
The Importance of N186 in the Alpha-1-Antitrypsin Shutter Region Is Revealed by the Novel Bologna Deficiency Variant

 , , , , ,
, , , , ,  and
and
Abstract
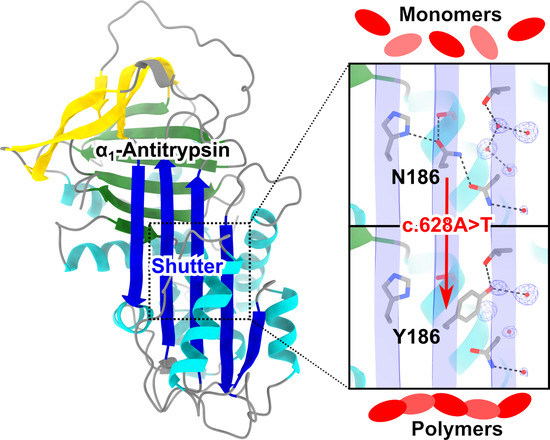
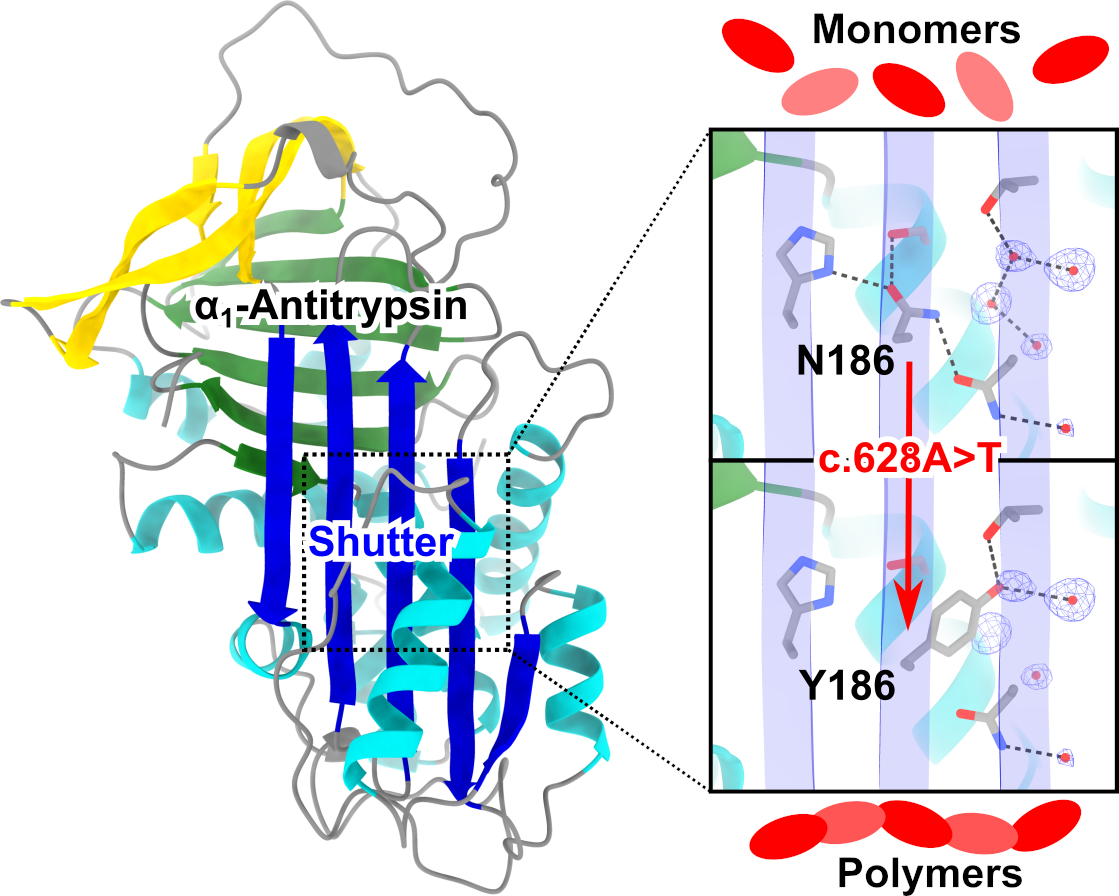
1. Introduction
2. Results
2.1. Identification and Clinical Profile of the Bologna AAT Variant
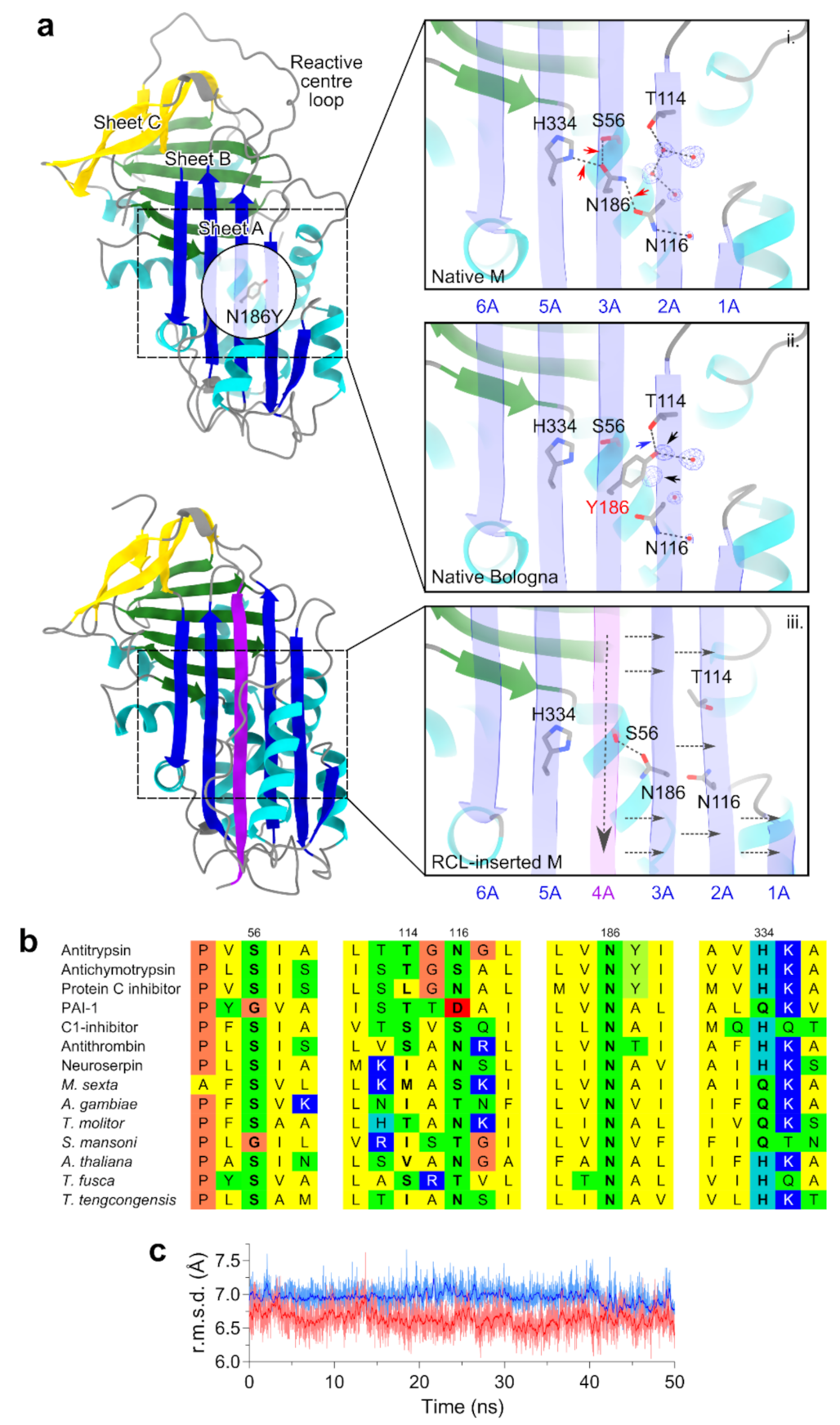
2.2. Structural Localization of the Bologna Mutation in the AAT Shutter Region
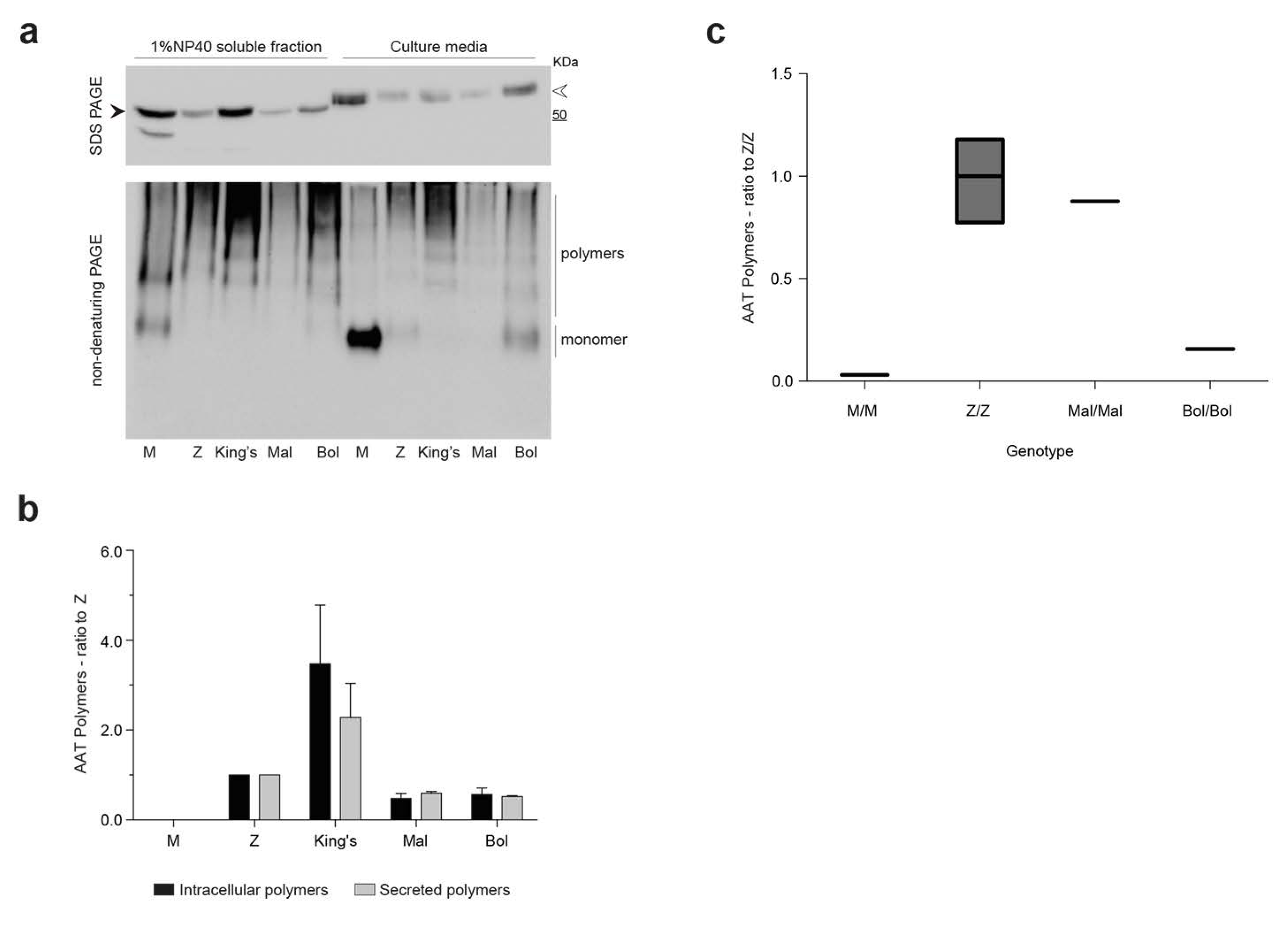
2.3. Intracellular Polymerization and Secretion Deficiency of the Bologna Variant Expressed in a Hepatoma Cell Line
2.4. Detection of Bologna AAT Polymers in Cell Culture Media and Plasma
2.5. Assessment of Intracellular Polymers of Bologna AAT by Immunofluorescence and Confocal Microscopy
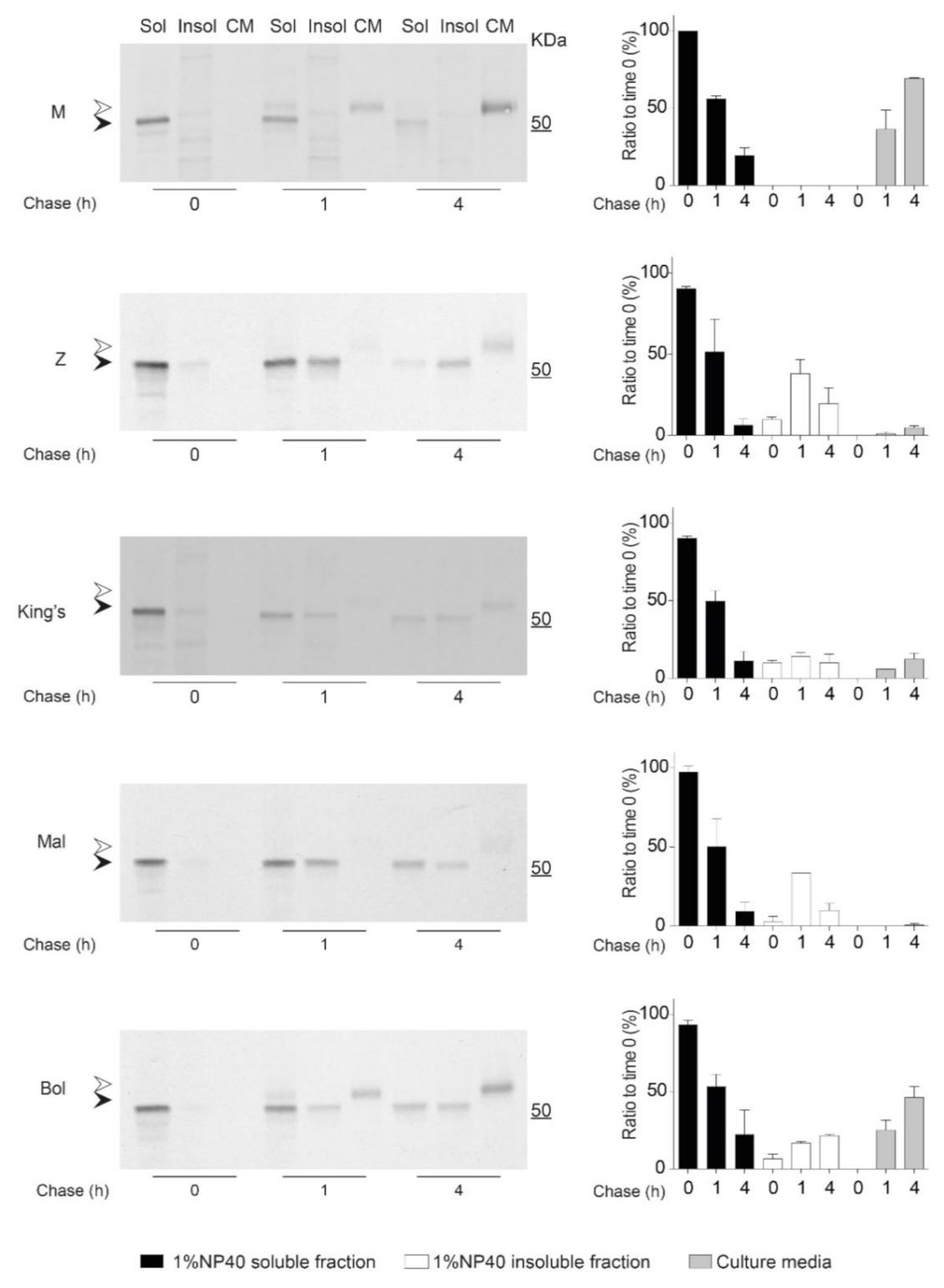
2.6. Kinetics of Accumulation and Secretion of the Bologna AAT Variant
3. Discussion
4. Materials and Methods
4.1. Genetic Analyses and Clinical Data
4.2. Expression Vectors
4.3. Cell Culture and Transfection
4.4. Cell Lysis and Fractionation
4.5. SDS-PAGE, Non-Denaturing PAGE and Immunoblots
4.6. Sandwich ELISA
4.7. Pulse-Chase Experiments
4.8. Immunofluorescence and Confocal Microscopy
4.9. Molecular Modelling, Dynamics and Sequence Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rutishauser, J.; Spiess, M. Endoplasmic reticulum storage diseases. Swiss Med. Wkly. 2002, 132, 211–222. [Google Scholar]
- Callea, F.; Brisigotti, M.; Fabbretti, G.; Bonino, F.; Desmet, V.J. Hepatic endoplasmic reticulum storage diseases. Liver 1992, 12, 357–362. [Google Scholar] [CrossRef]
- Lomas, D.A.; Evans, D.L.; Finch, J.T.; Carrell, R.W. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992, 357, 605–607. [Google Scholar] [CrossRef]
- Clark, V.C.; Marek, G.; Liu, C.; Collinsworth, A.; Shuster, J.; Kurtz, T.; Nolte, J.; Brantly, M. Clinical and histologic features of adults with alpha-1 antitrypsin deficiency in a non-cirrhotic cohort. J. Hepatol. 2018, 69, 1357–1364. [Google Scholar] [CrossRef]
- Patel, D.; Teckman, J.H. Alpha-1-antitrypsin deficiency liver disease. Clin. Liver Dis. 2018, 22, 643–655. [Google Scholar] [CrossRef]
- Strnad, P.; McElvaney, N.G.; Lomas, D.A. Alpha1-antitrypsin deficiency. N. Engl. J. Med. 2020, 382, 1443–1455. [Google Scholar] [CrossRef]
