
Hepatic and Extrahepatic Sources and Manifestations in Endoplasmic Reticulum Storage Diseases



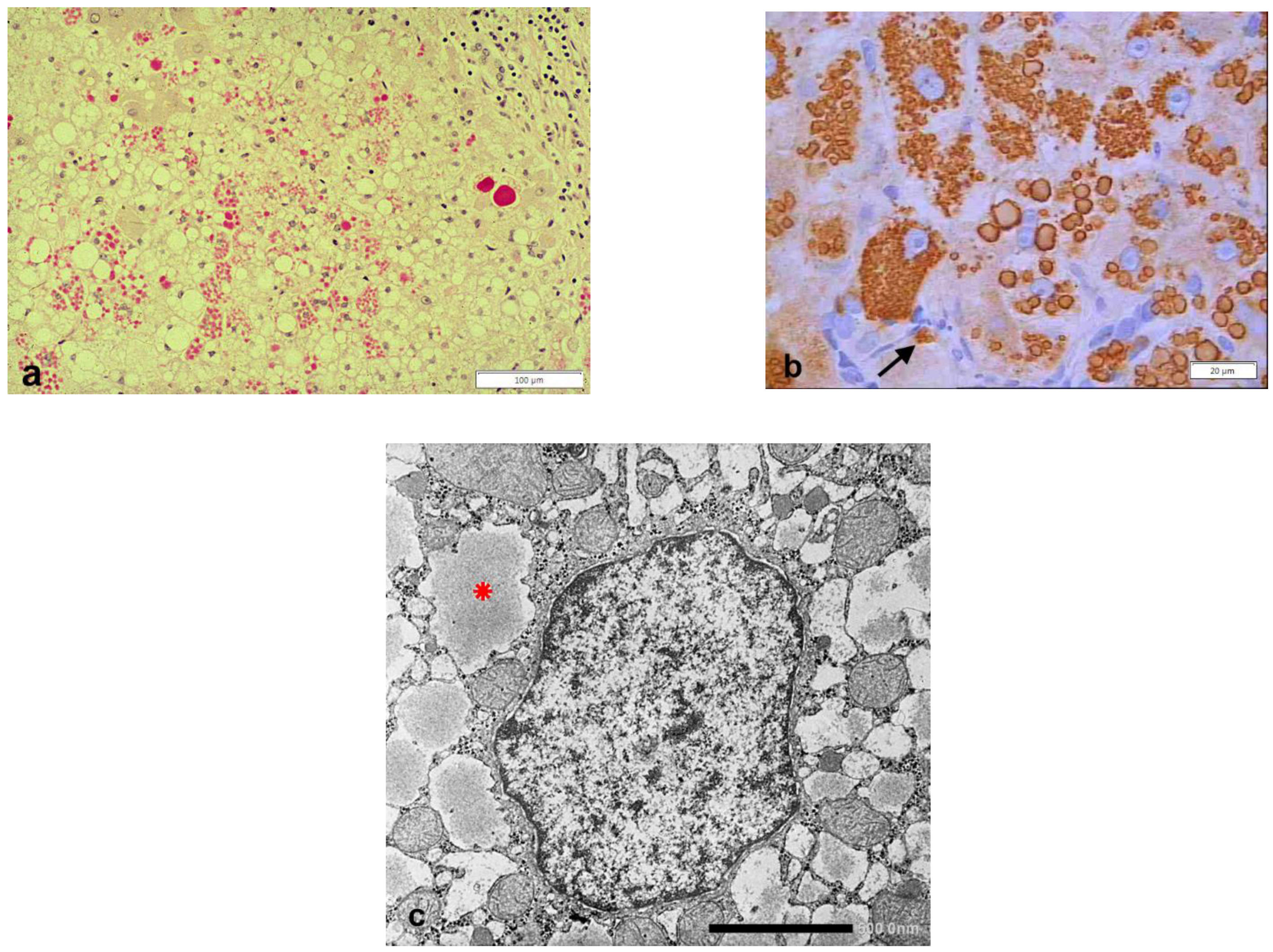{kind=link}
{kind=link}
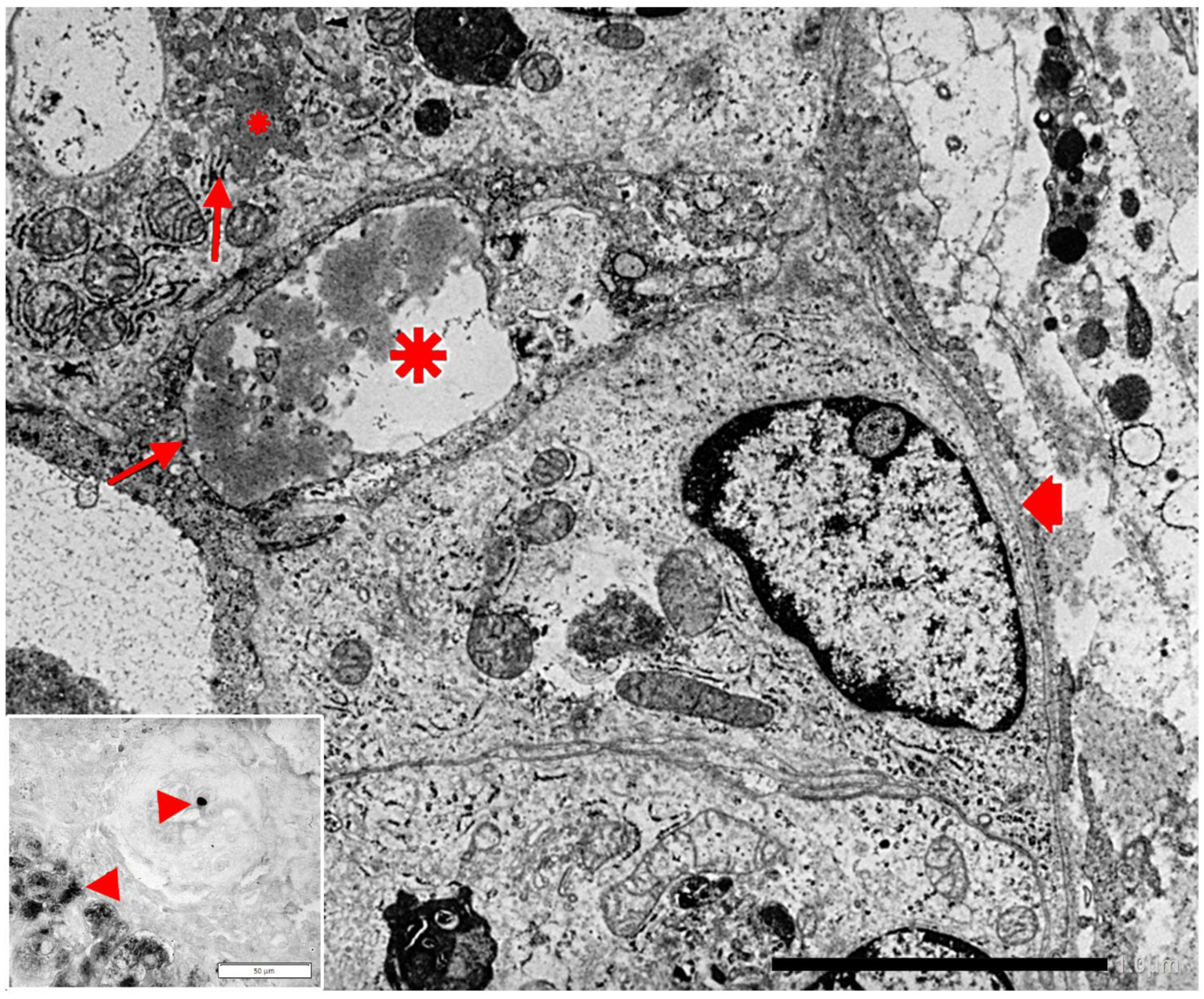{kind=link}
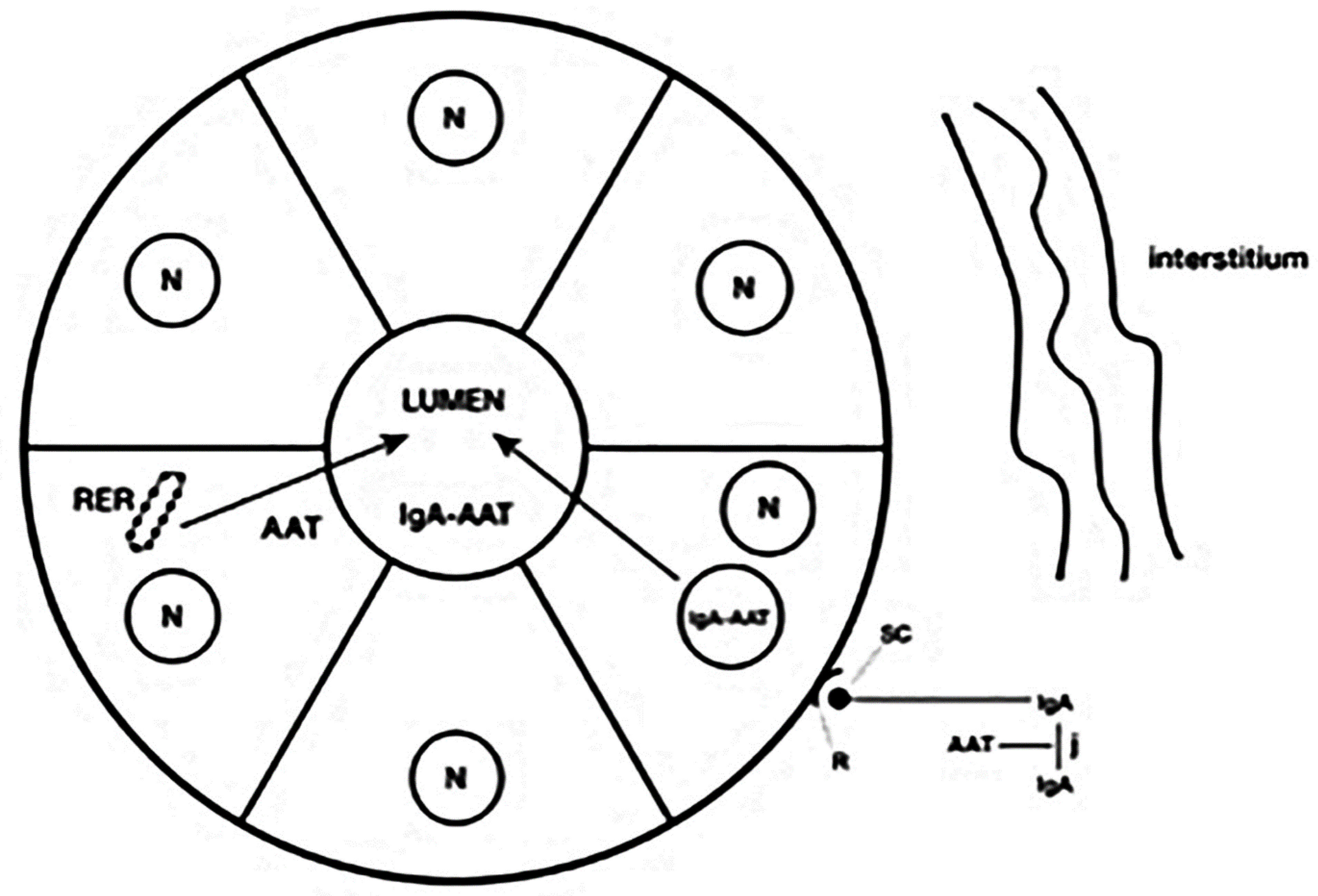{kind=link}
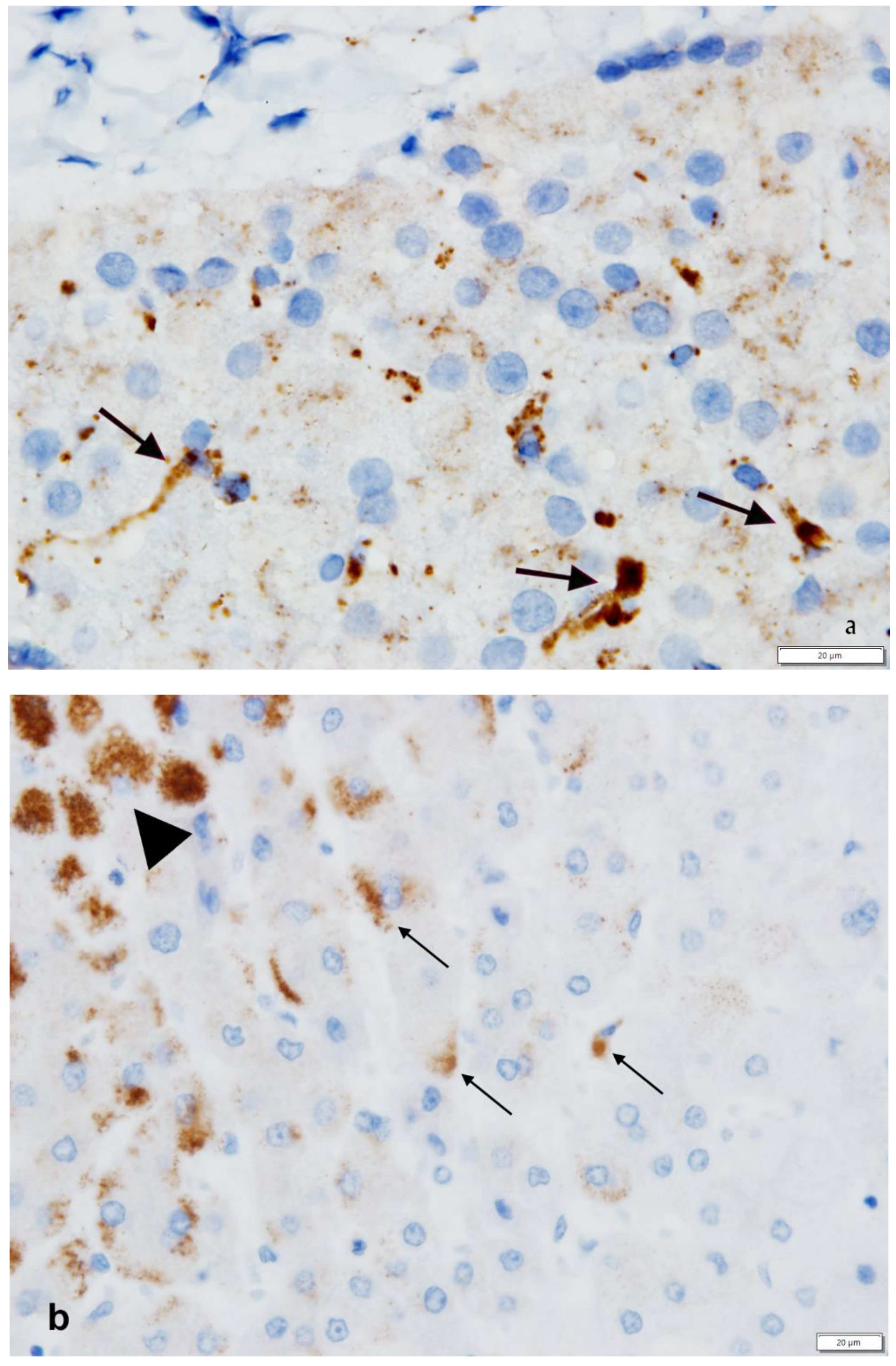{kind=link}
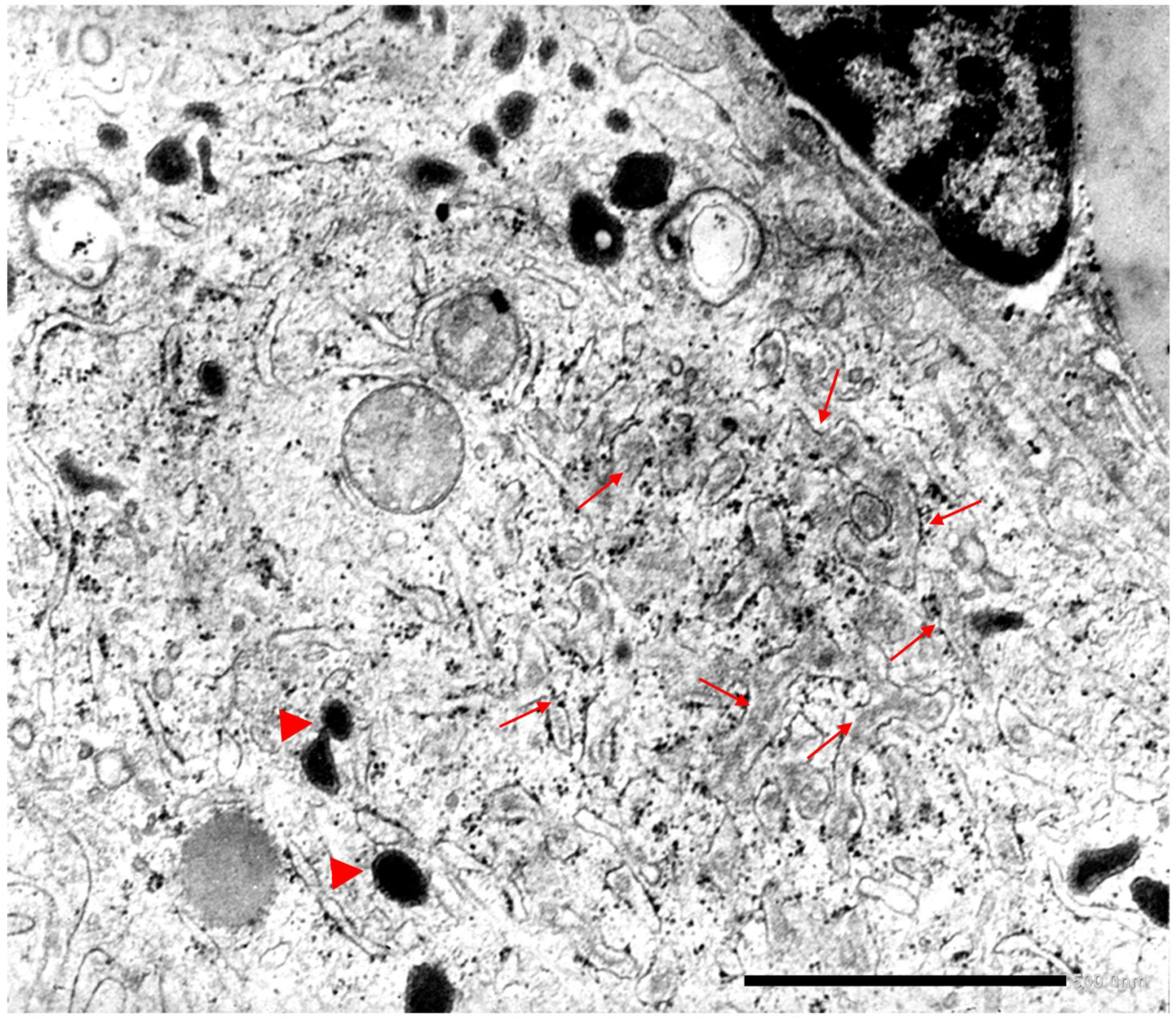{kind=link}
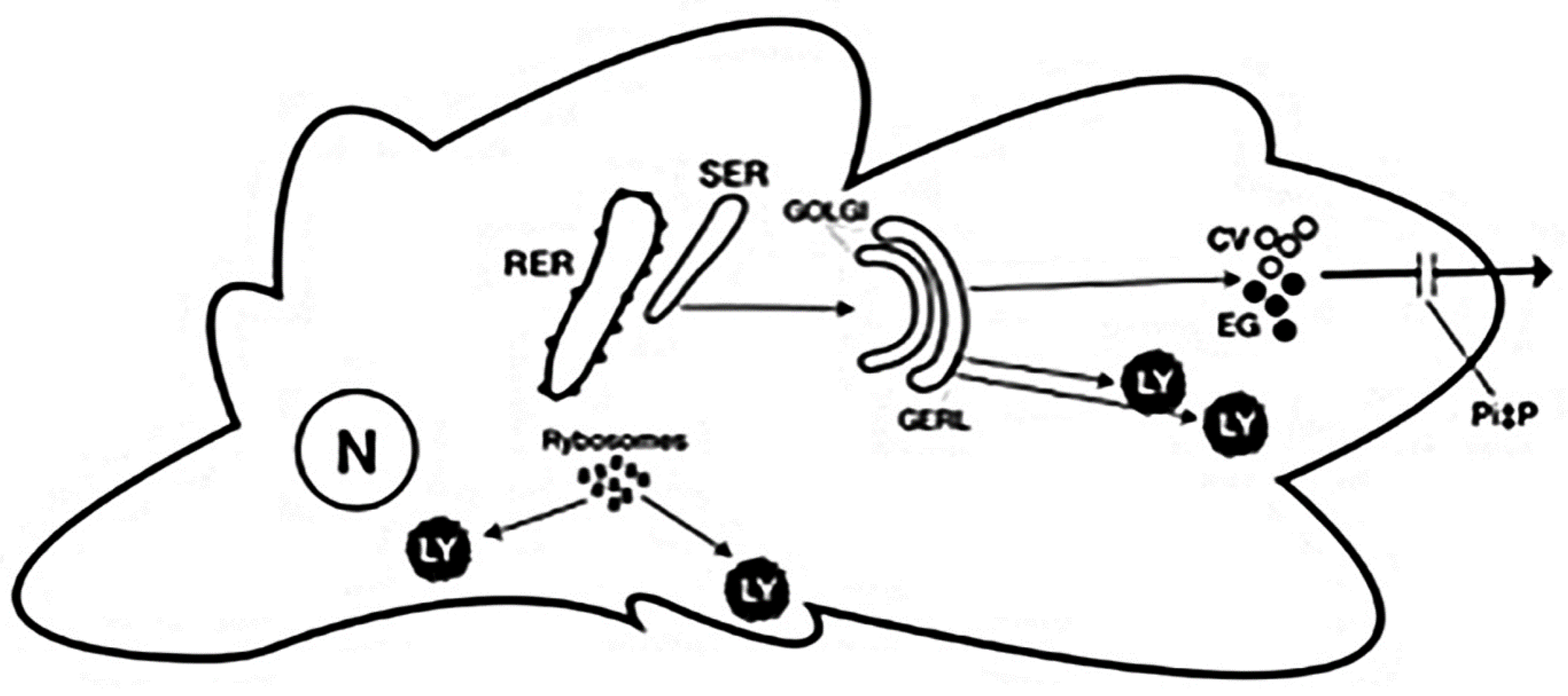{kind=link}
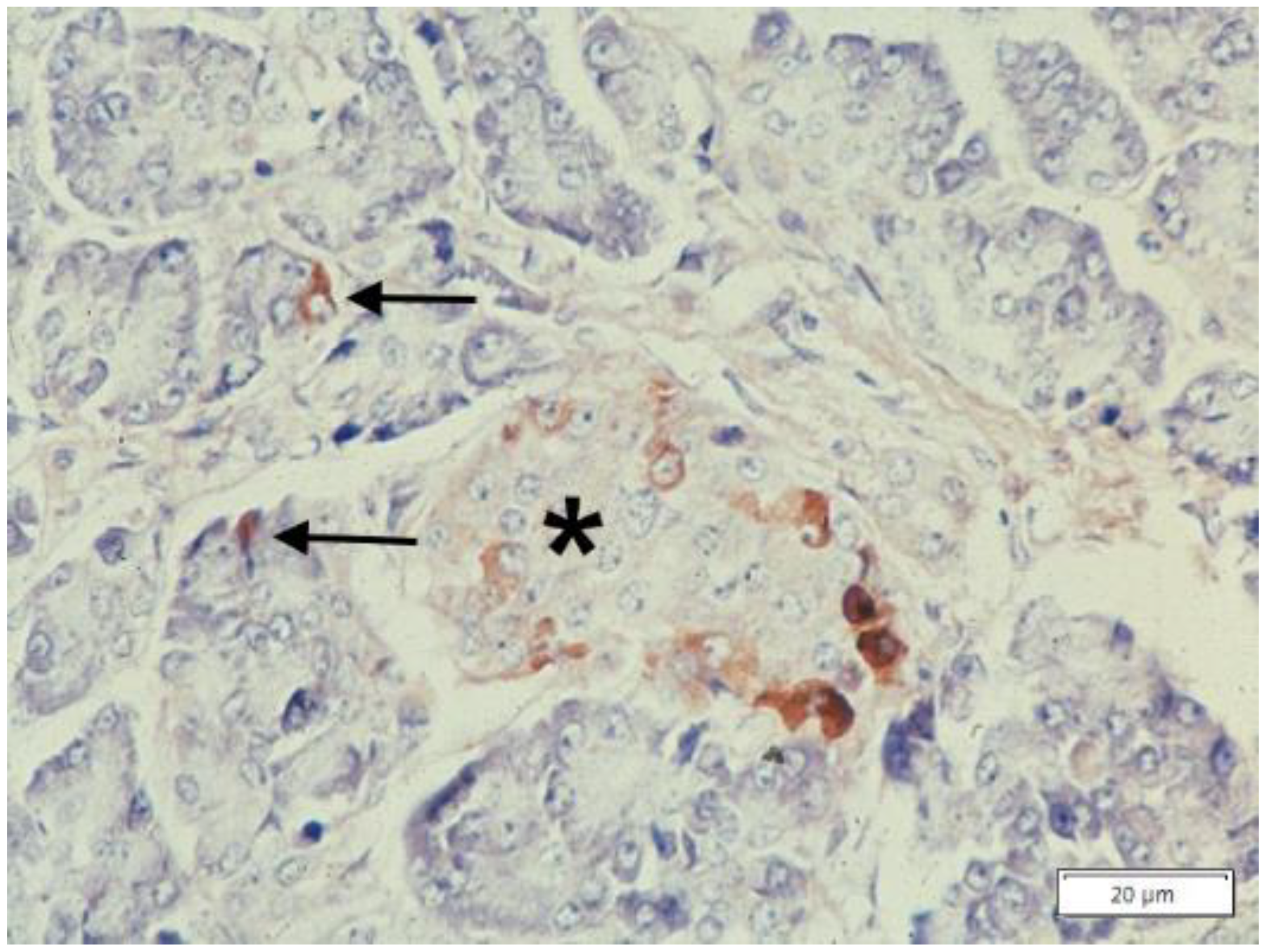{kind=link}
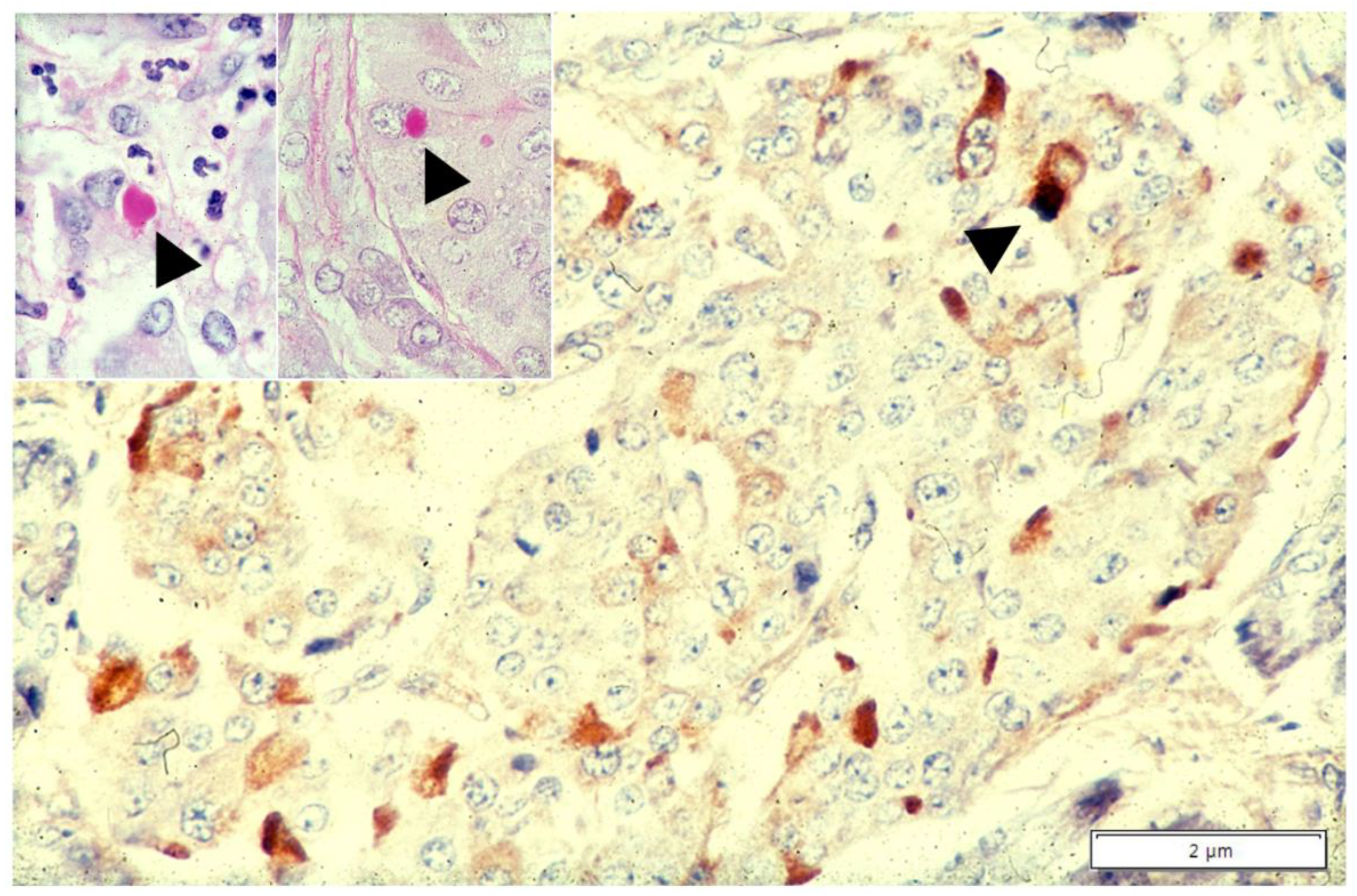{kind=link}
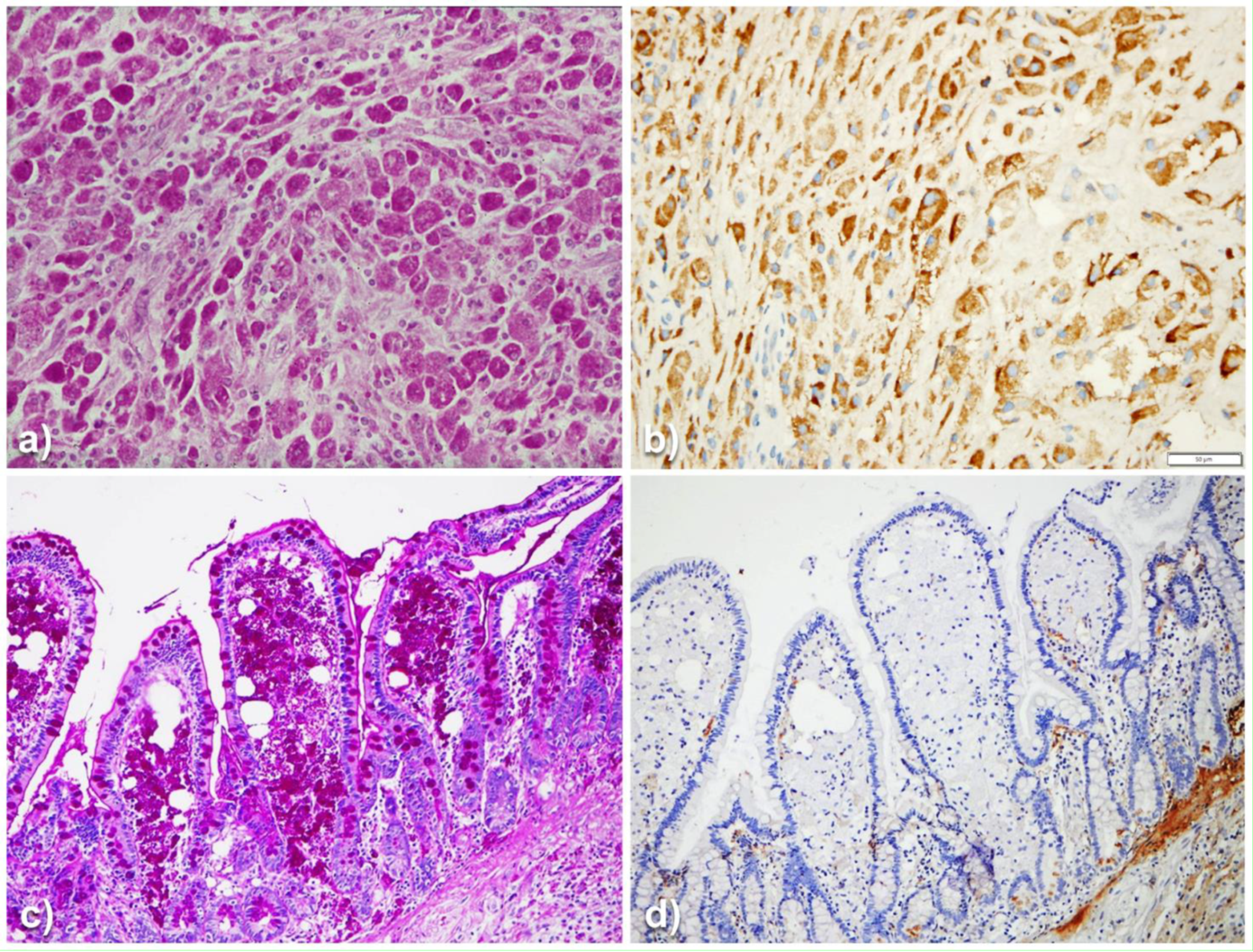{kind=link}
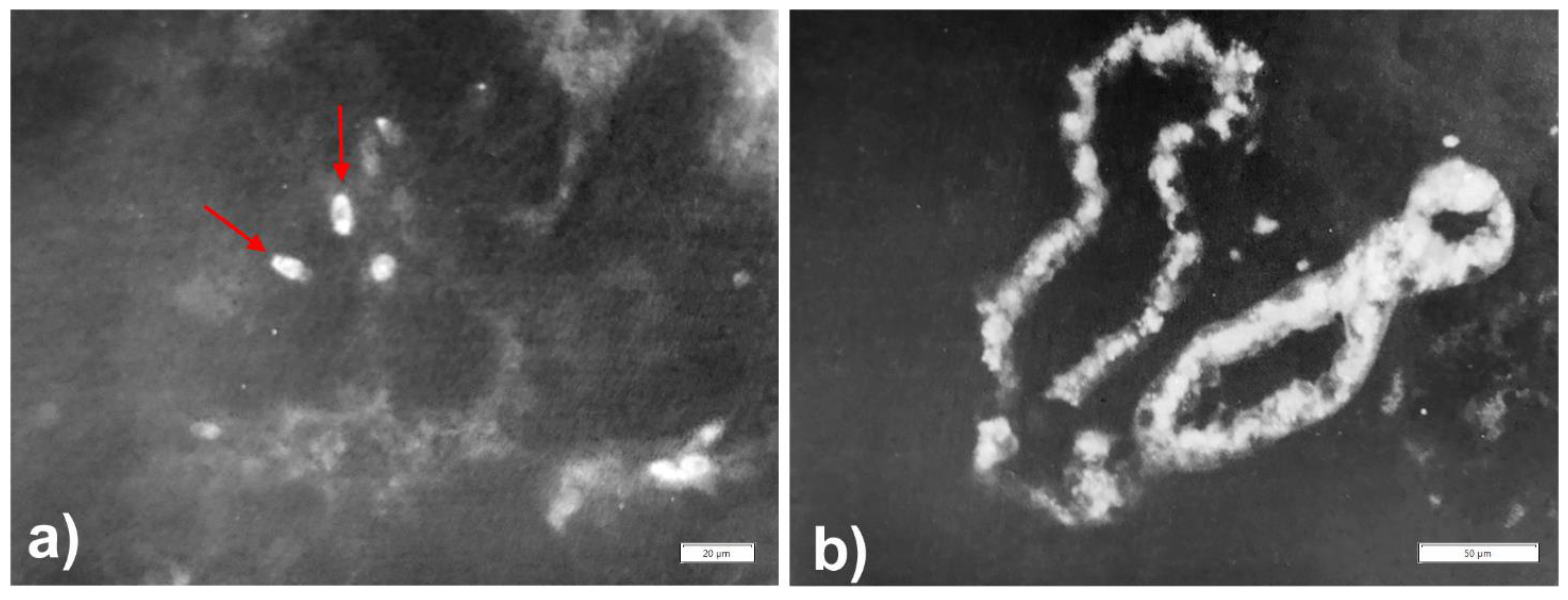{kind=link}
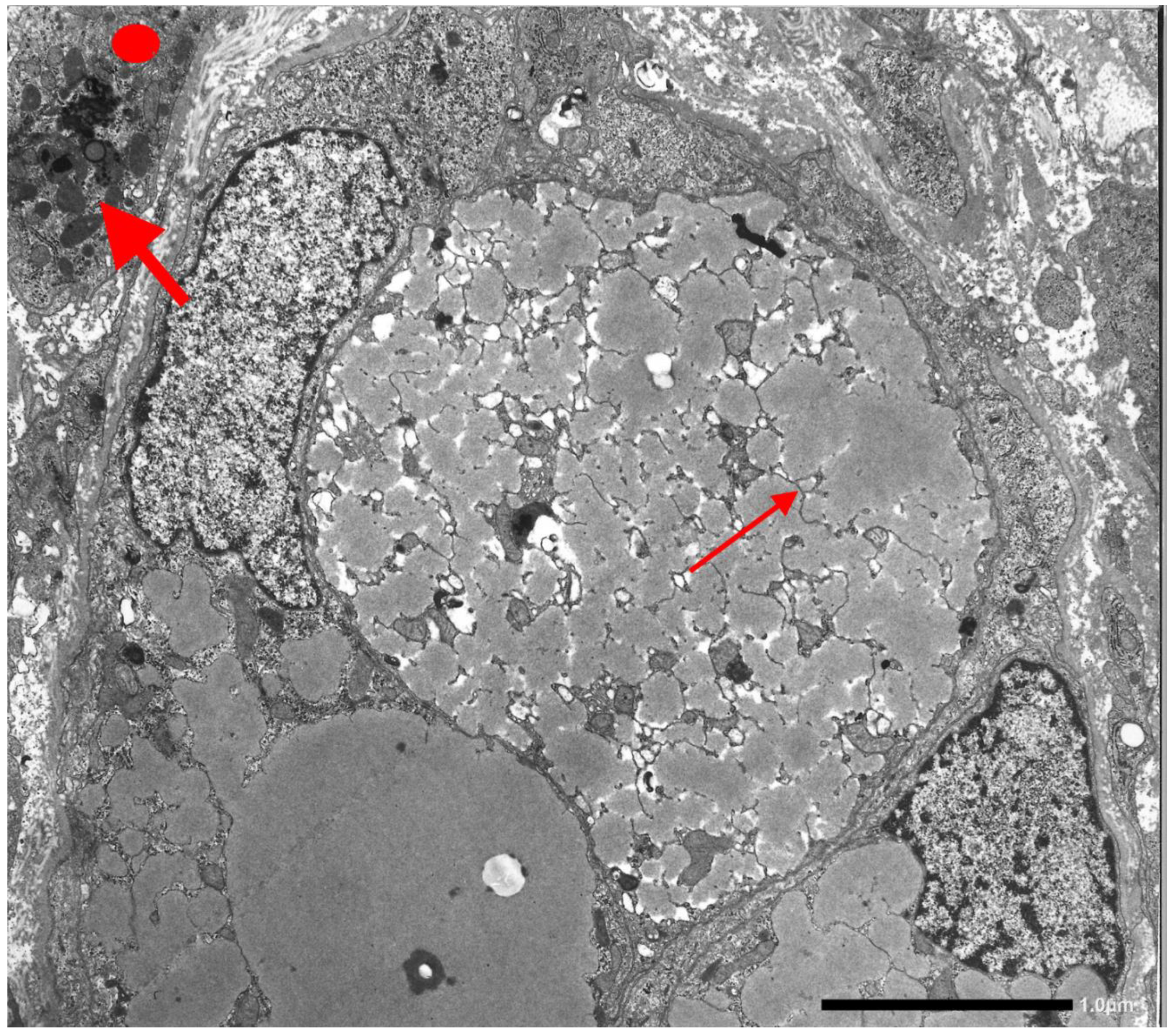{kind=link}
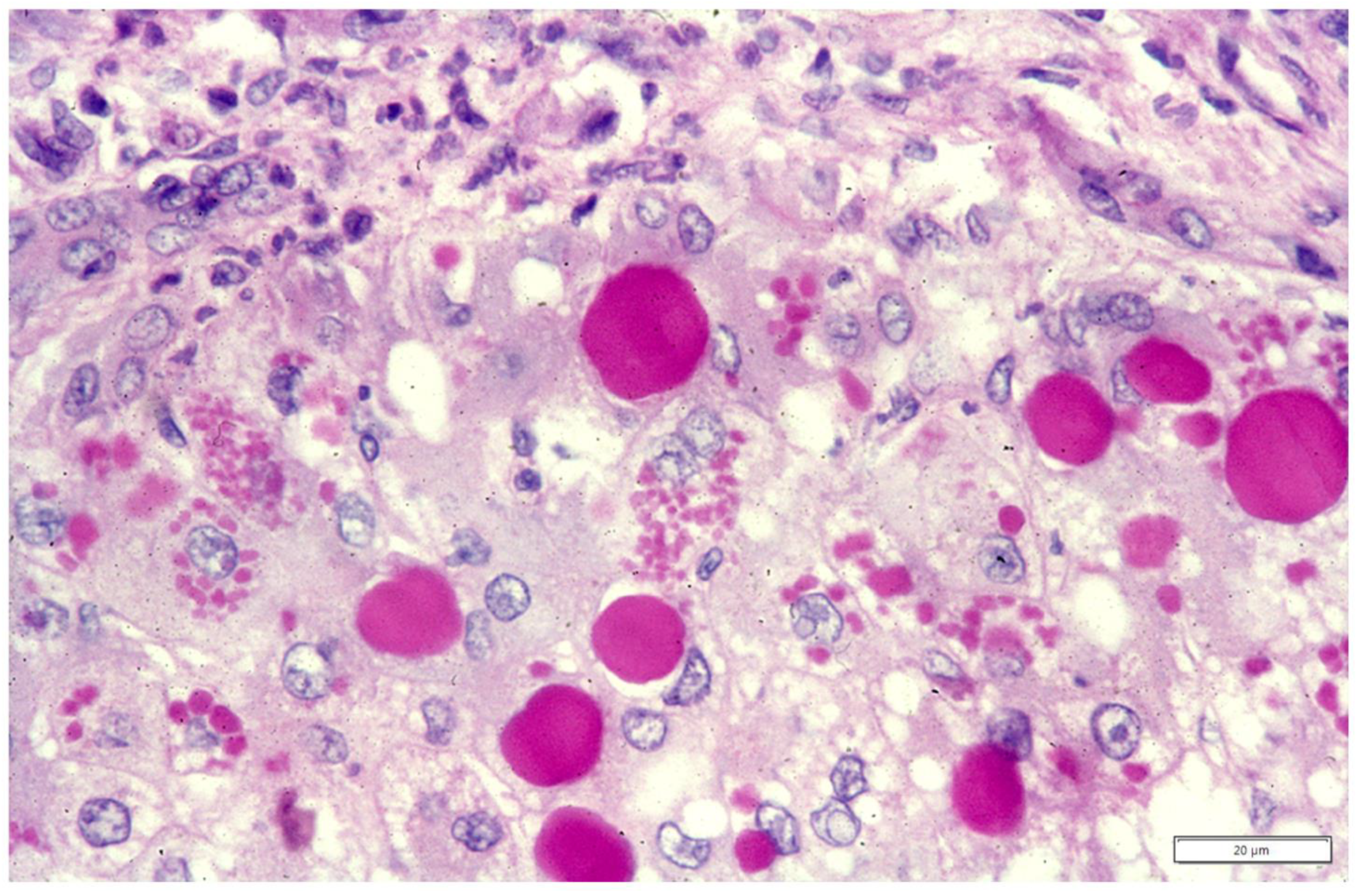{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Alpha-1-Antitryppsin Deficiency (AATD)
2.1. AAT: Source and Function
2.2. Hepatic Source and Function of AAT
2.3. Extrahepatic Source and Function of AAT
2.4. Hepatic Manifestations in AATD
2.5. Extrahepatic Manifestations of AATD
3. Hereditary Hypofibrinogenemia with Hepatic Storage (HHHS)
3.1. Fibrinogen Sources and Function
3.2. Hepatic Manifestations in HHHS
3.3. Extrahepatic Manifestations in HHHS
4. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Callea, F.; Brisigotti, M.; Fabbretti, G.; Bonino, F.; Desmet, V.J. Hepatic endoplasmic reticulum storage disease. Liver 1992, 12, 357–362. [Google Scholar] [CrossRef]
- Sharp, H.L.; Bridges, R.A.; Krivit, W.; Freier, E.F. Cirrhosis associated with alpha-1-antitrypsin deficiency: A previously unrecognized inherited disorder. J. Lab. Clin. Med. 1969, 73, 934–939. [Google Scholar] [PubMed]
- Eriksson, S. Pulmonary emphysema and alpha1-antitrypsin deficiency. Acta Med. Scand. 1964, 175, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, J. Heterozygous and hoNAmozygous alpha1-antitrypsin deficiency in patients with pulmonary emphysema. N. Engl. J. Med. 1969, 281, 279–284. [Google Scholar] [CrossRef]
