification in Drosophila midgut stem cells through AKT/TOR pathway. Mech. Ageing Dev. 2015, 149, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Marsin, A.S.; Bertrand, L.; Rider, M.H.; Deprez, J.; Beauloye, C.; Vincent, M.F.; Van den Berghe, G.; Carling, D.; Hue, L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr. Biol. 2000, 10, 1247–1255. [Google Scholar] [CrossRef]
- Russell, R.R., 3rd; Li, J.; Coven, D.L.; Pypaert, M.; Zechner, C.; Palmeri, M.; Giordano, F.J.; Mu, J.; Birnbaum, M.J.; Young, L.H. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Investig. 2004, 114, 495–503. [Google Scholar] [CrossRef]
- Xu, M.; Zhao, Y.T.; Song, Y.; Hao, T.P.; Lu, Z.Z.; Han, Q.D.; Wang, S.Q.; Zhang, Y.Y. α1-adrenergic receptors activate AMP-activated protein kinase in rat hearts. Sheng Li Xue Bao 2007, 59, 175–182. [Google Scholar] [PubMed]
- Pang, T.; Rajapurohitam, V.; Cook, M.A.; Karmazyn, M. Differential AMPK phosphorylation sites associated with phenylephrine vs. antihypertrophic effects of adenosine agonists in neonatal rat ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1382–H1390. [Google Scholar] [CrossRef]
- Horie, T.; Ono, K.; Nagao, K.; Nishi, H.; Kinoshita, M.; Kawamura, T.; Wada, H.; Shimatsu, A.; Kita, T.; Hasegawa, K. Oxidative stress induces GLUT4 translocation by activation of PI3-K/Akt and dual AMPK kinase in cardiac myocytes. J. Cell. Physiol. 2008, 215, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, L.; Ebihara, K.; Kusakabe, T.; Aotani, D.; Yamamoto-Kataoka, S.; Sakai, T.; Aizawa-Abe, M.; Yamamoto, Y.; Fujikura, J.; Hayashi, T.; et al. Leptin activates hepatic 5’-AMP-activated protein kinase through sympathetic nervous system and α1-adrenergic receptor: A potential mechanism for improvement of fatty liver in lipodystrophy by leptin. J. Biol. Chem. 2012, 287, 40441–40447. [Google Scholar] [CrossRef]
- Pulinilkunnil, T.; He, H.; Kong, D.; Asakura, K.; Peroni, O.D.; Lee, A.; Kahn, B.B. Adrenergic regulation of AMP-activated protein kinase in brown adipose tissue in vivo. J. Biol. Chem. 2011, 286, 8798–8809. [Google Scholar] [CrossRef] [PubMed]
- Minokoshi, Y.; Kim, Y.B.; Peroni, O.D.; Fryer, L.G.; Müller, C.; Carling, D.; Kahn, B.B. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002, 415, 339–343. [Google Scholar] [CrossRef]
- Kishi, K.; Yuasa, T.; Minami, A.; Yamada, M.; Hagi, A.; Hayashi, H.; Kemp, B.E.; Witters, L.A.; Ebina, Y. AMP-activated protein kinase is activated by the stimulations of G(q)-coupled receptors. Biochem. Biophys. Res. Commun. 2000, 276, 16–22. [Google Scholar] [CrossRef]
- Glund, S.; Deshmukh, A.; Long, Y.C.; Moller, T.; Koistinen, H.A.; Caidahl, K.; Zierath, Z.; Krook, A. Interleukin-6 directly increases glucose metabolism in resting human skeletal muscle. Diabetes 2007, 56, 1630–1637. [Google Scholar] [CrossRef]
- Cadaret, C.N.; Beede, K.A.; Riley, H.E.; Yates, D.T. Acute exposure of primary rat soleus muscle to zilpaterol HCl (β2 adrenergic agonist), TNFα, or IL-6 in culture increases glucose oxidation rates independent of the impact on insulin signaling or glucose uptake. Cytokine 2017, 96, 107–113. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef]
- Xiao, X.; Su, G.; Brown, S.N.; Chen, L.; Ren, J.; Zhao, P. Peroxisome proliferator-activated receptors gamma and alpha agonists stimulate cardiac glucose uptake via activation of AMP-activated protein kinase. J. Nutr. Biochem. 2010, 21, 621–626. [Google Scholar] [CrossRef]
- Huang, Q.; Huang, J.; Zeng, Z.; Luo, J.; Liu, P.; Chen, S.; Liu, B.; Pan, X.; Zang, L.; Zhou, S. Effects of ERK1/2/PPARα/SCAD signal pathways on cardiomyocyte hypertrophy induced by insulin-like growth factor 1 and phenylephrine. Life Sci. 2015, 124, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Kar, D.; Bandyopadhyay, A. Targeting Peroxisome Proliferator Activated Receptor α (PPAR α) for the Prevention of Mitochondrial Impairment and Hypertrophy in Cardiomyocytes. Cell. Physiol. Biochem. 2018, 49, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Hua, J.; Cai, W.; Zhan, Q.; Lai, W.; Zeng, Q.; Ren, H.; Xu, D. N-terminal truncated peroxisome proliferator-activated receptor-γ coactivator-1α alleviates phenylephrine-induced mitochondrial dysfunction and decreases lipid droplet accumulation in neonatal rat cardiomyocytes. Mol. Med. Rep. 2018, 18, 2142–2152. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Kim, H.S.; Seo, H.S.; Na, J.O.; Jang, Y.-N.; Han, Y.-M.; Kim, H.-M. Stimulation of α1-Adrenergic Receptor Ameliorates Cellular Functions of Multiorgans beyond Vasomotion through PPARδ. PPAR Res. 2020, 3785137. [Google Scholar] [CrossRef]
- Barger, P.M.; Kelly, D.P. PPAR signaling in the control of cardiac energy metabolism. Trends. Cardiovasc. Med. 2000, 10, 238–245. [Google Scholar] [CrossRef]
- Huss, J.M.; Kelly, D.P. Mitochondrial energy metabolism in heart failure: A question of balance. J. Clin. Investig. 2005, 115, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Burkart, E.M.; Sambandam, N.; Han, X.; Gross, R.W.; Courtois, M.; Gierasch, C.M.; Shoghi, K.; Welch, M.J.; Kelly, D.P. Nuclear receptors PPARβ/δ and PPARα direct distinct metabolic regulatory programs in the mouse heart. J. Clin. Investig. 2007, 117, 3930–3939. [Google Scholar] [CrossRef]
- Yang, Q.; Long, Q. PPARd, a Potential Therapeutic Target for Heart Disease. Nucl. Recept. Res. 2018, 5, 101375. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Ding, G.; Qin, Q.; Huang, Y.; Lewis, W.; He, N.; Evans, R.M.; Schneider, M.D.; Brako, F.A.; Xiao, Y.; et al. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-δ deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat. Med. 2004, 10, 1245–1250. [Google Scholar] [CrossRef]