- Lawrence, D.A.; Ginsburg, D.; Day, D.E.; Berkenpas, M.B.; Verhamme, I.M.; Kvassman, J.O.; Shore, J.D. Serpin-protease complexes are trapped as stable acyl-enzyme intermediates. J. Biol. Chem. 1995, 270, 25309–25312. [Google Scholar] [CrossRef]
- Huntington, J.A.; Read, R.J.; Carrell, R.W. Structure of a serpin-protease complex shows inhibition by deformation. Nature 2000, 407, 923–926. [Google Scholar] [CrossRef]
- Dafforn, T.R.; Mahadeva, R.; Elliott, P.R.; Sivasothy, P.; Lomas, D.A. A kinetic mechanism for the polymerization of alpha1-antitrypsin. J. Biol. Chem. 1999, 274, 9548–9555. [Google Scholar] [CrossRef]
- Irving, J.A.; Miranda, E.; Haq, I.; Perez, J.; Kotov, V.R.; Faull, S.V.; Motamedi-Shad, N.; Lomas, D.A. An antibody raised against a pathogenic serpin variant induces mutant-like behaviour in the wild-type protein. Biochem. J. 2015, 468, 99–108. [Google Scholar] [CrossRef]
- Jagger, A.M.; Waudby, C.A.; Irving, J.A.; Christodoulou, J.; Lomas, D.A. High-resolution ex vivo NMR spectroscopy of human Z α1-antitrypsin. Nat. Commun. 2020, 11, 6371. [Google Scholar] [CrossRef]
- Qu, D.; Teckman, J.H.; Omura, S.; Perlmutter, D.H. Degradation of a mutant secretory protein, α1-antitrypsin Z, in the endoplasmic reticulum requires proteasome activity. J. Biol. Chem. 1996, 271, 22791–22795. [Google Scholar] [CrossRef]
- Teckman, J.H.; Perlmutter, D.H. The endoplasmic reticulum degradation pathway for mutant secretory proteins alpha1-antitrypsin Z and S is distinct from that for an unassembled membrane protein. J. Biol. Chem. 1996, 271, 13215–13220. [Google Scholar] [CrossRef]
- Yamasaki, M.; Sendall, T.J.; Pearce, M.C.; Whisstock, J.C.; Huntington, J.A. Molecular basis of α1-antitrypsin deficiency revealed by the structure of a domain-swapped trimer. EMBO Rep. 2011, 12, 1011–1017. [Google Scholar] [CrossRef]
- Faull, S.V.; Elliston, E.L.K.; Gooptu, B.; Jagger, A.M.; Aldobiyan, I.; Redzej, A.; Badaoui, M.; Heyer-Chauhan, N.; Rashid, S.T.; Reynolds, G.M.; et al. The structural basis for Z α1-antitrypsin polymerization in the liver. Sci. Adv. 2020, 6, eabc1370. [Google Scholar] [CrossRef]
- Tan, L.; Dickens, J.A.; Demeo, D.L.; Miranda, E.; Perez, J.; Rashid, S.T.; Day, J.; Ordoñez, A.; Marciniak, S.J.; Haq, I.; et al. Circulating polymers in α1-antitrypsin deficiency. Eur. Respir. J. 2014, 43, 1501–1504. [Google Scholar] [CrossRef]
