
CX-5461 Enhances the Efficacy of APR-246 via Induction of DNA Damage and Replication Stress in Triple-Negative Breast Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
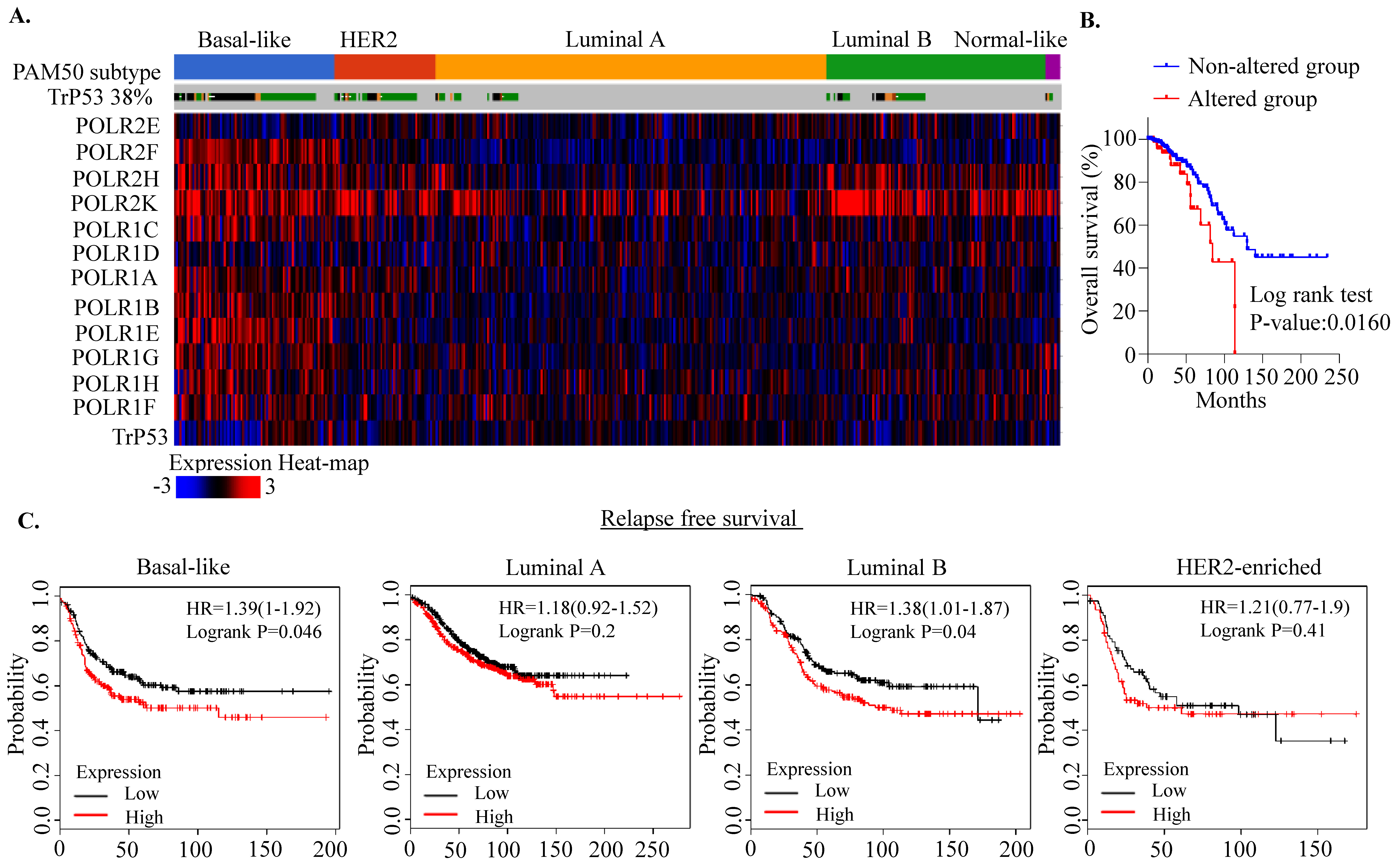
2.1. RNA Polymerase I Transcriptional Alteration Predicts Poor Clinical Outcome in Breast Cancer
2.2. APR-246 and CX-5461 Combination Enhances Growth Suppression Effect in a Panel of Breast Cancer Cell Lines
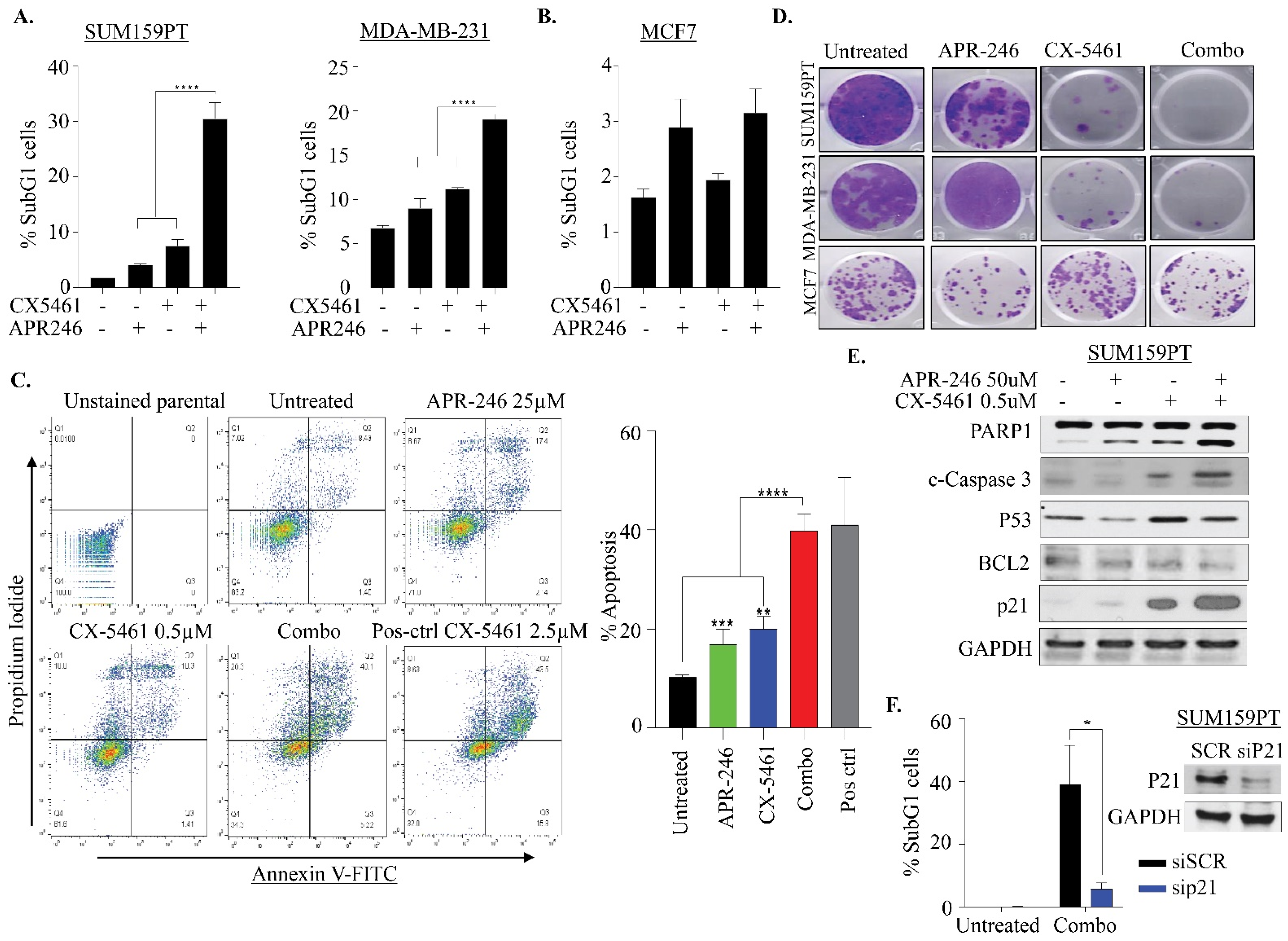
2.3. APR-246 and CX-5461 Combination Therapy Causes Apoptosis in a p21-Dependent Manner in TNBC Cells
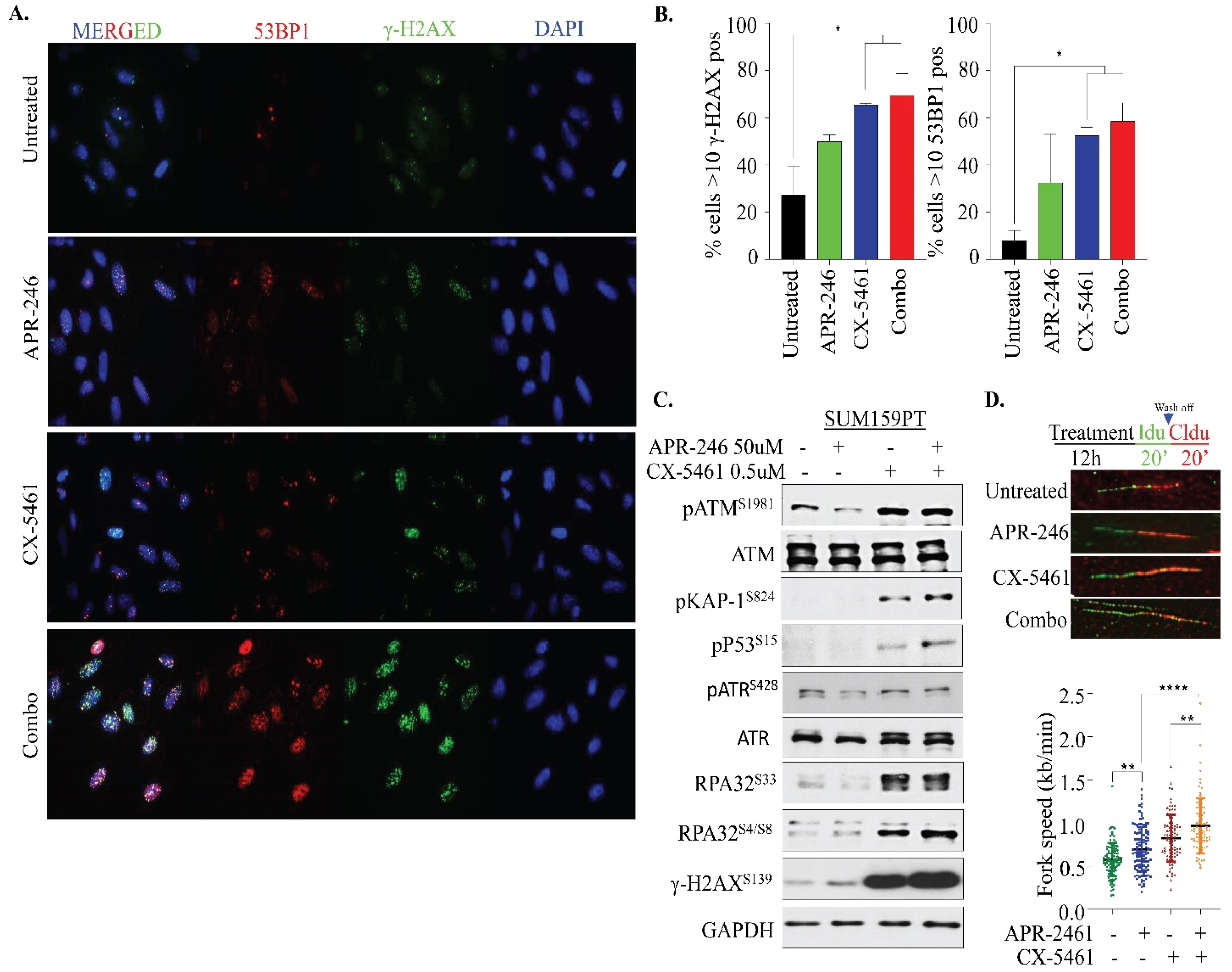
2.4. APR-246 and CX-5461 Combination Therapy Elicits DNA Damage and Replication Stress
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Immunoblotting
4.5. Cell Cycle Analysis
4.6. Colony Formation Assay
4.7. Immunofluorescence
4.8. AnnexinV/PI Staining
4.9. DNA Fiber Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalimutho, M.; Parsons, K.; Mittal, D.; Lopez, J.A.; Srihari, S.; Khanna, K.K. Targeted Therapies for Triple-Negative Breast Cancer: Combating a Stubborn Disease. Trends Pharmacol. Sci. 2015, 36, 822–846. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Reis-Filho, J.S.; Ellis, I.O. Basal-like breast cancer: A critical review. J. Clin. Oncol. 2008, 26, 2568–2581. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- De Giorgi, U.; Rosti, G.; Frassineti, L.; Kopf, B.; Giovannini, N.; Zumaglini, F.; Marangolo, M. High-dose chemotherapy for triple negative breast cancer. Ann. Oncol. 2006, 18, 202–203. [Google Scholar] [CrossRef] [PubMed]
- O’shaughnessy, J.; Osborne, C.; Pippen, J.E.; Yoffe, M.; Patt, D.; Rocha, C.; Koo, I.C.; Sherman, B.M.; Bradley, C. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N. Engl. J. Med. 2011, 364, 205–214. [Google Scholar] [CrossRef]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Carey, L.A.; Rugo, H.S.; Marcom, P.K.; Mayer, E.L.; Esteva, F.J.; Ma, C.X.; Liu, M.C.; Storniolo, A.M.; Rimawi, M.F.; Forero-Torres, A. TBCRC 001: Randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J. Clin. Oncol. 2012, 30, 2615. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359. [Google Scholar] [CrossRef] [PubMed]