- Li, P.; Luo, S.; Pan, C.; Cheng, X. Modulation of fatty acid metabolism is involved in the alleviation of isoproterenol-induced rat heart failure by fenofibrate. Mol. Med. Rep. 2015, 12, 7899–7906. [Google Scholar] [CrossRef][Green Version]
- Yuan, J.; Wu, J.; Han, Z.G. Fenofibrate improves energy metabolism and attenuates isoproterenol induced acute myocardial ischemic injury in rats via PPAR α activation. Zhonghua Xin Xue Guan Bing Za Zhi 2008, 36, 847–850. [Google Scholar]
- Zuo, X.; Peng, Z.; Moussalli, M.J.; Morris, J.S.; Broaddus, R.R.; Fischer, S.M.; Shureiqi, I. Targeted genetic disruption of peroxisome proliferator-activated receptor-delta and colonic tumorigenesis. J. Natl. Cancer Inst. 2009, 101, 762–767. [Google Scholar] [CrossRef]
- Zuo, X.; Xu, M.; Yu, J.; Wu, Y.; Moussalli, M.J.; Manyam, G.C.; Lee, S.I.; Lee, S.I.; Liang, S.; Gagea, M.; et al. Potentiation of colon cancer susceptibility in mice by colonic epithelial PPAR-δ/β overexpression. J. Natl. Cancer Inst. 2014, 106, dju052. [Google Scholar] [CrossRef]
- Xi, Y.; Zhang, Y.; Zhu, S.; Luo, Y.; Xu, P.; Huang, Z. PPAR-Mediated Toxicology and Applied Pharmacology. Cells 2020, 9, 352. [Google Scholar] [CrossRef]
- Wagner, N.; Wagner, K.D. PPAR Beta/Delta and the Hallmarks of Cancer. Cells 2020, 9, 1133. [Google Scholar] [CrossRef]
- Konstandi, M.; Kypreos, K.E.; Matsubara, T.; Xepapadaki, E.; Shah, Y.M.; Krausz, K.; Andriopoulou, C.E.; Kofinas, A.; Gonzalez, F.J. Adrenoceptor-related decrease in serum triglycerides is independent of PPARα activation. FEBS J. 2019, 286, 4328–4341. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Ilaiwy, A.; Montgomery, M.D.; Simpson, P.C.; Jensen, B.C. The α1A- adrenergic receptor agonist A61603 reduces cardiac polyunsaturated fatty acid and endocannabinoid metabolites associated with inflammation in vivo. Metabolomics 2016, 12, 155. [Google Scholar] [CrossRef] [PubMed]
- Burcelin, R.; Uldry, M.; Foretz, M.; Perrin, C.; Dacosta, A.; Nenniger-Tosato, M.; Seydoux, J.; Cotecchia, S.; Thorens, B. Impaired glucose homeostasis in mice lacking the α1b-adrenergic receptor subtype. J. Biol. Chem. 2004, 279, 1108–1115. [Google Scholar] [CrossRef]
- Zimmer, H.G.; Ibel, H.; Suchner, U. β-adrenergic agonists stimulate the oxidative pentose phosphate pathway in the rat heart. Circ. Res. 1990, 67, 1525–1534. [Google Scholar] [CrossRef]
- Zimmer, H.G.; Lankat-Buttgereit, B.; Kolbeck-Rühmkorff, C.; Nagano, T.; Zierhut, W. Effects of norepinephrine on the oxidative pentose phosphate pathway in the rat heart. Circ. Res. 1992, 71, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, H.G.; Irlbeck, M.; Kolbeck-Rühmkorff, C.K. Response of the rat heart to catecholamines and thyroid hormones. Mol. Cell. Biochem. 1995, 147, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Irlbeck, M.; Zimmer, H.G. The functional and metabolic responses of the heart to catecholamines are attenuated in diabetic rats. Cardioscience 1995, 6, 131–138. [Google Scholar] [PubMed]
- Giannattasio, C.; Cattaneo, B.M.; Seravalle, G.; Carugo, S.; Mangoni, A.A.; Grassi, G.; Zanchetti, A.; Mancia, G. α1-blocking properties of carvedilol during acute and chronic administration. J. Cardiovasc. Pharmacol. 1992, 19, S18–S22. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; O’Harrow, S.; Sponer, G.; Zimmer, H.G. Norepinephrine-induced changes in rat heart function, metabolism, and weight are antagonized by carvedilol. J. Cardiovasc. Pharmacol. 1993, 21, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.H.; Lee, C.O. Role of PKC in the effects of α1-adrenergic stimulation on Ca2+ transients, contraction and Ca2+ current in guinea-pig ventricular myocytes. Pflugers. Arch. 1999, 437, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Wier, W.G.; Morgan, K.G. α1-adrenergic signaling mechanisms in contraction of resistance arteries. Rev. Physiol. Biochem. Pharmacol. 2003, 150, 91–139. [Google Scholar]
- Villalba, N.; Stankevicius, E.; Garcia-Sacristán, A.; Simonsen, U.; Prieto, D. Contribution of both Ca2+ entry and Ca2+ sensitization to the α1-adrenergic vasoconstriction of rat penile small arteries. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1157–H1169. [Google Scholar] [CrossRef]
- Gutiérrez, A.; Contreras, C.; Sánchez, A.; Prieto, D. Role of Phosphatidylinositol 3-Kinase (PI3K), Mitogen-Activated Protein Kinase (MAPK), and Protein Kinase C (PKC) in Calcium Signaling Pathways Linked to the α1-Adrenoceptor in Resistance Arteries. Front. Physiol. 2019, 10, 55. [Google Scholar] [CrossRef] [PubMed]
- Fordyce, C.B.; Roe, M.T.; Ahmad, T.; Libby, P.; Borer, J.S.; Hiatt, W.R.; Bristow, M.R.; Packer, M.; Wasserman, S.M.; Braunstein, N.; et al. Cardiovascular drug development: Is it dead or just hibernating? J. Am. Coll. Cardiol. 2015, 65, 1567–1582. [Google Scholar] [CrossRef] [PubMed]
- Ruffolo, R.R., Jr.; Rice, P.J.; Patil, P.N.; Hamada, A.; Miller, D.D. Differences in the applicability of the Easson-Stedman hypothesis to the α1- and α2-adrenergic effects of phenethylamines and imidazolines. Eur. J. Pharmacol. 1983, 86, 471–475. [Google Scholar] [CrossRef]
- Ruffolo, R.R., Jr.; Waddell, J.E. Receptor interactions of imidazolines. IX. Cirazoline is an α1-adrenergic agonist and an α2-adrenergic antagonist. J. Pharmacol. Exp. Ther. 1982, 222, 29–36. [Google Scholar] [PubMed]
- Hieble, J.P.; DeMarinis, R.M.; Matthews, W.D. Evidence for and against heterogeneity of α1-adrenoceptors. Life Sci. 1986, 38, 1339–1350. [Google Scholar] [CrossRef]
- Ruffolo, R.R., Jr.; Yaden, E.L.; Waddell, J.E.; Dillard, R.D. Receptor interactions of imidazolines. VI. Significance of carbon bridge separating phenyl and imidazoline rings of tolazoline-like α-adrenergic imidazolines. J. Pharmacol. Exp. Ther. 1980, 214, 535–540. [Google Scholar] [PubMed]