- Elliott, P.R.; Bilton, D.; Lomas, D.A. Lung polymers in Z alpha1-antitrypsin deficiency-related emphysema. Am. J. Respir. Cell Mol. Biol. 1998, 18, 670–674. [Google Scholar] [CrossRef]
- Mahadeva, R.; Atkinson, C.; Li, Z.; Stewart, S.; Janciauskiene, S.; Kelley, D.G.; Parmar, J.; Pitman, R.; Shapiro, S.D.; Lomas, D.A.; et al. Polymers of Z alpha1-antitrypsin co-localize with neutrophils in emphysematous alveoli and are chemotactic in vivo. Am. J. Pathol. 2005, 166, 377–386. [Google Scholar] [CrossRef]
- Fra, A.; Cosmi, F.; Ordoñez, A.; Berardelli, R.; Perez, J.; Guadagno, N.A.; Corda, L.; Marciniak, S.J.; Lomas, D.A.; Miranda, E. Polymers of Z α1-antitrypsin are secreted in cell models of disease. Eur. Respir. J. 2016, 47, 1005–1009. [Google Scholar] [CrossRef]
- Ronzoni, R.; Heyer-Chauhan, N.; Fra, A.; Pearce, A.C.; Rüdiger, M.; Miranda, E.; Irving, J.; Lomas, D. The molecular species responsible for α1 -antitrypsin deficiency are suppressed by a small molecule chaperone. FEBS J. 2020, 288, 2222–2237. [Google Scholar] [CrossRef]
- Elliott, P.R.; Stein, P.E.; Bilton, D.; Carrell, R.W.; Lomas, D.A. Structural explanation for the deficiency of S alpha 1-antitrypsin. Nat. Struct. Biol. 1996, 3, 910–911. [Google Scholar] [CrossRef]
- McElvaney, G.N.; Sandhaus, R.A.; Miravitlles, M.; Turino, G.M.; Seersholm, N.; Wencker, M.; Stockley, R.A. Clinical considerations in individuals with α1-antitrypsin PI*SZ genotype. Eur. Respir. J. 2020, 55, 1902410. Available online: https://erj.ersjournals.com/content/55/6/1902410 (accessed on 31 March 2021). [CrossRef]
- Laffranchi, M.; Berardelli, R.; Ronzoni, R.; Lomas, D.A.; Fra, A. Heteropolymerization of α-1-antitrypsin mutants in cell models mimicking heterozygosity. Hum. Mol. Genet. 2018, 27, 1785–1793. [Google Scholar] [CrossRef]
- Laffranchi, M.; Elliston, E.L.; Miranda, E.; Perez, J.; Ronzoni, R.; Jagger, A.M.; Heyer-Chauhan, N.; Brantly, M.L.; Fra, A.; Lomas, D.A.; et al. Intrahepatic heteropolymerization of M and Z alpha-1-antitrypsin. JCI Insight 2020, 5, e135459. [Google Scholar] [CrossRef]