- Kalimutho, M.; Nones, K.; Srihari, S.; Duijf, P.H.G.; Waddell, N.; Khanna, K.K. Patterns of Genomic Instability in Breast Cancer. Trends Pharmacol. Sci. 2019, 40, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333. [Google Scholar] [CrossRef]
- Leroy, B.; Fournier, J.L.; Ishioka, C.; Monti, P.; Inga, A.; Fronza, G.; Soussi, T. The TP53 website: An integrative resource centre for the TP53 mutation database and TP53 mutant analysis. Nucleic Acids Res. 2012, 41, D962–D969. [Google Scholar] [CrossRef]
- Duffy, M.J.; Synnott, N.C.; McGowan, P.M.; Crown, J.; O’Connor, D.; Gallagher, W.M. p53 as a target for the treatment of cancer. Cancer Treat. Rev. 2014, 40, 1153–1160. [Google Scholar] [CrossRef]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.A.; Iwakuma, T.; Suh, Y.-A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42. [Google Scholar] [CrossRef]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282. [Google Scholar] [CrossRef]
- Bykov, V.; Selivanova, G.; Wiman, K. Small molecules that reactivate mutant p53. Eur. J. Cancer 2003, 39, 1828–1834. [Google Scholar] [CrossRef]
- Synnott, N.C.; Madden, S.F.; Bykov, V.J.N.; Crown, J.; Wiman, K.G.; Duffy, M.J. The Mutant p53-Targeting Compound APR-246 Induces ROS-Modulating Genes in Breast Cancer Cells. Transl. Oncol. 2018, 11, 1343–1349. [Google Scholar] [CrossRef]
- Peng, X.; Zhang, M.Q.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.J.; Arner, E.S.; Wiman, K.G. APR-246/PRIMA-1(MET) inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2017, 8, e2751. [Google Scholar] [CrossRef] [PubMed]
- Mohell, N.; Alfredsson, J.; Fransson, A.; Uustalu, M.; Bystrom, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.; Bjorklund, U.; Wiman, K.G. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015, 6, e1794. [Google Scholar] [CrossRef]