- Editorial: Pathogenesis of emphysema. Br. Med. J. 1974, 5007, 527–528.
- Laurell, C.B.; Eriksson, S. The electrophoretic α1-globulin pattern of serum in α1-antitrypsin deficiency. 1963. COPD 2013, 10 (Suppl. 1), 3–8. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Lieberman, J.; Gaidulis, L.; Ewing, C. Molecular abnormality of human alpha1-antitrypsin variant (Pi-ZZ) associated with plasma activity deficiency. Proc. Natl. Acad. Sci. USA 1976, 73, 1324–1328. [Google Scholar] [CrossRef] [PubMed]
- Fagerhol, M.K. Cox DW: The Pi polymorphism; genetic, biochemical and clinical aspects of human alpha1-antitrypsin. In Advances in Human Genetics; Harris, H., Hirschhorn, K., Eds.; Plenum Press: New York, NY, USA, 1981; Volume 11, pp. 1–62. [Google Scholar]
- Wallmark, A.; Alm, R.; Eriksson, S. Monoclonal antibody specific for the mutant PiZ alpha 1-antitrypsin and its application in an ELISA procedure for identification of PiZ gene carriers. Proc. Natl. Acad. Sci. USA 1984, 81, 5690–5693. [Google Scholar] [CrossRef]
- Callea, F. Immunohistochemical Study on alpha-1-antitrypsin. Ph.D. Thesis, KUL, Acco Leuven, Belgium, 1983; pp. 1–153. [Google Scholar]
- Feldmann, G.; Martin, J.P.; Serboue, R.; Popartz, C.R.; Perelman, R.; Netahanson, M.; Seringe, P.; Benhameu, J.P. The ultrastructure of hepatocytes in alpha-1-antitrypsin deficiency with the genotype Pi. Null. Gut 1975, 16, 796–799. [Google Scholar] [CrossRef]
- Brennan, S.O.; Wyatt, J.; Medicina, D.; Callea, F.; George, P.M. Fibrinogen Brescia: Hepatic endoplasmic reticulum storage and hypofibrinogenemia because of a gamma284 Gly-->Arg mutation. Am. J. Pathol. 2000, 157, 189–196. [Google Scholar] [CrossRef]
- Pfeifer, U.; Ormanns, W.; Klinge, O. Hepatocellular fibrinogen storage in familial hypofibrinogenemia. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1981, 36, 247–255. [Google Scholar] [CrossRef]