- Giacopuzzi, E.; Laffranchi, M.; Berardelli, R.; Ravasio, V.; Ferrarotti, I.; Gooptu, B.; Borsani, G.; Fra, A. Real-world clinical applicability of pathogenicity predictors assessed on SERPINA1 mutations in alpha-1-antitrypsin deficiency. Hum. Mutat. 2018, 39, 1203–1213. [Google Scholar] [CrossRef]
- Silva, D.; Oliveira, M.J.; Guimarães, M.; Lima, R.; Gomes, S.; Seixas, S. Alpha-1-antitrypsin (SERPINA1) mutation spectrum: Three novel variants and haplotype characterization of rare deficiency alleles identified in Portugal. Respir. Med. 2016, 116, 8–18. [Google Scholar] [CrossRef]
- Stein, P.E.; Carrell, R.W. What do dysfunctional serpins tell us about molecular mobility and disease? Nat. Struct. Biol. 1995, 2, 96–113. [Google Scholar] [CrossRef]
- Rodriguez-Frias, F.; Miravitlles, M.; Vidal, R.; Camos, S.; Jardi, R. Rare alpha-1-antitrypsin variants: Are they really so rare? Ther. Adv. Respir. Dis. 2012, 6, 79–85. [Google Scholar] [CrossRef]
- Canva, V.; Piotte, S.; Aubert, J.P.; Porchet, N.; Lecomte-Houcke, M.; Huet, G.; Zenjari, T.; Roumilhac, D.; Pruvot, F.-R.; Degand, P.; et al. Heterozygous M3Mmalton alpha1-antitrypsin deficiency associated with end-stage liver disease: Case report and review. Clin. Chem. 2001, 47, 1490–1496. [Google Scholar] [CrossRef]
- Figueira Gonçalves, J.M.; Martínez Bugallo, F.; Díaz Pérez, D.; Martín Martínez, M.D.; García-Talavera, I.; Pitti Pérez, R. Clinical manifestations of the Mmalton alpha-1 antitrypsin deficiency variant. Pulmonology 2018, 24, 48–49. [Google Scholar] [CrossRef]
- Joly, P.; Guillaud, O.; Hervieu, V.; Francina, A.; Mornex, J.-F.; Chapuis-Cellier, C. Clinical heterogeneity and potential high pathogenicity of the Mmalton Alpha 1 antitrypsin allele at the homozygous, compound heterozygous and heterozygous states. Orphanet J. Rare Dis. 2015, 10, 130. [Google Scholar] [CrossRef]
- Lomas, D.A.; Elliott, P.R.; Sidhar, S.K.; Foreman, R.C.; Finch, J.T.; Cox, D.W.; Whisstock, J.C.; Carrell, R. alpha 1-Antitrypsin Mmalton (Phe52-deleted) forms loop-sheet polymers in vivo. Evidence for the C sheet mechanism of polymerization. J. Biol. Chem. 1995, 270, 16864–16870. [Google Scholar] [CrossRef]
- Callea, F.; Giovannoni, I.; Francalanci, P.; Boldrini, R.; Faa, G.; Medicina, D.; Nobili, V.; Desmet, V.J.; Ishak, K.; Seyama, K.; et al. Mineralization of alpha-1-antitrypsin inclusion bodies in Mmalton alpha-1-antitrypsin deficiency. Orphanet J. Rare Dis. 2018, 13, 79. [Google Scholar] [CrossRef]
