
Functional Characterization of the Obesity-Linked Variant of the β3-Adrenergic Receptor


Abstract
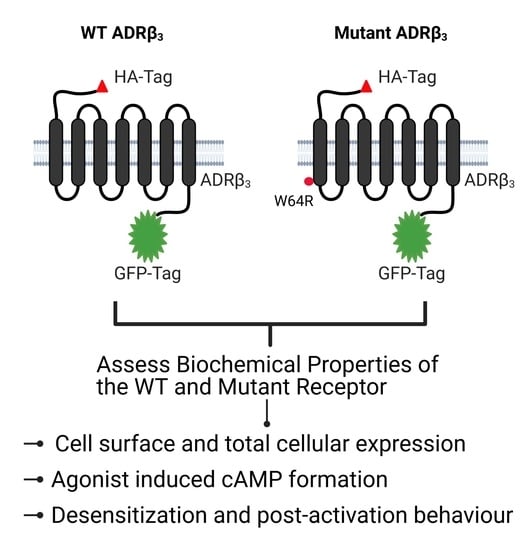
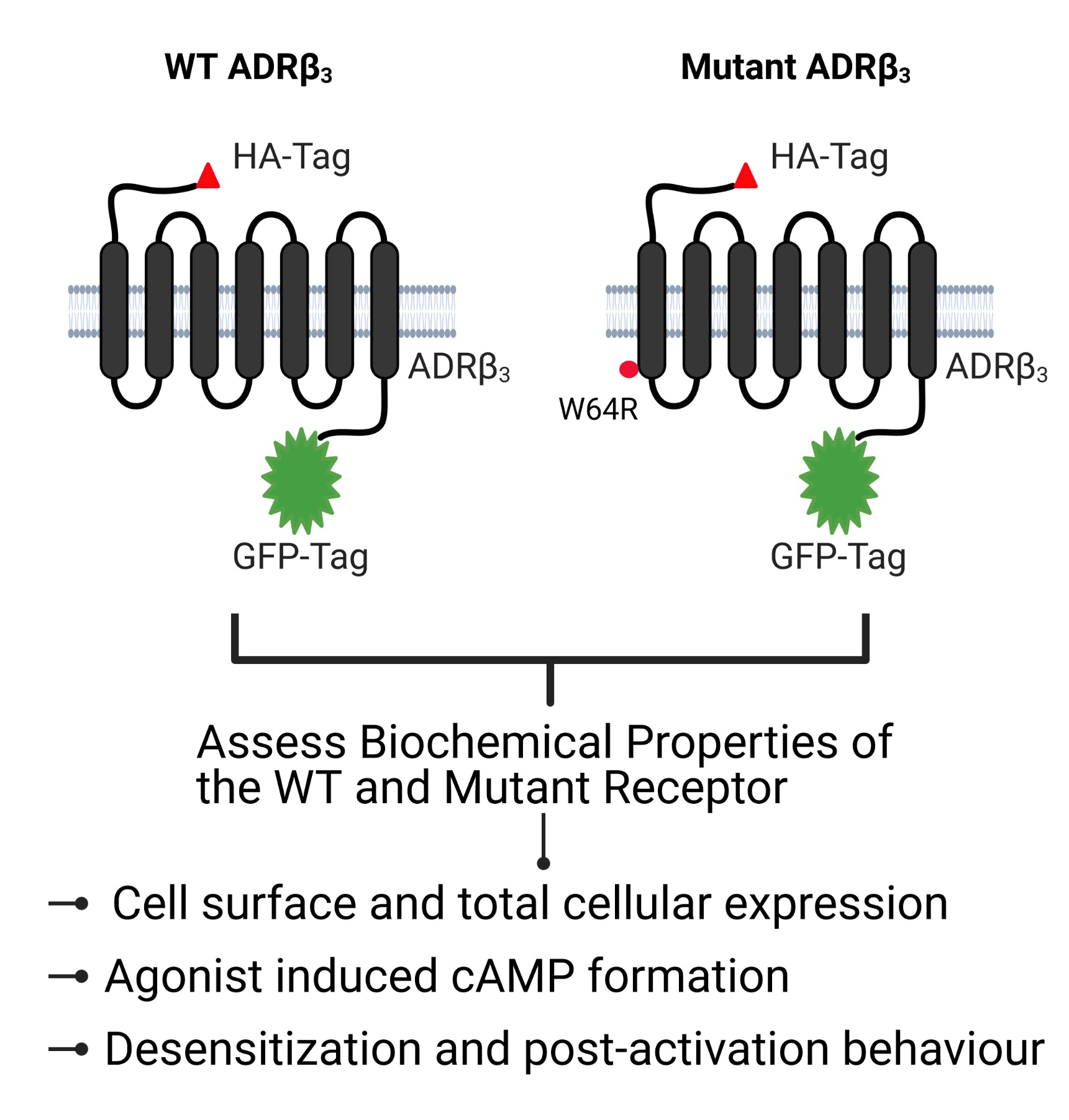{kind=link}
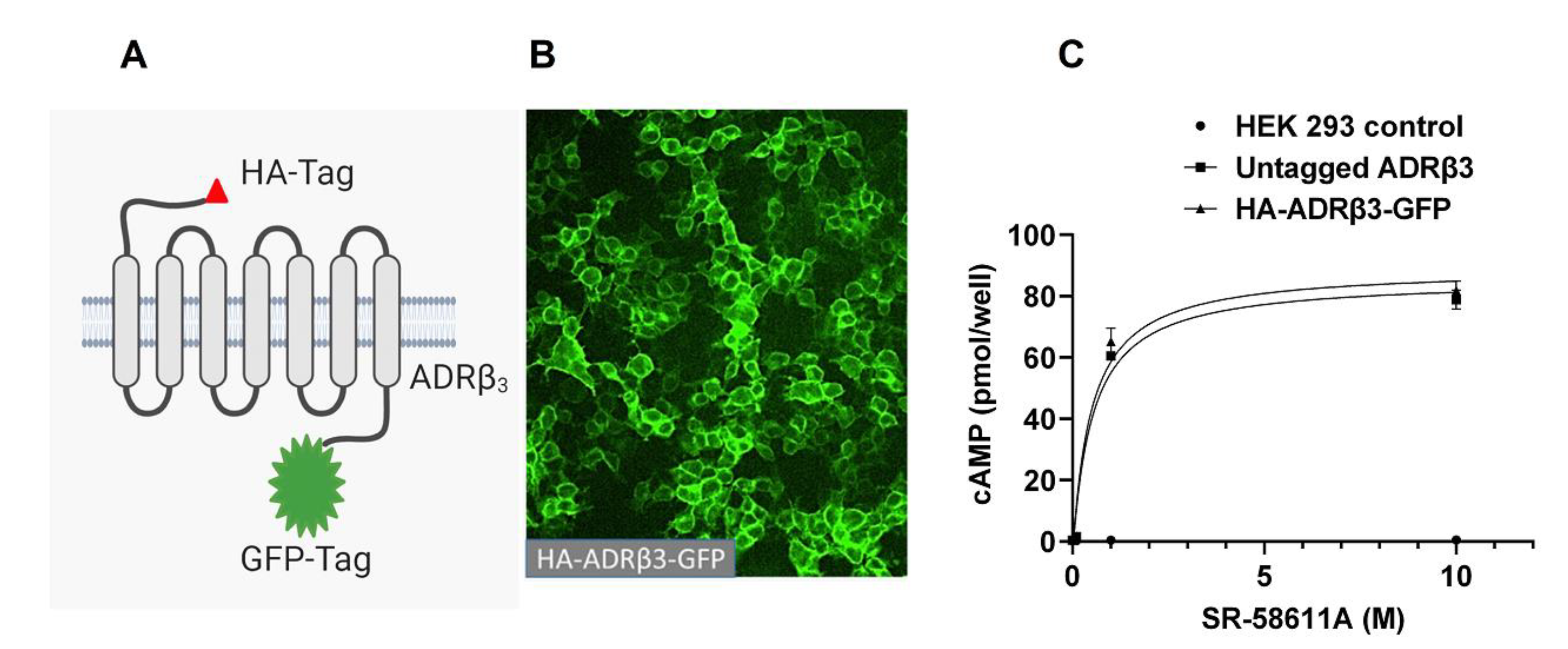{kind=link}
{kind=link}
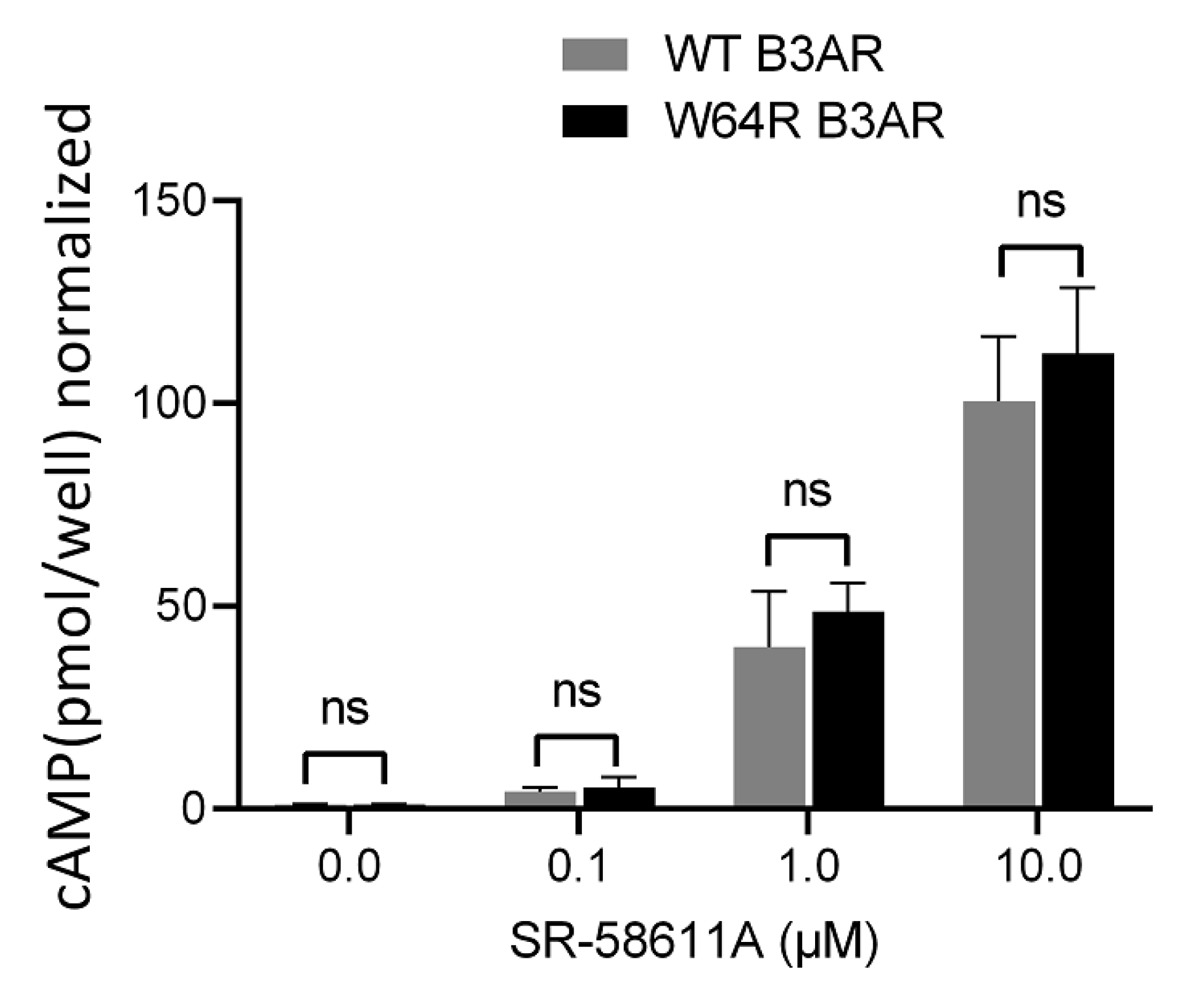{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. ADRβ3 (W64R) Variant Shows Normal Protein Expression and Subcellular Distribution
2.2. W64R Mutation Does Not Alter the Agonist-Induced cAMP Formation
2.3. Agonist-Induced Desensitization of ADRβ3 Does Not Involve Loss of Surface Receptor Expression
2.4. W64R Mutation Does Not Affect the Post-activation Behavior of ADRβ3
3. Discussion
4. Materials and Methods
4.1. Chemicals Reagents and Antibodies
4.2. DNA Reporter Constructs and Mutagenesis
4.3. Cell Culture and Generation of Stable cell Lines
4.4. cAMP Assay
4.5. Quantification of Cell Surface β3-Adrenergic Receptor
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Abboud, F.M. Concepts of adrenergic receptors. Med. Clin. N. Am. 1968, 52, 1009–1016. [Google Scholar] [CrossRef]
- Aurbach, G.D.; Spiegel, A.M.; Gardner, J.D. Beta-adrenergic receptors, cyclic AMP, and ion transport in the avian erythrocyte. Adv. Cycl. Nucleotide Res. 1975, 5, 117–132. [Google Scholar]