- Wehinger, H.; Klinge, O.; Alexandrkis, E.; Shurman, J.; Witt, J.; Seydevitz, H.H. Hereditary hypofibrinogenemia with fibrinogen storage in the liver. Eur. J. Pediatr. 1983, 141, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Callea, F.; De Vos, R.; Pinackat, J.; Favret, M.; Facchetti, F.; Fiaccavento, S.; Ascari, E.; Tortora, O.; Albertini, A.; Henschen, A.; et al. Hereditary hypofinogenemia with hepatic storage of fibrinogen: Anew endoplasmic reticulum storage diseases. In Fibrinogen 2, Biochemistry, Physiology and Clinical Relevance; Lowe, G.D.O., Ed.; Elsevier: Amsterdam, The Netherlands, 1987; pp. 75–78. [Google Scholar]
- Callea, F.; Desmet, V.J. The discovery of Endoplasmic reticulum storage diseases. Connection between a H&E slide and the brain. Int. J. Mol. Sci. 2021, 22, 2899. [Google Scholar]
- Lomas, D.A.; Evans, D.L.; Finch, J.T.; Carrell, R.W. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992, 357, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Morse, J.O. Alpha1-antitrypsin deficiency (first of two parts). N. Engl. J. Med. 1978, 299, 1045–1048. [Google Scholar] [CrossRef] [PubMed]
- Crawford, G.P.; Ogston, D. The influence of alpha-1-antitrypsin on plasmin, urokinase and Hageman factor cofactor. Biochim. Biophys. Acta 1974, 354, 107–113. [Google Scholar] [CrossRef]
- Breit, S.N.; Robinson, J.P.; Luckhurst, E.; Clark, P.; Penny, R. Immunoregulation by alpha 1 antitrypsin. J. Clin. Lab. Immunol. 1982, 7, 127–131. [Google Scholar] [PubMed]
- Hatcher, V.B.; Oberman, M.S.; Wertheim, M.S.; Rhee, C.Y.; Tsien, G.; Burk, P.G. The relationship between surface protease activity and the rate of cell proliferation in normal and transformed cells. Biochem. Biophys. Res. Commun. 1976, 76, 602–608. [Google Scholar] [CrossRef]
- Callea, F.; Fabbretti, G.; Bonetti, M.; Brisigotti, M.; Desmet, V.J. Alpha-1-Antitrypsin Deficiency in Extrahepatic Manifestations in Liver Diseases; Schmid, R., Ed.; Kluwer Academic Publisher: Dordrecht, The Netherland, 1993; pp. 315–330. [Google Scholar]