- Graham, A.; Kalsheker, N.A.; Newton, C.R.; Bamforth, F.J.; Powell, S.J.; Markham, A.F. Molecular characterisation of three alpha-1-antitrypsin deficiency variants: Proteinase inhibitor (Pi) nullcardiff (Asp256—Val); PiMmalton (Phe51—deletion) and PiI (Arg39—Cys). Hum. Genet. 1989, 84, 55–58. [Google Scholar] [CrossRef]
- Lomas, D.A.; Finch, J.T.; Seyama, K.; Nukiwa, T.; Carrell, R.W. Alpha 1-antitrypsin Siiyama (Ser53-->Phe). Further evidence for intracellular loop-sheet polymerization. J. Biol. Chem. 1993, 268, 15333–15335. [Google Scholar] [CrossRef]
- Seyama, K.; Nukiwa, T.; Takabe, K.; Takahashi, H.; Miyake, K.; Kira, S. Siiyama (serine 53 (TCC) to phenylalanine 53 (TTC)). A new alpha 1-antitrypsin-deficient variant with mutation on a predicted conserved residue of the serpin backbone. J. Biol. Chem. 1991, 266, 12627–12632. [Google Scholar] [CrossRef]
- Takabe, K.; Seyama, K.; Shinada, H.; Nouchi, T.; Miyahara, Y.; Nukiwa, T.; Miyake, K.; Tsukimoto, K.; Ichioka, M.; Marumo, F. A new variant of alpha-1-antitrypsin deficiency (Siiyama) associated with pulmonary emphysema. Intern. Med. 1992, 31, 702–707. [Google Scholar] [CrossRef]
- Miranda, E.; Pérez, J.; Ekeowa, U.I.; Hadzic, N.; Kalsheker, N.; Gooptu, B.; Portmann, B.; Belorgey, D.; Hill, M.; Chambers, S.; et al. A novel monoclonal antibody to characterize pathogenic polymers in liver disease associated with alpha1-antitrypsin deficiency. Hepatology 2010, 52, 1078–1088. [Google Scholar] [CrossRef]
- Brantly, M.L.; Wittes, J.T.; Vogelmeier, C.F.; Hubbard, R.C.; Fells, G.A.; Crystal, R.G. Use of a highly purified alpha 1-antitrypsin standard to establish ranges for the common normal and deficient alpha 1-antitrypsin phenotypes. Chest 1991, 100, 703–708. [Google Scholar] [CrossRef]
- Zhou, A.; Stein, P.E.; Huntington, J.A.; Carrell, R.W. Serpin polymerization is prevented by a hydrogen bond network that is centered on his-334 and stabilized by glycerol. J. Biol. Chem. 2003, 278, 15116–15122. [Google Scholar] [CrossRef]