- Knepper, S.M.; Buckner, S.A.; Brune, M.E.; DeBernardis, J.F.; Meyer, M.D.; Hancock, A.A. A-61603, a potent α1-adrenergic receptor agonist, selective for the α1A receptor subtype. J. Pharmacol. Exp. Ther. 1995, 274, 97–103. [Google Scholar]
- Minneman, K.P.; Theroux, T.L.; Hollinger, S.; Han, C.; Esbenshade, T.A. Selectivity of agonists for cloned α1-adrenergic receptor subtypes. Mol. Pharmacol. 1994, 46, 929–936. [Google Scholar]
- Waugh, D.J.J.; Gaivin, R.J.; Zuscik, M.J.; Gonzalez-Cabrera, P.; Ross, S.A.; Yun, J.; Perez, D.M. Phe308 and Phe312 in TM VII are major sites of α1-Adrenergic Receptor Antagonist Binding: Imidazoline Agonists Bind Like Antagonists. J. Biol. Chem. 2001, 276, 25366–25371. [Google Scholar] [CrossRef]
- Musselman, D.M.; Ford, A.P.; Gennevois, D.J.; Harbison, M.L.; Laurent, A.L.; Mokatrin, A.S.; Stoltz, R.R.; Blue, D.R. A randomized crossover study to evaluate Ro 115–1240, a selective α1A/L -adrenoceptor partial agonist in women with stress urinary incontinence. BJU Int. 2004, 93, 78–83. [Google Scholar] [CrossRef]
- Blue, D.R.; Daniels, D.V.; Gever, J.R.; Jett, M.F.; O’Yang, C.; Tang, H.M.; Williams, T.J.; Ford, A.P. Pharmacological characteristics of Ro 115–1240, a selective α1A/L-adrenoceptor partial agonist: A potential therapy for stress urinary incontinence. BJU Int. 2004, 93, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.A.; Broxton, N.; Merlin, J.; Sato, M.; Hutchinson, D.S.; Christopoulos, A.; Summers, R.J. Quantification of functional selectivity at the human α1A-adrenoceptor. Mol. Pharmacol. 2011, 79, 298–307. [Google Scholar] [CrossRef]
- Da Silva, E.D.; Sato, M.; Merlin, J.; Broxton, N.; Hutchinson, D.S.; Ventura, S.; Evans, B.A.; Summers, R.J. Factors influencing biased agonism in recombinant cells expressing the human α1A-adrenoceptor. Br. J. Pharmacol. 2017, 174, 2318–2333. [Google Scholar] [CrossRef] [PubMed]
- Bishop, M.J. Recent Advances in the Discovery of α1-Adrenoceptor Agonists. Curr. Top. Med. Chem. 2007, 7, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, A. Allosteric binding sites on cell-surface receptors: Novel targets for drug discovery. Nat. Rev. Drug Discov. 2002, 1, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Das, D.; Nakata, H.; Mitsuya, H. CCR5 inhibitors: Emergence, success, and challenges. Expert Opin. Emerg. Drugs. 2012, 17, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Wold, E.A.; Chen, J.; Cunningham, K.A.; Zhou, J. Allosteric Modulation of Class A GPCRs: Targets, Agents, and Emerging Concepts. J. Med. Chem. 2019, 62, 88–127. [Google Scholar] [CrossRef] [PubMed]
- Perez, D.M. Novel Positive Allosteric Modulators of the α1A-Adrenergic Receptor to Treat Alzheimer’s Disease. Brain Connect. 2021, 11, A1–A8. [Google Scholar]
- Kuschel, M.; Zhou, Y.Y.; Spurgeon, H.A.; Bartel, S.; Karczewski, P.; Zhang, S.J.; Krause, E.G.; Lakatta, E.G.; Xiao, R.P. β2-Adrenergic cAMP signaling is uncoupled from phosphorylation of cytoplasmic proteins in canine heart. Circulation 1999, 99, 2458–2465. [Google Scholar] [CrossRef]
- McConville, P.; Fishbein, K.W.; Lakatta, E.G.; Spencer, R.G. Differences in the bioenergetic response of the isolated perfused rat heart to selective β1- and β2-adrenergic receptor stimulation. Circulation 2003, 107, 2146–2152. [Google Scholar] [CrossRef]
- Goodwin, G.W.; Ahmad, F.; Taegtmeyer, H. Preferential oxidation of glycogen in isolated working rat heart. J. Clin. Investig. 1996, 97, 1409–1416. [Google Scholar] [CrossRef]
- Goodwin, G.W.; Ahmad, F.; Doenst, T.; Taegtmeyer, H. Energy provision from glycogen, glucose, and fatty acids on adrenergic stimulation of isolated working rat hearts. Am. J. Physiol. 1998, 274, H1239–H1247. [Google Scholar] [CrossRef] [PubMed]
- Nevzorova, J.; Bengtsson, T.; Evans, B.A.; Summers, R.J. Characterization of the β-adrenoceptor subtype involved in mediation of glucose transport in L6 cells. Br. J. Pharmacol. 2002, 137, 9–18. [Google Scholar] [CrossRef]
- Nevzorova, J.; Evans, B.A.; Bengtsson, T.; Summers, R.J. Multiple signalling pathways involved in β2-adrenoceptor-mediated glucose uptake in rat skeletal muscle cells. Br. J. Pharmacol. 2006, 147, 446–454. [Google Scholar] [CrossRef]
- McConville, P.; Lakatta, E.G.; Spencer, R.G. Greater glycogen utilization during β1- than β2-adrenergic receptor stimulation in the isolated perfused rat heart. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1828–E1835. [Google Scholar] [CrossRef] [PubMed]
- Andres, D.A.; Young, L.E.A.; Veeranki, S.; Hawkinson, T.R.; Levitan, B.M.; He, D.; Wang, C.; Satin, J.; Sun, R.C. Improved workflow for mass spectrometry-based metabolomics analysis of the heart. J. Biol. Chem. 2020, 295, 2676–2686. [Google Scholar] [CrossRef] [PubMed]
- McConville, P.; Spencer, R.G.; Lakatta, E.G. Temporal dynamics of inotropic, chronotropic, and metabolic responses during β1- and β2-AR stimulation in the isolated, perfused rat heart. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E412–E418. [Google Scholar] [CrossRef]
- Ahmad, T.; Miller, P.E.; McCullough, M.; Desai, N.R.; Riello, R.; Psotka, M.; Böhm, M.; Allen, L.A.; Teerlink, J.R.; Rosano, G.; et al. Why has positive inotropy failed in chronic heart failure? Lessons from prior inotrope trials. Eur. J. Heart Fail. 2019, 21, 1064–1078. [Google Scholar] [CrossRef] [PubMed]
- Lafontan, M.; Berlan, M. Fat cell adrenergic receptors and the control of white and brown fat cell function. J. Lipid Res. 1993, 34, 1057–1091. [Google Scholar] [CrossRef]
- Zhao, J.; Cannon, B.; Nedergaard, J. Thermogenesis is β3- but not β1-adrenergically mediated in rat brown fat cells, even after cold acclimation. Am. J. Physiol. 1998, 275, R2002–R2011. [Google Scholar] [CrossRef]