- Robinson, G.; Sutherland, E.W. On the relation of cyclic AMP to adrenergic receptors and sympathin. Adv. Cytopharmacol. 1971, 1, 263–272. [Google Scholar]
- Dawidek, G.M.B.; Robinson, M. Beta-adrenergic receptors in human anterior optic nerve: An autoradiographic study. Eye 1993, 7, 122–126. [Google Scholar] [CrossRef]
- Femminella, G.D.; Rengo, G.; Pagano, G.; de Lucia, C.; Komici, K.; Parisi, V.; Cannavo, A.; Liccardo, D.; Vigorito, C.; Filardi, P.P.; et al. Beta-adrenergic receptors and G protein-coupled receptor kinase-2 in Alzheimer’s disease: A new paradigm for prognosis and therapy? J. Alzheimers Dis. 2013, 34, 341–347. [Google Scholar] [CrossRef]
- Ghosh, P.M.; Shu, Z.-J.; Zhu, B.; Lu, Z.; Ikeno, Y.; Barnes, J.L.; Yeh, C.-K.; Zhang, B.-X.; Katz, M.S.; Kamat, A. Role of beta-adrenergic receptors in regulation of hepatic fat ac-cumulation during aging. J. Endocrinol. 2012, 213, 251–261. [Google Scholar] [CrossRef]
- Shiokawa, O.; Sadoshima, S.; Okada, Y.; Nagao, T.; Fujishima, M. Alpha- and beta-adrenergic receptors of noradrenergic inner-vation modulate the lower limits of cerebral and cerebellar blood flow autoregulation in spontaneously hypertensive