- Patschull, A.O.M.; Segu, L.; Nyon, M.P.; Lomas, D.A.; Nobeli, I.; Barrett, T.E.; Gooptu, B. Therapeutic target-site variability in α1-antitrypsin characterized at high resolution. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 1492–1497. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Fra, A.M.; Gooptu, B.; Ferrarotti, I.; Miranda, E.; Scabini, R.; Ronzoni, R.; Benini, F.; Corda, L.; Medicina, D.; Luisetti, M.; et al. Three new alpha1-antitrypsin deficiency variants help to define a C-terminal region regulating conformational change and polymerization. PLoS ONE 2012, 7, e38405. [Google Scholar] [CrossRef]
- Medicina, D.; Montani, N.; Fra, A.M.; Tiberio, L.; Corda, L.; Miranda, E.; Pezzini, A.; Bonetti, F.; Ingrassia, R.; Scabini, R.; et al. Molecular characterization of the new defective P(brescia) alpha1-antitrypsin allele. Hum. Mutat. 2009, 30, E771–E781. [Google Scholar] [CrossRef]
- Miranda, E.; Ferrarotti, I.; Berardelli, R.; Laffranchi, M.; Cerea, M.; Gangemi, F.; Haq, I.; Ottaviani, S.; Lomas, D.A.; Irving, J.A.; et al. The pathological Trento variant of alpha-1-antitrypsin (E75V) shows nonclassical behaviour during polymerization. FEBS J. 2017, 284, 2110–2126. [Google Scholar] [CrossRef]
- Ronzoni, R.; Berardelli, R.; Medicina, D.; Sitia, R.; Gooptu, B.; Fra, A.M. Aberrant disulphide bonding contributes to the ER retention of alpha1-antitrypsin deficiency variants. Hum. Mol. Genet. 2016, 25, 642–650. [Google Scholar] [CrossRef]
- Dickens, J.A.; Ordóñez, A.; Chambers, J.E.; Beckett, A.J.; Patel, V.; Malzer, E.; Dominicus, C.S.; Bradley, J.; Peden, A.A.; Prior, I.A.; et al. The endoplasmic reticulum remains functionally connected by vesicular transport after its fragmentation in cells expressing Z-α1-antitrypsin. FASEB J. 2016, 30, 4083–4097. [Google Scholar] [CrossRef]
- Fra, A.; D’Acunto, E.; Laffranchi, M.; Miranda, E. Cellular models for the serpinopathies. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; Volume 1826, pp. 109–121. [Google Scholar]
- Ordóñez, A.; Pérez, J.; Tan, L.; Dickens, J.A.; Motamedi-Shad, N.; Irving, J.A.; Haq, I.; Ekeowa, U.; Marciniak, S.J.; Miranda, E.; et al. A single-chain variable fragment intrabody prevents intracellular polymerization of Z α1-antitrypsin while allowing its antiproteinase activity. FASEB J. 2015, 29, 2667–2678. [Google Scholar] [CrossRef]
- Miranda, E.; Römisch, K.; Lomas, D.A. Mutants of neuroserpin that cause dementia accumulate as polymers within the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 28283–28291. [Google Scholar] [CrossRef]