- Barr, L.A.; Lambert, J.P.; Shimizu, Y.; Barouch, L.A.; Naqvi, N.; Calvert, J.W. Exercise training provides cardioprotection by activating and coupling endothelial nitric oxide synthase via a β3-adrenergic receptor-AMP-activated protein kinase signaling pathway. Med. Gas. Res. 2017, 7, 1–8. [Google Scholar]
- Moniotte, S.; Kobzik, L.; Feron, O.; Trochu, J.N.; Gauthier, C.; Balligand, J.L. Upregulation of β3-adrenoceptors and altered contractile response to inotropic amines in human failing myocardium. Circulation 2001, 103, 1649–1655. [Google Scholar] [CrossRef]
- Cheng, H.J.; Zhang, Z.S.; Onishi, K.; Ukai, T.; Sane, D.C.; Cheng, C.P. Upregulation of functional β3-adrenergic receptor in the failing canine myocardium. Circ. Res. 2001, 89, 599–606. [Google Scholar] [CrossRef]
- Treskatsch, S.; Feldheiser, A.; Rosin, A.T.; Sifringer, M.; Habazettl, H.; Mousa, S.A.; Shakibaei, M.; Schäfer, M.; Spies, C.D. A modified approach to induce predictable congestive heart failure by volume overload in rats. PLoS ONE 2014, 9, e87531. [Google Scholar]
- Kawaguchi, S.; Okada, M.; Ijiri, E.; Koga, D.; Watanabe, T.; Hayashi, K.; Kashiwagi, Y.; Fujita, S.; Hasebe, N. β3-Adrenergic receptor blockade reduces mortality in endotoxin-induced heart failure by suppressing induced nitric oxide synthase and saving cardiac metabolism. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H283–H294. [Google Scholar] [CrossRef] [PubMed]
- Ziskoven, C.; Grafweg, S.; Bolck, B.; Wiesner, R.J.; Jimenez, M.; Giacobino, J.P.; Bloch, W.; Schwinger, R.H.; Brixius, K. Increased Ca2+ sensitivity and protein expression of SERCA 2a in situations of chronic β3-adrenoceptor deficiency. Pflugers. Arch. 2007, 453, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Moens, A.L.; Leyton-Mange, J.S.; Niu, X.; Yang, R.; Cingolani, O.; Arkenbout, E.K.; Champion, H.C.; Bedja, D.; Gabrielson, K.L.; Chen, J.; et al. Adverse ventricular remodeling and exacerbated NOS uncoupling from pressure-overload in mice lacking the β3-adrenoreceptor. J. Mol. Cell. Cardiol. 2009, 47, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Aragon, J.P.; Condit, M.E.; Bhushan, S.; Predmore, B.L.; Patel, S.S.; Grinsfelder, D.B.; Gundewar, S.; Jha, S.; Calvert, J.W.; Barouch, L.A.; et al. β3-adrenoreceptor stimulation ameliorates myocardial ischemia-reperfusion injury via endothelial nitric oxide synthase and neuronal nitric oxide synthase activation. J. Am. Coll. Cardiol. 2011, 58, 2683–2691. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Watts, V.L.; Cingolani, O.H.; Sivakumaran, V.; Leyton-Mange, J.S.; Ellis, C.L.; Miller, K.L.; Vandegaer, K.; Bedja, D.; Gabrielson, K.L.; et al. Cardioprotective effect of β3-adrenergic receptor agonism: Role of neuronal nitric oxide synthase. J. Am. Coll. Cardiol. 2012, 59, 1979–1987. [Google Scholar] [CrossRef]
- Niu, X.; Zhao, L.; Li, X.; Xue, Y.; Wang, B.; Lv, Z.; Chen, J.; Sun, D.; Zheng, Q. β3-adrenoreceptor stimulation protects against myocardial infarction injury via eNOS and nNOS activation. PLoS ONE 2014, 9, e98713. [Google Scholar] [CrossRef]
- Belge, C.; Hammond, J.; Dubois-Deruy, E.; Manoury, B.; Hamelet, J.; Beauloye, C.; Markl, A.; Pouleur, A.C.; Bertrand, L.; Esfahani, H.; et al. Enhanced expression of β3-adrenoceptors in cardiac myocytes attenuates neurohormone-induced hypertrophic remodeling through nitric oxide synthase. Circulation 2014, 129, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Trappanese, D.M.; Liu, Y.; McCormick, R.C.; Cannavo, A.; Nanayakkara, G.; Baskharoun, M.M.; Jarrett, H.; Woitek, F.J.; Tillson, D.M.; Dillon, A.R.; et al. Chronic β1-adrenergic blockade enhances myocardial β3-adrenergic coupling with nitric oxide-cGMP signaling in a canine model of chronic volume overload: New insight into mechanisms of cardiac benefit with selective β1-blocker therapy. Basic Res. Cardiol. 2015, 110, 456. [Google Scholar] [CrossRef]
- Kamiya, M.; Asai, K.; Maejima, Y.; Shirakabe, A.; Murai, K.; Noma, S.; Komiyama, H.; Sato, N.; Mizuno, K.; Shimizu, W. β3-adrenergic receptor agonist prevents diastolic dysfunction in an angiotensin II-induced cardiomyopathy mouse model. J. Pharmacol. Exp. Ther. 2021, 376, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Finlin, B.S.; Memetimin, H.; Zhu, B.; Confides, A.L.; Vekaria, H.J.; El Khouli, R.H.; Johnson, Z.R.; Westgate, P.M.; Chen, J.; Morris, A.J.; et al. The β3-adrenergic receptor agonist mirabegron improves glucose homeostasis in obese humans. J. Clin. Investig. 2020, 130, 2319–2331. [Google Scholar] [CrossRef] [PubMed]
- Dehvari, N.; Sato, M.; Bokhari, M.H.; Kalinovich, A.; Ham, S.; Gao, J.; Nguyen, H.; Whiting, L.; Mukaida, S.; Merlin, J.; et al. The metabolic effects of mirabegron are mediated primarily by β3 -adrenoceptors. Pharmacol. Res. Perspect. 2020, 8, e00643. [Google Scholar] [CrossRef]
- Smith, S.A.; Levy, A.L.; Sennitt, M.V.; Simson, D.L.; Cawthorne, M.A. Effects of BRL 26830, a novel β-adrenoceptor agonist, on glucose tolerance, insulin sensitivity and glucose turnover in Zucker (fa/fa) rats. Biochem. Pharmacol. 1985, 34, 2425–2429. [Google Scholar] [CrossRef]
- Williams, C.A.; Shih, M.F.; Taberner, P.V. Sustained improvement in glucose homeostasis in lean and obese mice following chronic administration of the β3 agonist SR 58611A. Br. J. Pharmacol. 1999, 128, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Cawthorne, M.A.; Carroll, M.J.; Levy, A.L.; Lister, C.A.; Sennitt, M.V.; Smith, S.A.; Young, P. Effects of novel β-adrenoceptor agonists on carbohydrate metabolism: Relevance for the treatment of non-insulin-dependent diabetes. Int. J. Obes. 1984, 8, 93–102. [Google Scholar]
- Hao, L.; Scott, S.; Abbasi, M.; Zu, Y.; Khan, M.S.H.; Yang, Y.; Wu, D.; Zhao, L.; Wang, S. Beneficial Metabolic Effects of Mirabegron In Vitro and in High-Fat Diet-Induced Obese Mice. J. Pharmacol. Exp. Ther. 2019, 369, 419–427. [Google Scholar] [CrossRef]