- Palade, G. Intracellular aspects of the process of protein synthesis. Science 1975, 189, 347–358. [Google Scholar] [CrossRef]
- Sabatini, D.D.; Kreibich, G.; Morimoto, T.; Adesnik, M. Mechanisms for the incorporation of proteins in membranes and organelles. J. Cell. Biol. 1982, 92, 1–22. [Google Scholar] [CrossRef]
- Blobel, G. Regulation of intracellular protein traffic. Cold Spring Harb. Symp. Quant. Biol. 1982, 46 Pt 1, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Sharon, N.; Lys, H. Glycoproteins: Research booming on long-ignored ubiquitous compounds. Mol. Cell Biochem. 1982, 42, 167–187. [Google Scholar] [CrossRef]
- Callea, F.; Fevery, J.; Massi, G.; Lievens, C.; de Groote, J.; Desmet, V.J. Alpha-1-antitrypsin (AAT) and its stimulation in the liver of PiMZ phenotype individuals. A “recruitment-secretory block” (“R-SB”) phenomenon. Liver 1984, 4, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.M.; Koep, L.J.; Peters, R.L.; Schröter, G.P.; Weil, R., 3rd; Redeker, A.G.; Starzl, T.E. Liver transplantation for advanced liver disease with alpha-1-antitrypsin deficiency. N. Engl. J. Med. 1980, 302, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Kyaw-Myint, T.O.; Howell, A.M.; Murphy, G.M.; Anderson, C.M. Alpha-1-antitrypsin in duodenal fluid and gallbladder bile. Clin. Chim. Acta 1975, 59, 51–54. [Google Scholar] [CrossRef]
- Callea, F.; Fevery, J.; Massi, G.; de Groote, J.; Desmet, V.J. Storage of alpha-1-antitrypsin in intrahepatic bile duct cells in alpha-1-antitrypsin deficiency (Pi Z phenotype). Histopathology 1985, 9, 99–108. [Google Scholar] [CrossRef]
- Musiani, P.; Lauriola, L.; Piantelli, M. Inhibitory activity of alpha-1-antitrypsin bound to human IgA. Clin. Chim. Acta 1978, 85, 61–66. [Google Scholar] [CrossRef]
- Sung, J.Y.; Costerton, J.W.; Shaffer, E.A. Defense system in the biliary tract against bacterial infections. Dig. Dis. Sci. 1992, 37, 689–696. [Google Scholar] [CrossRef]