- Miranda, E.; MacLeod, I.; Davies, M.J.; Pérez, J.; Römisch, K.; Crowther, D.C.; Lomas, D.A. The intracellular accumulation of polymeric neuroserpin explains the severity of the dementia FENIB. Hum. Mol. Genet. 2008, 17, 1527–1539. [Google Scholar] [CrossRef]
- Haq, I.; Irving, J.A.; Saleh, A.D.; Dron, L.; Regan-Mochrie, G.L.; Motamedi-Shad, N.; Hurst, J.R.; Gooptu, B.; Lomas, D.A. Deficiency mutations of alpha-1 antitrypsin. Effects on folding, function, and polymerization. Am. J. Respir. Cell Mol. Biol. 2016, 54, 71–80. [Google Scholar] [CrossRef]
- Hansen, M.; Busse, M.N.; Andreasen, P.A. Importance of the amino-acid composition of the shutter region of plasminogen activator inhibitor-1 for its transitions to latent and substrate forms. Eur. J. Biochem. 2001, 268, 6274–6283. [Google Scholar] [CrossRef]
- Krem, M.M.; Di Cera, E. Conserved Ser residues, the shutter region, and speciation in serpin evolution. J. Biol. Chem. 2003, 278, 37810–37814. [Google Scholar] [CrossRef]
- Irving, J.A.; Pike, R.N.; Lesk, A.M.; Whisstock, J.C. Phylogeny of the serpin superfamily: Implications of patterns of amino acid conservation for structure and function. Genome Res. 2000, 10, 1845–1864. [Google Scholar] [CrossRef]
- Ferrarotti, I.; Thun, G.A.; Zorzetto, M.; Ottaviani, S.; Imboden, M.; Schindler, C.; von Eckardstein, A.; Rohrer, L.; Rochat, T.; Russi, E.W.; et al. Serum levels and genotype distribution of A1-antitrypsin in the general population. Thorax 2012, 67, 669–674. [Google Scholar] [CrossRef]
- Ferrarotti, I.; Scabini, R.; Campo, I.; Ottaviani, S.; Zorzetto, M.; Gorrini, M.; Luisetti, M. Laboratory diagnosis of alpha1-antitrypsin deficiency. Transl. Res. 2007, 150, 267–274. [Google Scholar] [CrossRef]
- Belorgey, D.; Irving, J.A.; Ekeowa, U.I.; Freeke, J.; Roussel, B.D.; Miranda, E.; Pérez, J.; Robinson, C.V.; Marciniak, S.J.; Crowther, D.C.; et al. Characterisation of serpin polymers in vitro and in vivo. Methods 2011, 53, 255–266. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38, 27–28. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; Mackerell, A.D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Konagurthu, A.S.; Whisstock, J.C.; Stuckey, P.J.; Lesk, A.M. MUSTANG: A multiple structural alignment algorithm. Proteins 2006, 64, 559–574. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Genotype | AAT 1 | CRP 2 | Age 3 | Clinical Presentation |