- Wang, Z.H.; Li, Y.F.; Guo, Y.Q. β3-Adrenoceptor activation attenuates atherosclerotic plaque formation in ApoE−/− mice through lowering blood lipids and glucose. Acta. Pharmacol. Sin. 2013, 34, 1156–1163. [Google Scholar] [CrossRef][Green Version]
- Shi, S.T.; Li, Y.F.; Guo, Y.Q.; Wang, Z.H. Effect of β3-adrenoceptor stimulation on the levels of ApoA-I, PPARα, and PPARγ in apolipoprotein E-deficient mice. J. Cardiovasc. Pharmacol. 2014, 64, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Bundgaard, H.; Axelsson, A.; Hartvig Thomsen, J.; Sorgaard, M.; Kofoed, K.F.; Hasselbalch, R.; Fry, N.A.; Valeur, N.; Boesgaard, S.; Gustafsson, F.; et al. The first-in-man randomized trial of a β3 adrenoceptor agonist in chronic heart failure: The BEAT-HF trial. Eur. J. Heart Fail. 2017, 19, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Pouleur, A.C.; Anker, S.; Brito, D.; Brosteanu, O.; Hasenclever, D.; Casadei, B.; Edelmann, F.; Filippatos, G.; Gruson, D.; Ikonomidis, I.; et al. Rationale and design of a multicentre, randomized, placebo-controlled trial of mirabegron, a β3-adrenergic receptor agonist on left ventricular mass and diastolic function in patients with structural heart disease β3-left ventricular hypertrophy (b3-LVH). Esc Heart Fail. 2018, 5, 830–841. [Google Scholar] [CrossRef]
- Böhm, M.; Maack, C. Treatment of heart failure with b-blockers. Mechanisms and results. Basic Res. Cardiol. 2000, 95, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Katz, A.M. Changing strategies in the management of heart failure. J. Am. Coll. Cardiol. 1989, 13, 513–523. [Google Scholar] [CrossRef]
- Hwang, I.C. Myocardial Efficiency: A Reliable Load-independent Parameter of Cardiac Performance? J. Cardiovasc. Imaging 2020, 28, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Bing, R.J.; Siegel, A.; Ungar, I.; Gilbert, M. Metabolism of the human heart. II. Studies on fat, ketone and amino acid metabolism. Am. J. Med. 1954, 16, 504–515. [Google Scholar] [CrossRef]
- Opie, L.H. Effect of β-adrenergic blockade on biochemical and metabolic response to exercise. Am. J. Cardiol. 1985, 55, 95D–100D. [Google Scholar] [CrossRef]
- McLeod, A.A.; Brown, J.E.; Kitchell, B.B.; Sedor, F.A.; Kuhn, C.; Shand, D.G.; Williams, R.S. Hemodynamic and metabolic responses to exercise after adrenoceptor blockade in humans. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1984, 56, 716–722. [Google Scholar] [CrossRef]
- Verstappen, F.T.; van Baak, M.A. Exercise capacity, energy metabolism, and β-adrenoceptor blockade. Comparison between a β1-selective and a non-selective β blocker. Eur. J. Appl. Physiol. Occup. Physiol. 1987, 56, 712–718. [Google Scholar] [CrossRef]
- Hansen, O.; Johansson, B.W.; Nilsson-Ehle, P. Metabolic, electrocardiographic, and hemodynamic responses to increased circulating adrenaline: Effects of selective and nonselective β-adrenoceptor blockade. Angiology 1990, 41, 175–188. [Google Scholar] [CrossRef]
- Sarafidis, P.A.; Bakris, G.L. Antihypertensive treatment with β-blockers and the spectrum of glycaemic control. QJM 2006, 99, 431–436. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Deacon, S.P. The effects of atenolol and propranolol upon lipolysis. Br. J. Clin. Pharmacol. 1978, 5, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Zmudka, K.; Dubiel, J.; Pieniazek, P.; Dudek, D.; Kocurek, A.; Trebacz, J.; Grodecki, J.; Flameng, W.; de Geest, H. Influence of an early adrenergic blockade on thrombotic infarct size and myocardial metabolism. J. Physiol. Pharmacol. 1998, 49, 333–352. [Google Scholar] [PubMed]
- Panchal, A.R.; Stanley, W.C.; Kerner, J.; Sabbah, H.N. β-receptor blockade decreases carnitine palmitoyl transferase I activity in dogs with heart failure. J. Card. Fail. 1998, 4, 121–126. [Google Scholar] [CrossRef]
- Igarashi, N.; Nozawa, T.; Fujii, N.; Suzuki, T.; Matsuki, A.; Nakadate, T.; Igawa, A.; Inoue, H. Influence of β-adrenoceptor blockade on the myocardial accumulation of fatty acid tracer and its intracellular metabolism in the heart after ischemia–reperfusion injury. Circ. J. 2006, 70, 1509–1514. [Google Scholar] [CrossRef][Green Version]
- Wallhaus, T.R.; Taylor, M.; DeGrado, T.R.; Russell, D.C.; Stanko, P.; Nickles, R.J.; Stone, C.K. Myocardial free fatty acid and glucose use after carvedilol treatment in patients with congestive heart failure. Circulation 2001, 103, 2441–2446. [Google Scholar] [CrossRef]
- Sharma, V.; Dhillon, P.; Wambolt, R.; Parsons, H.; Brownsey, R.; Allard, M.F.; McNeill, J.H. Metoprolol improves cardiac function and modulates cardiac metabolism in the streptozotocin-diabetic rat. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1609–H1620. [Google Scholar] [CrossRef]
- Bøttcher, M.; Refsgaard, J.; Gøtzsche, O.; Andreasen, F.; Nielsen, T.T. Effect of carvedilol on microcirculatory and glucose metabolic regulation in patients with congestive heart failure secondary to ischemic cardiomyopathy. Am. J. Cardiol. 2002, 89, 1388–1393. [Google Scholar] [CrossRef]
- Podbregar, M.; Voga, G. Effect of selective and nonselective β-blockers on resting energy production rate and total body substrate utilization in chronic heart failure. J. Card Fail. 2002, 8, 369–378. [Google Scholar] [CrossRef]
- Bakris, G.L.; Fonseca, V.; Katholi, R.E.; McGill, J.B.; Messerki, F.H.; Phillips, R.A.; Raskin, P.; Wright, J.T., Jr.; Oakes, R.; Lukas, M.A.; et al. Metabolic effects of carvedilol vs metoprolol in patients with type 2 diabetes mellitus and hypertension A randomized controlled trial. J. Am. Med. Assoc. 2004, 292, 2227–2236. [Google Scholar] [CrossRef]