- Drygin, D.; Rice, W.G.; Grummt, I. The RNA polymerase I transcription machinery: An emerging target for the treatment of cancer. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 131–156. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Schneekloth, J.S., Jr.; Panov, K.I.; Hannan, K.M.; Hannan, R.D. Targeting the RNA Polymerase I Transcription for Cancer Therapy Comes of Age. Cells 2020, 9, 266. [Google Scholar] [CrossRef]
- Haddach, M.; Schwaebe, M.K.; Michaux, J.; Nagasawa, J.; O’Brien, S.E.; Whitten, J.P.; Pierre, F.; Kerdoncuff, P.; Darjania, L.; Stansfield, R.; et al. Discovery of CX-5461, the First Direct and Selective Inhibitor of RNA Polymerase I, for Cancer Therapeutics. ACS Med. Chem. Lett. 2012, 3, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P. Anticancer activity of CX-3543: A direct inhibitor of rRNA biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef]
- Drygin, D.; Lin, A.; Bliesath, J.; Ho, C.B.; O’Brien, S.E.; Proffitt, C.; Omori, M.; Haddach, M.; Schwaebe, M.K.; Siddiqui-Jain, A. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011, 71, 1418–1430. [Google Scholar] [CrossRef]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Dos Santos, N.; Silvester, J.; Wei, V.; Garcia, J. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef]
- Russell, J.; Zomerdijk, J.C. The RNA polymerase I transcription machinery. Biochem. Soc. Symp. 2006, 73, 1203–1216. [Google Scholar] [CrossRef]
- Marcotte, R.; Brown, K.R.; Suarez, F.; Sayad, A.; Karamboulas, K.; Krzyzanowski, P.M.; Sircoulomb, F.; Medrano, M.; Fedyshyn, Y.; Koh, J.L.Y.; et al. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2012, 2, 172–189. [Google Scholar] [CrossRef] [PubMed]
- Bruno, P.M.; Lu, M.; Dennis, K.A.; Inam, H.; Moore, C.J.; Sheehe, J.; Elledge, S.J.; Hemann, M.T.; Pritchard, J.R. The primary mechanism of cytotoxicity of the chemotherapeutic agent CX-5461 is topoisomerase II poisoning. Proc. Natl. Acad. Sci. USA 2020, 117, 4053–4060. [Google Scholar] [CrossRef] [PubMed]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Quin, J.; Chan, K.T.; Devlin, J.R.; Cameron, D.P.; Diesch, J.; Cullinane, C.; Ahern, J.; Khot, A.; Hein, N.; George, A.J.; et al. Inhibition of RNA polymerase I transcription initiation by CX-5461 activates non-canonical ATM/ATR signaling. Oncotarget 2016, 7, 49800–49818. [Google Scholar] [CrossRef]