- Grün, M.; Liehr, H.; Rasenack, U. Significance of endotoxaemia in experimental “galactosamine-hepatitis” in the rat. Acta Hepatogastroenterol. (Stuttg) 1977, 24, 64–81. [Google Scholar]
- Labrune, P.; Odièvre, M.; Alagille, D. Influence of sex and breastfeeding on liver disease in alpha 1-antitrypsin deficiency. Hepatology 1989, 10, 122. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.B. Interrelationships between the human alveolar macrophages and alpha-1-antitrypsin. J. Clin. Investig. 1973, 52, 2793–2799. [Google Scholar] [CrossRef]
- Isaacson, P.; Jones, D.B.; Judd, M.A. Alpha 1-antitrypsin in human macrophages. Lancet 1979, 2, 964–965. [Google Scholar] [CrossRef]
- Benitez-Bibriesca, L.; Freyre Horta, R. immunofluorescent localization of alpha-1-antitrypsin in human polymorphonuclears. Life Sci. 1978, 21, 99–104. [Google Scholar] [CrossRef]
- Benitez-Bibriesca, L.; Freyre Horta, R.; De La Vega, G. Alpha-1-antrypsin in human mast cells. Immunofluorescent localization. Life Sci. 1973, 13, 631–638. [Google Scholar] [CrossRef]
- Geboes, K.; Ray, M.B.; Rutgeerts, P.; Callea, F.; Desmet, V.J.; Vantrappen, G. Morphological identification of alpha-1-antitrypsin in the human small intestine. Histopathology 1982, 6, 55–60. [Google Scholar] [CrossRef]
- Molmenti, E.P.; Perlmutter, D.H.; Rubin, D.C. Cell-specific expression of alpha 1-antitrypsin in human intestinal epithelium. J. Clin. Investig. 1993, 92, 2022–2034. [Google Scholar] [CrossRef] [PubMed]
- Ray, M.B.; Geboes, K.; Callea, F.; Desmet, V.J. Alpha-1-antitrypsin immunoreactivity in gastric carcinoid. Histopathology 1982, 6, 289–297. [Google Scholar] [CrossRef]
- Ray, M.B.; Desmet, V.J.; Gepts, W. alpha-1-Antitrypsin immunoreactivity in islet cells of adult human pancreas. Cell Tissue Res. 1977, 185, 63–68. [Google Scholar] [CrossRef]