rats. Gerontology 1989, 35, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Thandroyen, F.T.; Muntz, K.H.; Buja, L.M.; Willerson, J.T. Alterations in beta-adrenergic receptors, adenylate cyclase, and cyclic AMP concentrations during acute myocardial ischemia and reperfusion. Circulation 1990, 82, 1975523. [Google Scholar]
- Collins, S.; Surwit, R.S. The beta-adrenergic receptors and the control of adipose tissue metabolism and thermogenesis. Recent Prog. Horm. Res. 2001, 56, 309–328. [Google Scholar] [CrossRef]
- Insel, P.A.; Hammond, H.K. Beta-adrenergic receptors in heart failure. J. Clin. Investig. 1993, 92, 2564. [Google Scholar] [CrossRef]
- Lowell, B.B.; Bachman, E.S. Beta-Adrenergic receptors, diet-induced thermogenesis, and obesity. J. Biol. Chem. 2003, 278, 29385–29388. [Google Scholar] [CrossRef]
- Reuter, H.; Porzig, H. Beta-adrenergic receptors and responses in the heart. Postgrad. Med. J. 1981, 57, 62–70. [Google Scholar]
- Robidoux, J.; Martin, T.L.; Collins, S. Beta-adrenergic receptors and regulation of energy expenditure: A family affair. Ann. Rev. Pharmacol. Toxicol. 2004, 44, 297–323. [Google Scholar] [CrossRef] [PubMed]