- Al-Hesayen, A.; Azevedo, E.R.; Floras, J.S.; Hollingshead, S.; Lopaschuk, G.D.; Parker, J.D. Selective versus nonselective β-adrenergic receptor blockade in chronic heart failure: Differential effects on myocardial energy substrate utilization. Eur. J. Heart Fail 2005, 7, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Basat, O.; Ucak, S.; Seber, S.; Oztekin, E.; Altuntas, Y. After myocardial infarction carvedilol improves insulin resistance compared to metoprolol. Clin. Res. Cardiol. 2006, 95, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, V.A. Effects of β-blockers on glucose and lipid metabolism. Curr. Med. Res. Opin. 2010, 26, 615–629. [Google Scholar] [CrossRef]
- De Peuter, O.R.; Verberne, H.J.; Kok, W.E.; van den Bogaard, B.; Schaap, M.; Nieuwland, R.; Meijers, J.C.; Somsen, G.A.; Bakx, A.; Kamphuisen, P.W. Differential effects of nonselective versus selective β-blockers on cardiac sympathetic activity and hemostasis in patients with heart failure. J. Nucl. Med. 2013, 54, 1733–1739. [Google Scholar] [CrossRef] [PubMed]
- Järvisalo, J.O.; Saris, N.-E.L. Action of propranolol on mitochondrial functions—Effects on energized ion fluxes in the presence of valinomycin. Biochem. Pharmacol. 1975, 24, 1701–1705. [Google Scholar]
- Komai, H.; Berkoff, H.A. Effects of quinidine and propranolol on energy transduction in beef heart mitochondria. Biochem. Pharmacol. 1979, 28, 1501–1504. [Google Scholar] [CrossRef]
- Bhayana, V.; Alto, L.E.; Dhalla, N.S. The effects of β-adrenergic receptor blockers on heart mitochondrial metabolism. Gen. Pharmacol. Vasc. System. 1980, 11, 271–274. [Google Scholar] [CrossRef]
- Kametani, R.; Miura, T.; Harada, N.; Shibuya, M.; Wang, R.; Tan, H.; Fukagawa, Y.; Kawamura, S.; Matsuzaki, M. Carvedilol inhibits mitochondrial oxygen consumption and superoxide production during calcium overload in isolated heart mitochondria. Circ. J. 2006, 70, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Goldhammer, E.; Maor, I.; Shnitzer, S.; Lanir, A.; Abinader, E.G. The early antioxidant effect of carvedilol predicts the clinical course in congestive heart failure patients. J. Cardiovasc. Med. 2007, 8, 453–456. [Google Scholar] [CrossRef]
- Noack, E.; Greeff, K. Inhibition of calcium transport in mitochondria by -receptor blocking substances and its reactivation by phospholipids. Experientia 1971, 27, 810–811. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Lee, S.L. Comparison of the actions of acebutolol, practolol and propranolol on calcium transport by heart microsomes and mitochondria. Br. J. Pharmac. 1976, 57, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Palaniyandi, S.S.; Qi, X.; Yogalingam, G.; Ferreira, J.C.B.; Mochly-Rosen, D. Regulation of mitochondrial processes: A target for heart failure. Drug Discov. Today Dis. Mech. 2010, 7, 1–14. [Google Scholar] [CrossRef]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Staudt, A.; Mobini, R.; Fu, M.; Grosse, Y.; Stangl, V.; Stangl, K.; Thiele, A.; Baumann, G.; Felix, S.B. β1-Adrenoceptor antibodies induce positive inotropic response in isolated cardiomyocytes. Eur. J. Pharmacol. 2001, 423, 115–119. [Google Scholar] [CrossRef]
- Wallukat, G.; Müller, J.; Podlowski, S.; Nissen, E.; Morwinski, R.; Hetzer, R. Agonist-like β-adrenoceptor antibodies in heart failure. Am. J. Cardiol. 1999, 83, 75H–79H. [Google Scholar] [CrossRef]
- Magnusson, Y.; Wallukat, G.; Waagstein, F.; Hjalmarson, A.; Hoebeke, J. Autoimmunity in idiopathic dilated cardiomyopathy. Characterization of antibodies against the β1-adrenoceptor with positive chronotropic effect. Circulation 1994, 89, 2760–2767. [Google Scholar] [CrossRef] [PubMed]
- Staudt, Y.; Mobini, R.; Fu, M.; Felix, S.B.; Kühn, J.P.; Staudt, A. β1-adrenoceptor antibodies induce apoptosis in adult isolated cardiomyocytes. Eur. J. Pharmacol. 2003, 466, 1–6. [Google Scholar] [CrossRef]
- Shi, L.; Liu, J.; Zhang, Y.; Chen, M.; Liu, J. β1-adrenoceptor antibodies induce myocardial apoptosis via inhibiting PGC-1α-related pathway. BMC Cardiovasc. Disord. 2020, 20, 269. [Google Scholar] [CrossRef]
- Wen, J.; Wang, J.; Li, P.; Wang, R.; Wang, J.; Zhou, X.; Zhang, L.; Li, H.; Wei, S.; Cai, H.; et al. Protective effects of higenamine combined with (6)-gingerol against doxorubicin-induced mitochondrial dysfunction and toxicity in H9c2 cells and potential mechanisms. Biomed. Pharmacother. 2019, 115, 108881. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Jiang, Q.; Yin, J.; Chen, J.; Ma, X.; Wu, M.; Liu, G.; Yao, K.; Tan, B.; Yin, Y. Mitochondria-Targeted Antioxidants: A Step towards Disease Treatment. Oxidative Med. Cell. Longev. 2020, 8837893. [Google Scholar] [CrossRef]
- Kiyuna, L.A.; Albuquerque, R.P.E.; Chen, C.H.; Mochly-Rosen, D.; Ferreira, J.C.B. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free Radic. Biol. Med. 2018, 129, 155–168. [Google Scholar] [CrossRef]
- Eichhorn, E.J.; Bedotto, J.B.; Malloy, C.R.; Hatfield, B.A.; Deitchman, D.; Brown, M.; Willard, J.E.; Grayburn, P.A. Effect of β-adrenergic blockade on myocardial function and energetics in congestive heart failure. Improvements in hemodynamic, contractile, and diastolic performance with bucindolol. Circulation 1990, 82, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, E.J.; Heesch, C.M.; Barnett, J.H.; Alvarez, L.G.; Fass, S.M.; Grayburn, P.A.; Hatfield, B.A.; Marcoux, L.G.; Malloy, C.R. Effect of metoprolol on myocardial function and energetics in patients with nonischemic dilated cardiomyopathy A randomized, double-blind, placebo-controlled study. J. Am. Coll. Cardiol. 1994, 24, 1310–1320. [Google Scholar] [CrossRef]
- Andersson, B.; Lomsky, M.; Waagstein, F. The link between acute haemodynamic adrenergic beta-blockade and long-term effects in patients with heart failure. A study on diastolic function, heart rate and myocardial metabolism following intravenous metoprolol. Eur. Heart J. 1993, 14, 1375–1385. [Google Scholar] [CrossRef]