- Perlmutter, D.H.; Cole, F.S.; Kilbridge, P.; Rossing, T.H.; Colten, H.R. Expression of the alpha 1-proteinase inhibitor gene in human monocytes and macrophages. Proc. Natl. Acad. Sci. USA 1985, 82, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Mornex, J.F.; Chytil-Weir, A.; Martinet, Y.; Courtney, M.; LeCocq, J.P.; Crystal, R.G. Expression of the alpha-1-antitrypsin gene in mononuclear phagocytes of normal and alpha-1-antitrypsin-deficient individuals. J. Clin. Investig. 1986, 77, 1952–1961. [Google Scholar] [CrossRef]
- Callea, F.; Fevery, J.; Desmet, V.J. Simultaneous alpha-1-antitrypsin accumulation in liver and pancreas. Hum. Pathol. 1984, 15, 293–295. [Google Scholar] [CrossRef]
- Callea, F.; Van Damme, B.; Desmet, V.J. Alpha-1-antitrypsin in Malakoplakia. Vircows Arch. (Pathol. Anat.) 1982, 395, 1–8. [Google Scholar] [CrossRef]
- Lewin, K.J.; Harrel, G.S.; Lee, A.S.; Crowley, L.G. Malakoplakia. An electron microscopic study demonstration of bacilliform organisms in malakoplakia macrophages. Gastroenterology 1974, 66, 28. [Google Scholar] [CrossRef]
- Lou, T.Y.; Teplitz, C. Malakoplakia: Pathogenesis and ultrastructural morphogenesis. A problem of altered macrophage (phagolysosomal) response. Hum. Pathol. 1974, 5, 191. [Google Scholar] [CrossRef]
- Moos, V.; Schneider, T. Changing paradigms in Whipple’s disease and infection with Tropherima wippleii. Eur. J. Clin. Microbiol. Infect. 2011, 30, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Chung, D.Y.; Kim, E.F.; Cho, N.H. Ultrastructural evidence of the evolutionary process in Malakoplakia. Histol. Histopathol. 2020, 35, 177–184. [Google Scholar] [PubMed]
- Fritz, H.; Heimburger, N.; Meier, M.; Arnhold, M.; Zaneveld, L.J.; Schumacher, G.F. Humanakrosin: Zur Kinetik der Hemmung durch Human-Serum inhibitoren [Human acrosin: Kinetics of the inhibition by inhibitors from human sera]. Hoppe Seylers Z. Physiol. Chem. 1972, 353, 1953–1956. [Google Scholar] [CrossRef] [PubMed]
- Bloom & Fawcett Textbook of Histology; W.B. Saunders Co.: Philadekphia, PA, USA, 1968; pp. 685–709.