- Khot, A.; Brajanovski, N.; Cameron, D.P.; Hein, N.; Maclachlan, K.H.; Sanij, E.; Lim, J.; Soong, J.; Link, E.; Blombery, P.; et al. First-in-Human RNA Polymerase I Transcription Inhibitor CX-5461 in Patients with Advanced Hematologic Cancers: Results of a Phase I Dose-Escalation Study. Cancer Discov. 2019, 9, 1036–1049. [Google Scholar] [CrossRef]
- Lehmann, S.; Bykov, V.J.; Ali, D.; Andren, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef]
- Liang, Y.; Besch-Williford, C.; Cook, M.T.; Belenchia, A.; Brekken, R.A.; Hyder, S.M. APR-246 alone and in combination with a phosphatidylserine-targeting antibody inhibits lung metastasis of human triple-negative breast cancer cells in nude mice. Breast Cancer 2019, 11, 249–259. [Google Scholar] [CrossRef]
- Liang, Y.; Mafuvadze, B.; Besch-Williford, C.; Hyder, S.M. A combination of p53-activating APR-246 and phosphatidylserine-targeting antibody potently inhibits tumor development in hormone-dependent mutant p53-expressing breast cancer xenografts. Breast Cancer 2018, 10, 53–67. [Google Scholar] [CrossRef]
- Menichini, P.; Monti, P.; Speciale, A.; Cutrona, G.; Matis, S.; Fais, F.; Taiana, E.; Neri, A.; Bomben, R.; Gentile, M.; et al. Antitumor Effects of PRIMA-1 and PRIMA-1(Met) (APR246) in Hematological Malignancies: Still a Mutant P53-Dependent Affair? Cells 2021, 10, 98. [Google Scholar] [CrossRef]
- Fransson, Å.; Glaessgen, D.; Alfredsson, J.; Wiman, K.G.; Bajalica-Lagercrantz, S.; Mohell, N. Strong synergy with APR-246 and DNA-damaging drugs in primary cancer cells from patients with TP53 mutant High-Grade Serous ovarian cancer. J. Ovarian Res. 2016, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Deben, C.; Lardon, F.; Wouters, A.; de Beeck, K.O.; Van den Bossche, J.; Jacobs, J.; Van Der Steen, N.; Peeters, M.; Rolfo, C.; Deschoolmeester, V. APR-246 (PRIMA-1MET) strongly synergizes with AZD2281 (olaparib) induced PARP inhibition to induce apoptosis in