- Galie, N.; Branzi, A.; Magnani, G.; Melandri, G.; Caldarera, I.; Rapezzi, C.; Grattoni, C.; Magnani, B. Effect of enoximone alone and in combination with metoprolol on myocardial function and energetics in severe congestive heart failure: Improvement in hemodynamic and metabolic profile. Cardiovasc. Drugs Ther. 1993, 7, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Beanlands, R.S.; Nahmias, C.; Gordon, E.; Coates, G.; de Kemp, R.; Firnau, G.; Fallen, E. The effects of β1-blockade on oxidative metabolism and the metabolic cost of ventricular work in patients with left ventricular dysfunction: A double-blind, placebo-controlled, positron-emission tomography study. Circulation 2000, 102, 2070–2075. [Google Scholar] [CrossRef]
- Sanchez-Roman, I.; Gomez, J.; Naudi, A.; Ayala, V.; Portero-Otín, M.; Lopez-Torres, M.; Pamplona, R.; Barja, G. The β-blocker atenolol lowers the longevity-related degree of fatty acid unsaturation, decreases protein oxidative damage and increases ERK signaling in the heart of C57BL/6 mice. Rejuvenation Res. 2010, 13, 683–693. [Google Scholar] [CrossRef]
- Gómez, A.; Sánchez-Roman, I.; Gomez, J.; Cruces, J.; Mate, I.; Lopez-Torres, M.; Naudi, A.; Portero-Otin, M.; Pamplona, R.; De la Fuente, M.; et al. Lifelong treatment with atenolol decreases membrane fatty acid unsaturation and oxidative stress in heart and skeletal muscle mitochondria and improves immunity and behavior, without changing mice longevity. Aging Cell. 2014, 13, 551–560. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Port, J.D.; Asano, K.; Chidiak, P.; Bouvier, M.; Dutcher, D.; Roden, R.L.; Minobe, W.; Tremmel, K.D.; Bristow, M.R. Cardiac adrenergic receptor effects of carvedilol. Eur. Heart J. 1996, 17, 8–16. [Google Scholar] [CrossRef]
- ALLHAT Collaborative Research Group. Major cardiovascular events in hypertensive patients randomized to doxazosin vs chlorthalidone: The antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT). JAMA 2000, 283, 1967–1975. [Google Scholar] [CrossRef]
- Molenaar, P.; Christ, T.; Ravens, U.; Kaumann, A. Carvedilol blocks β2- more than β1-adrenoceptors in human heart. Cardiovasc. Res. 2006, 69, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, P.; Christ, T.; Berk, E.; Engel, A.; Gillette, K.T.; Galindo-Tovar, A.; Ravens, U.; Kaumann, A.J. Carvedilol induces greater control of β2- than β1-adrenoceptor-mediated inotropic and lusitropic effects by PDE3, while PDE4 has no effect in human failing myocardium. Naunyn. Schmiedebergs. Arch. Pharmacol. 2014, 387, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Koshimizu, T.A.; Tsujimoto, G.; Hirasawa, A.; Kitagawa, Y.; Tanoue, A. Carvedilol selectively inhibits oscillatory intracellular calcium changes evoked by human α1D- and α1B-adrenergic receptors. Cardiovasc. Res. 2004, 63, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Iaccarino, G.; Keys, J.R.; Rapacciuolo, A.; Shotwell, K.F.; Lefkowitz, R.J.; Rockman, H.A.; Koch, W.J. Regulation of myocardial βARK1 expression in catecholamine-induced cardiac hypertrophy in transgenic mice overexpressing α1B-adrenergic receptors. J. Am. Coll. Cardiol. 2001, 38, 534–540. [Google Scholar] [CrossRef]
- Kukin, M.L.; Kalman, J.; Charney, R.H.; Levy, D.K.; Buchholz-Varley, C.; Ocampo, O.N.; Eng, C. Prospective, randomized comparison of effect of long-term treatment with metoprolol or carvedilol on symptoms, exercise, ejection fraction, and oxidative stress in heart failure. Circulation 1999, 99, 2645–2651. [Google Scholar] [CrossRef]
- Lysko, P.G.; Webb, C.L.; Gu, J.L.; Ohlstein, E.H.; Ruffolo, R.R., Jr.; Yue, T.L. A comparison of carvedilol and metoprolol antioxidant activities in vitro. J. Cardiovasc. Pharmacol. 2000, 36, 277–281. [Google Scholar] [CrossRef]
- Arumanayagam, M.; Chan, S.; Tong, S.; Sanderson, J.E. Antioxidant properties of carvedilol and metoprolol in heart failure: A double-blind randomized controlled trial. J. Cardiovasc. Pharmacol. 2001, 37, 48–54. [Google Scholar] [CrossRef]
- Yasunari, K.; Maeda, K.; Nakamura, M.; Watanabe, T.; Yoshikawa, J.; Asada, A. Effects of carvedilol on oxidative stress in polymorphonuclear and mononuclear cells in patients with essential hypertension. Am. J. Med. 2004, 116, 460–465. [Google Scholar] [CrossRef]
- Kveiborg, B.; Christiansen, B.; Major-Petersen, A.; Torp-Pedersen, C. Metabolic Effects of β-Adrenoceptor Antagonists with Special Emphasis on Carvedilol. Am. J. Cardiovasc. Drugs 2006, 6, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Yao, A.; Kohmoto, O.; Oyama, T.; Sugishita, Y.; Shimizu, T.; Harada, K.; Matsui, H.; Komuro, I.; Nagai, R.; Matsuo, H.; et al. Characteristic effects of α1-β1,2-adrenergic blocking agent, carvedilol, on [Ca2+]i in ventricular myocytes compared with those of timolol and atenolol. Circ. J. 2003, 67, 83–90. [Google Scholar] [CrossRef][Green Version]
- Heesch, C.; Marcoux, L.; Hatfield, B.; Eichhorn, E.J. Hemodynamic and energetic comparison of carvedilol and metoprolol for the treatment of congestive heart failure. Am. J. Cardiol. 1995, 75, 360–364. [Google Scholar] [CrossRef]
- Onay-Besikci, A.; Suzmecelik, E.; Ozcelikay, A.T. Carvedilol suppresses fatty acid oxidation and stimulates glycolysis in C2C12 cells. Can. J. Physiol. Pharmacol. 2012, 90, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Toda, N. Vasodilating β-adrenoceptor blockers as cardiovascular therapeutics. Pharmacol. Ther. 2003, 100, 215–234. [Google Scholar] [CrossRef]