- Zaneveld, L.J.D.; Dragole, B.M.; Schumacher, G.F.B. Acrosomal proteinase and proteinase inhibitor of human spermatozoa. Science 1972, 177, 702. [Google Scholar] [CrossRef]
- Ishibashi, H.; Shibata, K.; Okubo, H.; Tsuda-Kawamura, K.; Yanase, Y. Distribution of alpha 1-antitrypsin in normal, granuloma, and tumor tissues in rats. J. Lab. Clin. Med. 1978, 91, 576–583. [Google Scholar] [PubMed]
- Callea, F.; Fevery, J.; De Groote, J.; Desmet, V.J. Detection of Pi Z phenotype individuals by alpha-1-antitrypsin (AAT) immunohistochemistry in paraffin-embedded liver tissue specimens. J. Hepatol. 1986, 2, 389–401. [Google Scholar] [CrossRef]
- Callea, F.; Brisigotti, M.; Faa, G.; Lucini, L.; Eriksson, S. Identification of PiZ gene products in liver tissue by a monoclonal antibody specific for the Z mutant of alpha 1-antitrypsin. J. Hepatol. 1991, 12, 372–376. [Google Scholar] [CrossRef]
- Janciauskiene, S.; Eriksson, S.; Callea, F.; Mallya, M.; Zhou, A.; Seyama, K.; Hata, S.; Lomas, D.A. Differential detection of PAS-positive inclusions formed by the Z, Siiyama, and Mmalton variants of alpha1-antitrypsin. Hepatology 2004, 40, 1203–1210. [Google Scholar] [CrossRef]
- Callea, F.; Giovannoni, I.; Francalanci, P.; Boldrini, R.; Faa, G.; Medicina, D.; Nobili, V.; Desmet, V.J.; Ishak, K.; Seyama, K.; et al. Mineralization of alpha-1-antitrypsin inclusion bodies in Mmalton alpha-1-antitrypsin deficiency. Orphanet J. Rare Dis. 2018, 13, 79. [Google Scholar] [CrossRef]
- Lomas, D.A. The selective advantage of alpha-1-ntitrypsin deficiency. AJRCCM 2006, 173, 1072–1077. [Google Scholar]
- Hultcrantz, R.; Mingarelli, S. Ultrastructure liver pathology in patients with minimal liver disease and alpha-1-antitripsin deficiency: A comparison between heterozygotes and homozygotes. Hepatology 1884, 4, 937–945. [Google Scholar] [CrossRef]
- Perlmutter, D.H. Alpha-1-antitrypsin deficiency: Importance of proteasomal and autophagic degradative pathways in disposal of liver disease-associated protein aggregates. Annu. Rev. Med. 2011, 62, 333–345. [Google Scholar] [CrossRef]
- Feng, Y.; Klionsky, D.J. Receptors make the pathway choice for protein degradation. Autophagy 2017, 13, 1617–1618. [Google Scholar] [CrossRef]
- Janciauskiene, S.; Dominaitiene, R.; Sternby, N.H.; Piitulainen, E.; Eriksson, S. Detection of circulating and endothelial cell polymers of Z and wild type alpha 1-antitrypsin by a monoclonal antibody. J. Biol. Chem. 2002, 277, 26540–26546. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Perez, J.; Mela, M.; Miranda, E.; Surling, K.B.; Bouhani, F.N.; De Meo, D.; Haq, I.; Irving, J.A.; Ordóñez, A.; et al. Characterizing the association of latency with alpha-1-antitrypsin polymerization using a novel monoclonal antibody. Int. J. Biochem. Cell Biol. 2015, 58, 81–91. [Google Scholar] [CrossRef]
- Starzl, T.E.; Porter, K.A.; Terblanche, J. Interorgan communications: With Particular Reference to Hepatotrophic Factors and Intrinsic Liver Growth Factors; Communications of Liver Cells; Popper, H., Ed.; MTP Press Division Kluwer Academic Publishers: Norwel, MA, USA, 1980; p. 93. [Google Scholar]
- Eriksson, S.; Moestrup, T.; Hägerstrand, I. Liver, lung and malignant disease in heterozygous (Pi MZ) alpha1-antitrypsin deficiency. Acta Med. Scand. 1975, 198, 243–247. [Google Scholar] [CrossRef]
- Callea, F. Natural history of hepatocellular carcinoma as viewed by the pathologist. Appl. Pathol. 1988, 6, 105–116. [Google Scholar]
- Giovannoni, I.; Callea, F.; Stefanelli, M.; Mariani, R.; Santorelli, F.M.; Francalanci, P. Alpha-1-antitrypsin deficiency: From genoma to liver disease. PiZ mouse as model for the development of liver pathology in human. Liver Int. 2015, 35, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Kvittingen, E.A.; Rootwelt, H.; Berger, R.; Brandtzaeg, P. Self-induced correction of the genetic defect in tyrosinemia type I. J. Clin. Investig. 1994, 94, 1657–1661. [Google Scholar] [CrossRef] [PubMed]
- Francalanci, P.; Santorelli, F.M.; Saccani, S.; Bonetti, M.F.; Medicina, D.; Coni, P.; Faa, G.; Callea, F. Z and Mmalton-1-antitrypsin deficiency-associated hepatocellular carcinoma: A genetic study. Liver Int. 2009, 29, 1593–1596. [Google Scholar] [CrossRef]
- Thoolen, B.; Maronpot, R.B.; Harada, T. Proliferative and non- proliferative lesions of the rat and mouse hepatobiliary system. Toxicol. Pathology 2010, 38 (Suppl.), 5S–81S. [Google Scholar] [CrossRef]
- Rudnick, D.A.; Liao, Y.; An, J.K.; Muglia, L.J.; Perlmutter, D.H.; Teckman, J.H. Analyses of hepatocellular proliferation in a mouse model of alpha-1-antitrypsin deficiency. Hepatology 2004, 39, 1048–1055. [Google Scholar] [CrossRef]
- Carlson, J.A.; Rogers, B.B.; Sifers, R.N.; Finegold, M.J.; Clift, S.M.; De Mayo, F.J.; Bullock, D.W.; Woo, S.L. Accumulation of PiZ alpha 1-antitrypsin causes liver damage in transgenic mice. J. Clin. Investig. 1989, 83, 1183–1190. [Google Scholar] [CrossRef]
- Geller, S.A.; Nichols, W.S.; Kim, S.; Tolmachoff, T.; Lee, S.; Dycaico, M.J.; Felts, K.; Sorge, J.A. Hepatocarcinogenesis is the sequel to hepatitis in Z#2 alpha 1-antitrypsin transgenic mice: Histopathological and DNA ploidy studies. Hepatology 1994, 19, 389–397. [Google Scholar]
- Marcus, N.J.; Brunt, E.M.; Blomekamp, K. Characteristics of hepatocellular carcinoma in a murine model of alpha-1-antitrypsin deficiency. Hepatol. Res. 2010, 40, 641–653. [Google Scholar] [CrossRef]
- Elzouki, A.N.; Segelmark, M.; Wieslander, J.; Eriksson, S. Strong link between the alpha 1-antitrypsin PiZ allele and Wegener’s granulomatosis. J. Intern. Med. 1994, 236, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Esnault, V.L.; Testa, A.; Audrain, M.; Rogé, C.; Hamidou, M.; Barrier, J.H.; Sesboüé, R.; Martin, J.P.; Lesavre, P. Alpha 1-antitrypsin genetic polymorphism in ANCA-positive systemic vasculitis. Kidney Int. 1993, 43, 1329–1332. [Google Scholar] [CrossRef]
- Greulich, T.; Nell, C.; Hohmann, D.; Grebe, M.; Janciauskiene, S.; Koczulla, A.R.; Vogelmeier, C.F. The prevalence of diagnosed α1-antitrypsin deficiency and its comorbidities: Results from a large population-based database. Eur. Respir. J. 2017, 49, 1600154. [Google Scholar] [CrossRef]
- Callea, F.; Gregorini, G.; Sinico, A.; Consalez, G.G.; Bossolasco, M.; Salvidio, G.; Radice, A.; Tira, P.; Candiano, G.; Rossi, G.; et al. alpha 1-Antitrypsin (AAT) deficiency and ANCA-positive systemic vasculitis: Genetic and clinical implications. Eur. J. Clin. Investig. 1997, 27, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.; Travis, J. Structural evidence for methionine at the reactive site of human alpha-1-proteinase inhibitor. J. Biol. Chem. 1978, 253, 7142–7144. [Google Scholar] [CrossRef]
- Carp, H.; Janoff, A. Possible mechanisms of emphysema in smokers. In vitro suppression of serum elastase-inhibitory capacity by fresh cigarette smoke and its prevention by antioxidants. Am. Rev. Respir. Dis. 1978, 118, 617–621. [Google Scholar]
- Gadek, J.E.; Fells, G.A.; Zimmerman, R.L.; Rennard, S.I.; Crystal, R.G. Antielastases of the human alveolar structures. Implications for the protease-antiprotease theory of emphysema. J. Clin. Investig. 1981, 68, 889–898. [Google Scholar] [CrossRef]
- Strnad, P.; McElvaney, N.G.; Lomas, D.A. Alpha1-Antitrypsin Deficiency. N. Engl. J. Med. 2020, 382, 1443–1455. [Google Scholar] [CrossRef] [PubMed]
- Bazzan, E.; Tinè, M.; Biondini, D.; Benetti, R.; Baraldo, S.; Turato, G.; Fagiuoli, S.; Sonzogni, A.; Rigobello, C.; Rea, F.; et al. α1-Antitrypsin Polymerizes in Alveolar Macrophages of Smokers With and Without α1-Antitrypsin Deficiency. Chest 2018, 154, 607–616. [Google Scholar] [CrossRef]
- Santiago, J.V.; Dew, T.A.; Haymond, M.; Williamson, J.R.; Kilo, C.; Kipnis, D.M.; Pierce, J.A. Carbohydrate intolerance and relative insulin deficiency in alpha-1-anti-trypsin deficiency. J. Clin. Investig. 1974, 53, 70. [Google Scholar]
- Callea, F.; Geboes, K.; Goddeeris, P.; Desmet, V.J. Endocrine pancreas and alpha-1-anti-trypsin deficiency. In The Endocrines and the Liver; Langer, M., Chiandussi, L., Chopra, I.J., Martini, L., Eds.; Academic Press: London, UK; New York, NY, USA, 1982. [Google Scholar]
- Koulmanda, M.; Bhasin, M.; Hoffman, L.; Fan, Z.; Qipo, A.; Shi, H.; Bonner-Weir, S.; Putheti, P.; Degauque, N.; Libermann, T.A.; et al. Curative and beta cell regenerative effects of alpha1-antitrypsin treatment in autoimmune diabetic NOD mice. Proc. Natl. Acad. Sci. USA 2008, 105, 16242–16247. [Google Scholar] [CrossRef] [PubMed]