|---|---|---|---|---|---|
| 1.1. Proband | Z/Bologna | 0.23 | 0.001 | 50 | Emphysema, bronchiectasis, diabetes, liver steatosis |
| 1.2. Son | M1/Z | 0.67 | 0.007 | 16 | Healthy |
| 1.3. Daughter | M1/Bologna | 1.49 | 0.009 | 14 | Healthy |
| 2.1. Proband | Bologna/Bologna | 0.34 | 0.004 | 44 | Emphysema |
| 2.2. Son | M1/Bologna | 0.60 | 0.001 | 14 | Healthy |
| 2.3. Daughter | M1/Bologna | 0.72 | 0.001 | 8 | Healthy |
| 3.1. Proband | Bologna/Bologna | 0.28 | 0.003 | 39 | Emphysema |
| 4.1. Proband | M1/Bologna | 0.95 | 0.006 | 74 | Emphysema, chronic bronchitis |
| 5.1. Proband | M1/Bologna | 0.86 | 0.001 | 58 | Hepatitis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ronzoni, R.; Ferrarotti, I.; D’Acunto, E.; Balderacchi, A.M.; Ottaviani, S.; Lomas, D.A.; Irving, J.A.; Miranda, E.; Fra, A. The Importance of N186 in the Alpha-1-Antitrypsin Shutter Region Is Revealed by the Novel Bologna Deficiency Variant. Int. J. Mol. Sci. 2021, 22, 5668. https://doi.org/10.3390/ijms22115668
Ronzoni R, Ferrarotti I, D’Acunto E, Balderacchi AM, Ottaviani S, Lomas DA, Irving JA, Miranda E, Fra A. The Importance of N186 in the Alpha-1-Antitrypsin Shutter Region Is Revealed by the Novel Bologna Deficiency Variant. International Journal of Molecular Sciences. 2021; 22(11):5668. https://doi.org/10.3390/ijms22115668
Chicago/Turabian StyleRonzoni, Riccardo, Ilaria Ferrarotti, Emanuela D’Acunto, Alice M. Balderacchi, Stefania Ottaviani, David A. Lomas, James A. Irving, Elena Miranda, and Annamaria Fra. 2021. "The Importance of N186 in the Alpha-1-Antitrypsin Shutter Region Is Revealed by the Novel Bologna Deficiency Variant" International Journal of Molecular Sciences 22, no. 11: 5668. https://doi.org/10.3390/ijms22115668
APA StyleRonzoni, R., Ferrarotti, I., D’Acunto, E., Balderacchi, A. M., Ottaviani, S., Lomas, D. A., Irving, J. A., Miranda, E., & Fra, A. (2021). The Importance of N186 in the Alpha-1-Antitrypsin Shutter Region Is Revealed by the Novel Bologna Deficiency Variant. International Journal of Molecular Sciences, 22(11), 5668. https://doi.org/10.3390/ijms22115668