non-small cell lung cancer cell lines. Cancer Lett. 2016, 375, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Perdrix, A.; Najem, A.; Saussez, S.; Awada, A.; Journe, F.; Ghanem, G.; Krayem, M. PRIMA-1 and PRIMA-1Met (APR-246): From Mutant/Wild Type p53 Reactivation to Unexpected Mechanisms Underlying Their Potent Anti-Tumor Effect in Combinatorial Therapies. Cancers 2017, 9, 172. [Google Scholar] [CrossRef] [PubMed]
- Bywater, M.J.; Poortinga, G.; Sanij, E.; Hein, N.; Peck, A.; Cullinane, C.; Wall, M.; Cluse, L.; Drygin, D.; Anderes, K.; et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 2012, 22, 51–65. [Google Scholar] [CrossRef]
- Ismael, M.; Webb, R.; Ajaz, M.; Kirkby, K.J.; Coley, H.M. The Targeting of RNA Polymerase I Transcription Using CX-5461 in Combination with Radiation Enhances Tumour Cell Killing Effects in Human Solid Cancers. Cancers 2019, 11, 1429. [Google Scholar] [CrossRef] [PubMed]
- Zandi, R.; Selivanova, G.; Christensen, C.L.; Gerds, T.A.; Willumsen, B.M.; Poulsen, H.S. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. 2011, 17, 2830–2841. [Google Scholar] [CrossRef]
- Grellety, T.; Laroche-Clary, A.; Chaire, V.; Lagarde, P.; Chibon, F.; Neuville, A.; Italiano, A. PRIMA-1(MET) induces death in soft-tissue sarcomas cell independent of p53. BMC Cancer 2015, 15, 684. [Google Scholar] [CrossRef]
- Russo, D.; Ottaggio, L.; Foggetti, G.; Masini, M.; Masiello, P.; Fronza, G.; Menichini, P. PRIMA-1 induces autophagy in cancer cells carrying mutant or wild type p53. Biochim. Biophys. Acta 2013, 1833, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, Y.; Zhao, J.; Fan, S.; Wang, L.; Li, X. CX-5461 induces autophagy and inhibits tumor growth via mammalian target of rapamycin-related signaling pathways in osteosarcoma. Onco. Targets Ther. 2016, 9, 5985–5997. [Google Scholar] [CrossRef]
- Negi, S.S.; Brown, P. rRNA synthesis inhibitor, CX-5461, activates ATM/ATR pathway in acute lymphoblastic leukemia, arrests cells in G2 phase and induces apoptosis. Oncotarget 2015, 6, 18094–18104. [Google Scholar] [CrossRef] [PubMed]