- De Moerloose, P.; Casini, A.; Neerman-Arbez, M. Congenital Fibrinogen Disorders: An update. Semin. Thromb. Hemost. 2013, 39, 585–595. [Google Scholar]
- Burcu, G.; Bellacchio, E.; Sag, E.; Cebi, A.H.; Saygin, I.; Bahadir, A.; Yilmaz, G.; Corbeddu, M.; Cakir, M.; Callea, F. Structural Characteristics in the γ Chain Variants Associated with Fibrinogen Storage Disease Suggest the Underlying Pathogenic Mechanism. Int. J. Mol. Sci. 2020, 21, 5139. [Google Scholar] [CrossRef]
- Bellacchio, E. Mutations Causing Mild or No Structural Damage in Interfaces of Multimerization of the Fibrinogen γ-Module More Likely Confer Negative Dominant Behaviors. Int. J. Mol. Sci. 2020, 21, 9016. [Google Scholar] [CrossRef]
- Lee, M.J.; Venick, R.; Bhuta, S.; Li, X.; Wang, H.L. Hepatic Fibrinogen Storage Disease in a Patient with Hypofibrinogenemia: Report of a Case with a Missense Mutation of the FGA Gene. Semin. Liver Dis. 2015, 35, 439–443. [Google Scholar] [CrossRef]
- Galanakis, D.; Spitzer, S.; Scharrer, I. Unusual A alpha 16Arg-->Cys dysfibrinogenaemic family: Absence of normal A alpha-chains in fibrinogen from two of four heterozygous siblings. Blood Coagul. Fibrinolysis. 1993, 4, 67–71. [Google Scholar] [CrossRef]
- Mosesson, M.W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 2005, 3, 1894–1904. [Google Scholar] [CrossRef]
- Redman, C.M.; Xia, H. Fibrinogen biosynthesis. Assembly, intracellular degradation, and association with lipid synthesis and secretion. Ann. N. Y. Acad. Sci. 2001, 936, 480–495. [Google Scholar] [CrossRef] [PubMed]
- Kopito, R.R.; Ron, D. Conformational disease. Nat. Cell Biol. 2000, 2, 524–530. [Google Scholar] [CrossRef]
- Louache, F.; Debili, N.; Cramer, E.; Breton-Gorius, J.; Vainchenker, W. Fibrinogen is not synthesized by human megakaryocytes. Blood 1991, 77, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Maghzal, G.J.; Brennan, S.O.; Homer, V.M.; George, P.M. The molecular mechanisms of congenital hypofibrinogenaemia. Cell Mol. Life Sci. 2004, 61, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Callea, F.; Giovannoni, I.; Sari, S.; Aksu, A.U.; Esendagly, G.; Dalgic, B.; Boldrini, R.; Akyol, G.; Francalanci, P.; Bellacchio, E. A novel fibrinogen gamma chain mutation (c.1096C>G; p.His340Asp), fibrinogen Ankara, causing hypofibrinogenaemia and hepatic storage. Pathology 2017, 49, 534–537. [Google Scholar] [CrossRef]
- Callea, F.; Giovannoni, I.; Sari, S.; Guldal, E.; Dalgic, B.; Akyol, G.; Sogo, T.; Al-Hussaini, A.; Maggiore, G.; Bartuli, A.; et al. Fibrinogen Gamma Chain Mutations Provoke Fibrinogen and Apolipoprotein B Plasma Deficiency and Liver Storage. Int. J. Mol. Sci. 2017, 18, 2717. [Google Scholar] [CrossRef]
- Francalanci, P.; Santorelli, F.M.; Talini, I.; Boldrini, R.; Devito, R.; Camassei, F.D.; Maggiore, G.; Callea, F. Severe liver disease in early childhood due to fibrinogen storage and de novo gamma375Arg-->Trp gene mutation. J. Pediatr. 2006, 148, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Sogo, T.; Nagasaka, H.; Komatsu, H.; Inui, A.; Miida, T.; Callea, F.; Francalanci, P.; Hirano, K.; Kitamura, H.; Yorifuji, T.; et al. Fibrinogen storage disease caused by Aguadilla mutation presenting with hypobeta-lipoproteinemia and considerable liver disease. J. Pediatr. Gastroenterol. Nutr. 2009, 49, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussaini, A.; Altalhi, A.; El Hag, I.; AlHussaini, H.; Francalanci, P.; Giovannoni, I.; Callea, F. Hepatic fibrinogen storage disease due to the fibrinogen γ375 Arg → Trp mutation "fibrinogen Aguadilla" is present in Arabs. Saudi. J. Gastroenterol. 2014, 20, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Sari, S.; Yilmaz, G.; Gonul, I.I.; Dalgic, B.; Akyol, G.; Giovannoni, I.; Francalanci, P.; Callea, F. Fibrinogen storage disease and cirrhosis associated with hypobetalipoproteinemia owing to fibrinogen Aguadilla in a Turkish child. Liver Int. 2015, 35, 2501–2505. [Google Scholar] [CrossRef]
- Asselta, R.; Paraboschi, E.M.; Duga, S. Hereditary Hypofibrinogenemia with Hepatic Storage. Int. J. Mol. Sci. 2020, 21, 7830. [Google Scholar] [CrossRef]
- Picken, M.M.; Linke, R.P. Nephrotic syndrome due to an amyloidogenic mutation in fibrinogen A alpha chain. J. Am. Soc. Nephrol. 2009, 20, 1681–1685. [Google Scholar] [CrossRef]
- Chapman, J.; Dogan, A. Fibrinogen alpha amyloidosis: Insights from proteomics. Expert Rev. Proteomics 2019, 16, 783–793. [Google Scholar] [CrossRef]
- Stangou, A.J.; Banner, N.R.; Hendry, B.M.; Rela, M.; Portmann, B.; Wendon, J.; Monaghan, M.; Maccarthy, P.; Buxton-Thomas, M.; Mathias, C.J.; et al. Hereditary fibrinogen A alpha-chain amyloidosis: Phenotypic characterization of a systemic disease and the role of liver transplantation. Blood 2010, 115, 2998–3007. [Google Scholar] [CrossRef]
- Carrell, R.W.; Lomas, D.A. Conformational disease. Lancet 1997, 350, 134–138. [Google Scholar] [CrossRef]
- Majno, G.; Joris, I. Cells Tissues and Disease; Oxford University Press: Oxford, UK, 2004. [Google Scholar]
- Ishak, H.G.; Sharp, H.L.; Schwarzenberg, S.J. Metabolic Errors and Liver Disease in Pathology of the Liver RNM; MacSween, B.A.D., Portmann, B.C., Ishak, K.G., Scheuer, P.J., Anthony, P.P., Eds.; Churchill Livingstone: Edimburgh, UK, 2003. [Google Scholar]
- Carlson, J.; Eriksson, S. Chronic ‘cryptogenic’ liver disease and malignant hepatoma in intermediate alpha 1-antitrypsin deficiency identified by a Pi Z-specific monoclonal antibody. Scand J. Gastroenterol. 1985, 20, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Pittschieler, K. Liver disease and heterozygous alpha-1-antitrypsin deficiency. Acta Paediatr. Scand. 1991, 80, 323–327. [Google Scholar] [CrossRef]
- Eigenbrodt, M.L.; McCashland, T.M.; Dy, R.M.; Clark, J.; Galati, J. Heterozygous alpha 1-antitrypsin phenotypes in patients with end stage liver disease. Am. J. Gastroenterol. 1997, 92, 602–607. [Google Scholar]
- Rawlings, W., Jr.; Moss, J.; Cooper, H.S.; Hamilton, S.R. Hepatocellular carcinoma and partial deficiency of alpha-1 antitrypsin (MZ). Ann. Intern. Med. 1974, 81, 771–773. [Google Scholar] [CrossRef]
- Campbell, K.M.; Arya, G.; Ryckman, F.C.; Alonso, M.; Tiao, G.; Balistreri, W.F.; Bezerra, J.A. High prevalence of alpha-1-antitrypsin heterozygosity in children with chronic liver disease. J. Pediatr. Gastroenterol. Nutr. 2007, 44, 99–103. [Google Scholar] [CrossRef]
- Lieberman, J.; Silton, R.M.; Agliozzo, C.M.; McMahon, J. Hepatocellular carcinoma and intermediate alpha1-antitrypsin deficiency (MZ phenotype). Am. J. Clin. Pathol. 1975, 64, 304–310. [Google Scholar] [CrossRef]
- Wewers, M.D.; Gadel, J.E.; Keugh, R.A.; Fells, G.A.; Cristal, R.G. Evaluation of danazol for patients with Pi ZZ alpha-1-antitrypsin deficiency. Am. J. Resp. Dis. 1986, 134, 476–480. [Google Scholar]
- Zhang, X.; Pham, K.; Li, D.; Schutte, R.J.; Gonzalo, D.H.; Zhang, P.; Oshins, R.; Tan, W.; Brantly, M.; Liu, C.; et al. A Novel Small Molecule Inhibits Intrahepatocellular Accumulation of Z-Variant Alpha 1-Antitrypsin in Vitro and in Vivo. Cells 2019, 8, 1586. [Google Scholar] [CrossRef]
- Lomas, D.A.; Irving, J.A.; Arico-Muendel, C.; Belyaskaya, S.; Brewster, A.; Brown, M.; Chung Chun-Wa Dave, H.; Denis, A.; Dodic, N.; Dossang, A.; et al. Development of a small molecule that corrects misfolding and increases secretion of Z alpha-1-antitrypsin. EMBO Molec. Med. 2021, 13, 13267. [Google Scholar] [CrossRef]


















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Callea, F.; Francalanci, P.; Giovannoni, I. Hepatic and Extrahepatic Sources and Manifestations in Endoplasmic Reticulum Storage Diseases. Int. J. Mol. Sci. 2021, 22, 5778. https://doi.org/10.3390/ijms22115778
Callea F, Francalanci P, Giovannoni I. Hepatic and Extrahepatic Sources and Manifestations in Endoplasmic Reticulum Storage Diseases. International Journal of Molecular Sciences. 2021; 22(11):5778. https://doi.org/10.3390/ijms22115778
Chicago/Turabian StyleCallea, Francesco, Paola Francalanci, and Isabella Giovannoni. 2021. "Hepatic and Extrahepatic Sources and Manifestations in Endoplasmic Reticulum Storage Diseases" International Journal of Molecular Sciences 22, no. 11: 5778. https://doi.org/10.3390/ijms22115778
APA StyleCallea, F., Francalanci, P., & Giovannoni, I. (2021). Hepatic and Extrahepatic Sources and Manifestations in Endoplasmic Reticulum Storage Diseases. International Journal of Molecular Sciences, 22(11), 5778. https://doi.org/10.3390/ijms22115778