- Grandinetti, V.; Carlos, F.P.; Antonio, E.L.; de Oliveira, H.A.; Dos Santos, L.; Yoshizaki, A.; Mansano, B.; Silva, F.A.; Porte, L.A.; Albuquerque-Pontes, G.M.; et al. Photobiomodulation therapy combined with carvedilol attenuates post-infarction heart failure by suppressing excessive inflammation and oxidative stress in rats. Sci. Rep. 2019, 9, 9425. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, S.; Haruyama, A.; Inami, S.; Arikawa, T.; Saito, F.; Watanabe, R.; Sakuma, M.; Abe, S.; Nakajima, T.; Tanaka, A.; et al. Effects of carvedilol vs bisoprolol on inflammation and oxidative stress in patients with chronic heart failure. J. Cardiol. 2020, 75, 140–147. [Google Scholar] [CrossRef]
- Gomes, K.M.; Bechara, L.R.; Lima, V.M.; Ribeiro, M.A.; Campos, J.C.; Dourado, P.M.; Kowaltowski, A.J.; Mochly-Rosen, D.; Ferreira, J.C. Aldehydic load and aldehyde dehydrogenase 2 profile during the progression of post-myocardial infarction cardiomyopathy: Benefits of Alda. Int. J. Cardiol. 2015, 179, 129–138. [Google Scholar] [CrossRef]
- Jacob, S.; Rett, K.; Wicklmayr, M.; Agrawal, B.; Augustin, H.J.; Dietze, G.J. Differential effect of chronic treatment with two b-blocking agents on insulin sensitivity: The carvedilol-metoprolol study. J. Hypertens. 1996, 14, 489–494. [Google Scholar] [CrossRef]
- Giugliano, D.; Acampora, R.; Marfella, R.; De Rosa, N.; Ziccardi, P.; Ragone, R.; De Angelis, L.; D’Onofrio, F. Metabolic and cardiovascular effects of carvedilol and atenolol in non-insulin-dependent diabetes mellitus and hypertension. A randomized, controlled trial. Ann. Intern. Med. 1997, 126, 955–959. [Google Scholar] [CrossRef]
- Scolletta, S.; Biagioli, B. Energetic myocardial metabolism and oxidative stress: Let’s make them our friends in the fight against heart failure. Biomed. Pharmacother. 2010, 64, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Beadle, R.M.; Frenneaux, M. Modification of myocardial substrate utilization: A new therapeutic paradigm in cardiovascular disease. Heart 2010, 96, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Li, X.; Ren, D.; Tan, Y.; Chen, J.; Yang, L.; Chen, R.; Li, J.; Zhu, P. The cardioprotective effects of carvedilol on ischemia and reperfusion injury by AMPK signaling pathway. Biomed. Pharmacother. 2019, 117, 109106. [Google Scholar] [CrossRef]
- Chabowski, A.; Momken, I.; Coort, S.L.; Calles-Escandon, J.; Tandon, N.N.; Glatz, J.F.; Luiken, J.J.; Bonen, A. Prolonged AMPK activation increases the expression of fatty acid transporters in cardiac myocytes and perfused hearts. Mol. Cell Biochem. 2006, 288, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Kudo, N.; Barr, A.J.; Barr, R.L.; Desai, S.; Lopaschuk, G.D. High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5’-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J. Biol. Chem. 1995, 270, 17513–17520. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Kewalramani, G.; Qi, D.; Pulinilkunnil, T.; Ghosh, S.; Abrahani, A.; Wambolt, R.; Allard, M.; Innis, S.M.; Rodrigues, B. β-Agonist stimulation produces changes in cardiac AMPK and coronary lumen LPL only during increased workload. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1120–E1127. [Google Scholar] [CrossRef]
- Bussey, C.T.; Thaung, H.P.A.; Hughes, G.; Bahn, A.; Lamberts, R.R. Cardiac b-adrenergic responsiveness of obese Zucker rats: The role of AMPK. Exp. Physiol. 2018, 103, 1067–1075. [Google Scholar] [CrossRef]
- Wang, J.; Song, Y.; Li, H.; Shen, Q.; Shen, J.; An, X.; Wu, J.; Zhang, J.; Wu, Y.; Xiao, H.; et al. Exacerbated cardiac fibrosis induced by β-adrenergic activation in old mice due to decreased AMPK activity. Clin. Exp. Pharmacol. Physiol. 2016, 43, 1029–1037. [Google Scholar] [CrossRef]
- Dubois-Deruy, E.; Gelinas, R.; Beauloye, C.; Esfahani, H.; Michel, L.Y.M.; Dessy, C.; Bertrand, L.; Balligand, J.L. β3-adrenoreceptors protect from hypertrophic remodelling through AMP-activated protein kinase and autophagy. Esc. Heart Fail 2020, 7, 920–932. [Google Scholar] [CrossRef]



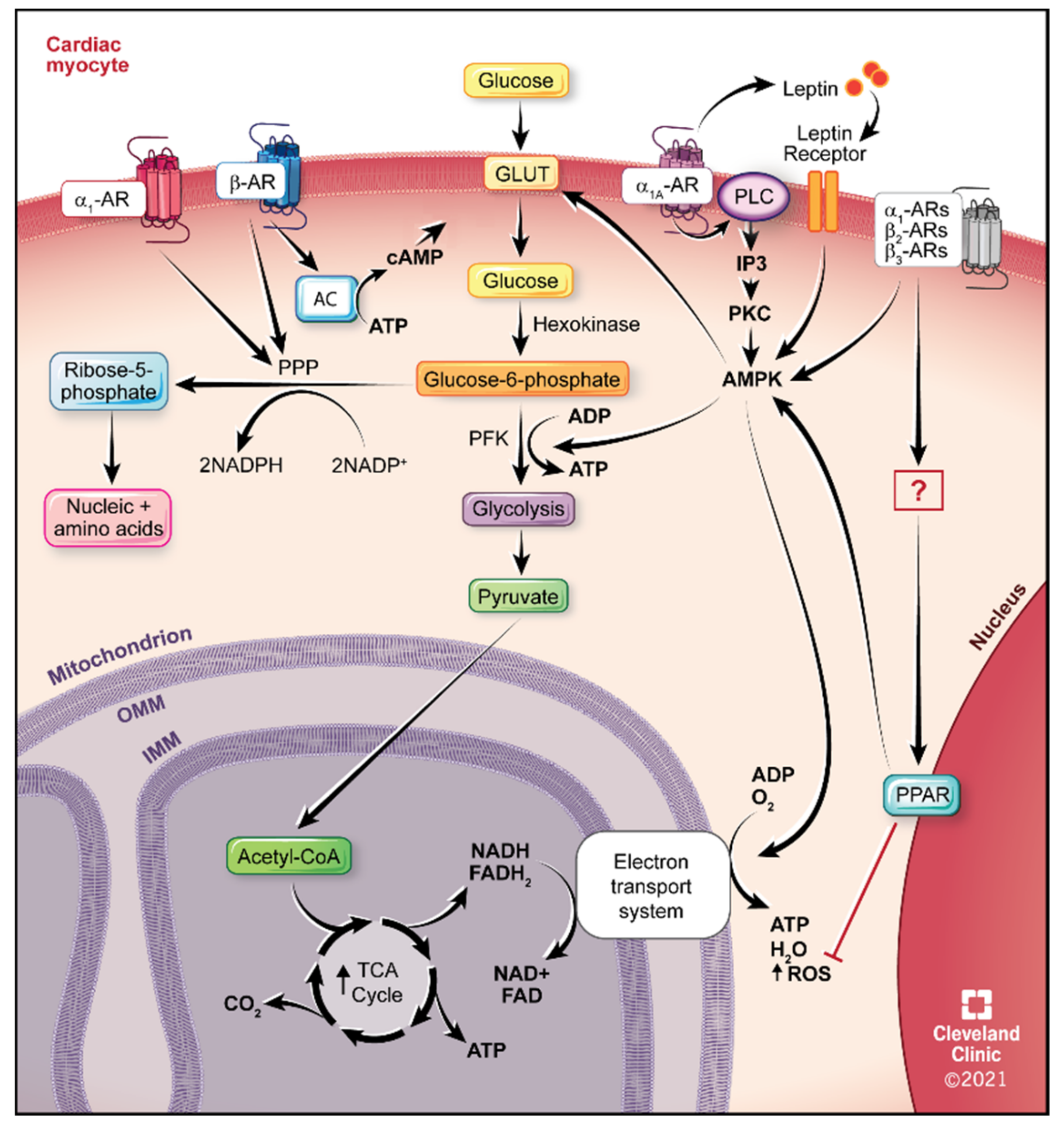{kind=link}
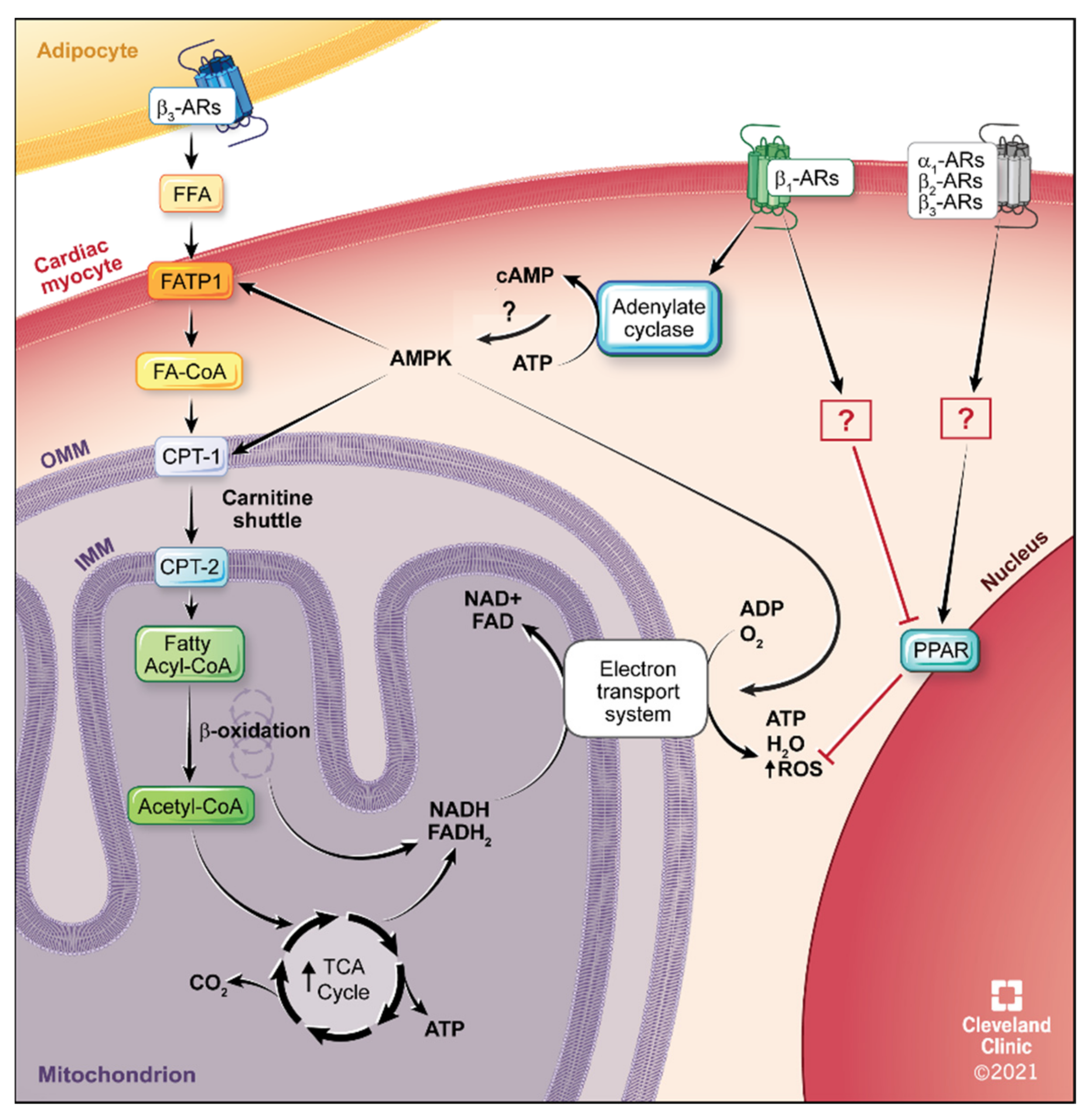{kind=link}
| Subtype | Signal Transduction | Tissue Distribution | Physiological Function |
|---|---|---|---|
| α1A | Gq/G11/PLC/PKC/ DAG/IP3/Ca+2 | Cardiac myocyte Vascular smooth muscle | Positive inotropy, chronotropy, cardiac hypertrophy, contraction smooth muscle, blood pressure |
| α1B | Gq/G11/PLC/PKC/ DAG/IP3/Ca+2 | Cardiac myocyte Vascular smooth muscle | Negative inotropy, cardiac hypertrophy, contraction smooth muscle, blood pressure |
| α1D | Gq/G11/PLC/PKC/ DAG/IP3/Ca+2 | Coronary arteries Vascular smooth muscle | Contraction smooth muscle, blood pressure |
| α2A α2B α2C | Gi/inhibit AC/ cAMP/PKA | Not in any cardiac tissue Vascular smooth muscle | NE release- Sympathetic nerve endings |
| β1 | Gs/AC/cAMP/PKA Ca+2 channel | Cardiac myocyte | Positive inotropy, chronotropy, cardiac hypertrophy |
| β2 | Gs/AC/cAMP/PKA Ca+2 channel Gi/inhibit AC/ cAMP/PKA | Cardiac myocyte Vascular smooth muscle | Cardiac hypertrophy Relaxation smooth muscle |
| β3 | Gs/Gi/AC/cAMP/PKA NO | Cardiac myocyte | Negative inotropy |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez, D.M. Targeting Adrenergic Receptors in Metabolic Therapies for Heart Failure. Int. J. Mol. Sci. 2021, 22, 5783. https://doi.org/10.3390/ijms22115783
Perez DM. Targeting Adrenergic Receptors in Metabolic Therapies for Heart Failure. International Journal of Molecular Sciences. 2021; 22(11):5783. https://doi.org/10.3390/ijms22115783
Chicago/Turabian StylePerez, Dianne M. 2021. "Targeting Adrenergic Receptors in Metabolic Therapies for Heart Failure" International Journal of Molecular Sciences 22, no. 11: 5783. https://doi.org/10.3390/ijms22115783
APA StylePerez, D. M. (2021). Targeting Adrenergic Receptors in Metabolic Therapies for Heart Failure. International Journal of Molecular Sciences, 22(11), 5783. https://doi.org/10.3390/ijms22115783