- Sanij, E.; Hannan, K.M.; Xuan, J.; Yan, S.; Ahern, J.E.; Trigos, A.S.; Brajanovski, N.; Son, J.; Chan, K.T.; Kondrashova, O.; et al. CX-5461 activates the DNA damage response and demonstrates therapeutic efficacy in high-grade serous ovarian cancer. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Xuan, J.; Brajanovski, N.; Tancock, M.R.C.; Madhamshettiwar, P.B.; Simpson, K.J.; Ellis, S.; Kang, J.; Cullinane, C.; Sheppard, K.E.; et al. The RNA polymerase I transcription inhibitor CX-5461 cooperates with topoisomerase 1 inhibition by enhancing the DNA damage response in homologous recombination-proficient high-grade serous ovarian cancer. Br. J. Cancer 2021, 124, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, B.M.; Couch, F.B.; Feigerle, J.T.; Bhaskara, S.; Hiebert, S.W.; Cortez, D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011, 25, 1320–1327. [Google Scholar] [CrossRef]
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High speed of fork progression induces DNA replication stress and genomic instability. Nature 2018, 559, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Genois, M.-M.; Gagné, J.-P.; Yasuhara, T.; Jackson, J.; Saxena, S.; Langelier, M.-F.; Ahel, I.; Bedford, M.T.; Pascal, J.M.; Vindigni, A.; et al. CARM1 regulates replication fork speed and stress response by stimulating PARP1. Mol. Cell 2021, 81, 784–800.e8. [Google Scholar] [CrossRef]
- Kalimutho, M.; Sinha, D.; Mittal, D.; Srihari, S.; Nanayakkara, D.; Shafique, S.; Raninga, P.; Nag, P.; Parsons, K.; Khanna, K.K. Blockade of PDGFRbeta circumvents resistance to MEK-JAK inhibition via intratumoral CD8(+) T-cells infiltration in triple-negative breast cancer. J. Exp. Clin. Cancer Res. 2019, 38, 85. [Google Scholar] [CrossRef]
- Kalimutho, M.; Sinha, D.; Jeffery, J.; Nones, K.; Srihari, S.; Fernando, W.C.; Duijf, P.H.; Vennin, C.; Raninga, P.; Nanayakkara, D.; et al. CEP55 is a determinant of cell fate during perturbed mitosis in breast cancer. EMBO Mol. Med. 2018, 10, e8566. [Google Scholar] [CrossRef] [PubMed]
- Kalimutho, M.; Bain, A.L.; Mukherjee, B.; Nag, P.; Nanayakkara, D.M.; Harten, S.K.; Harris, J.L.; Subramanian, G.N.; Sinha, D.; Shirasawa, S.; et al. Enhanced dependency of KRAS-mutant colorectal cancer cells on RAD51-dependent homologous recombination repair identified from genetic interactions in Saccharomyces cerevisiae. Mol. Oncol. 2017, 11, 470–490. [Google Scholar] [CrossRef]
- Schwab, R.A.; Niedzwiedz, W. Visualization of DNA replication in the vertebrate model system DT40 using the DNA fiber technique. J. Vis. Exp. JoVE 2011, 56, e3255. [Google Scholar] [CrossRef]
- Henry-Mowatt, J.; Jackson, D.; Masson, J.Y.; Johnson, P.A.; Clements, P.M.; Benson, F.E.; Thompson, L.H.; Takeda, S.; West, S.C.; Caldecott, K.W. XRCC3 and Rad51 modulate replication fork progression on damaged vertebrate chromosomes. Mol. Cell 2003, 11, 1109–1117. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makhale, A.; Nanayakkara, D.; Raninga, P.; Khanna, K.K.; Kalimutho, M. CX-5461 Enhances the Efficacy of APR-246 via Induction of DNA Damage and Replication Stress in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2021, 22, 5782. https://doi.org/10.3390/ijms22115782
Makhale A, Nanayakkara D, Raninga P, Khanna KK, Kalimutho M. CX-5461 Enhances the Efficacy of APR-246 via Induction of DNA Damage and Replication Stress in Triple-Negative Breast Cancer. International Journal of Molecular Sciences. 2021; 22(11):5782. https://doi.org/10.3390/ijms22115782
Chicago/Turabian StyleMakhale, Ashwini, Devathri Nanayakkara, Prahlad Raninga, Kum Kum Khanna, and Murugan Kalimutho. 2021. "CX-5461 Enhances the Efficacy of APR-246 via Induction of DNA Damage and Replication Stress in Triple-Negative Breast Cancer" International Journal of Molecular Sciences 22, no. 11: 5782. https://doi.org/10.3390/ijms22115782
APA StyleMakhale, A., Nanayakkara, D., Raninga, P., Khanna, K. K., & Kalimutho, M. (2021). CX-5461 Enhances the Efficacy of APR-246 via Induction of DNA Damage and Replication Stress in Triple-Negative Breast Cancer. International Journal of Molecular Sciences, 22(11), 5782. https://doi.org/10.3390/ijms22115782