- Stiles, G.L.; Caron, M.G.; Lefkowitz, R.J. Beta-adrenergic receptors: Biochemical mechanisms of physiological regulation. Physiol. Rev. 1984, 64, 661–743. [Google Scholar] [CrossRef]
- Molinoff, P.B.; Aarons, R.D. Effects of drugs on beta-adrenergic receptors on human lymphocytes. J. Cardiovasc. Pharmacol. 1983, 5, S63–S67. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S.; Yamaoka, K. Antidepressant drugs and beta-adrenergic receptors. Jpn. J. Psychopharmacol. 1988, 8, 437–441. [Google Scholar]
- Pearce, C.J.; Wallin, J.D. Labetalol and other agents that block both alpha- and beta-adrenergic receptors. Clevel. Clin. J. Med. 1994, 61, 80–82. [Google Scholar]
- Emorine, L.J.; Feve, B.; Pairault, J.; Briend-Sutren, M.M.; Marullo, S.; Delavier-Klutchko, C.; Strosberg, D.A. Structural basis for functional di-versity of beta 1-, beta 2- and beta 3-adrenergic receptors. Biochem. Pharmacol. 1991, 41, 853–859. [Google Scholar] [CrossRef]
- Emorine, L.J.; Feve, B.; Pairault, J.; Briend-Sutren, M.M.; Nahmias, C.; Marullo, S.; Delavier-Klutchko, C.; Strosberg, D.A. The human beta 3-adrenergic receptor: Relationship with atypical receptors. Am. J. Clin. Nutr. 1992, 55, 215S–218S. [Google Scholar] [CrossRef]
- Coman, O.A.; Păunescu, H.; Ghiţă, I.; Coman, L.; Bădărăru, A.; Fulga, I. Beta 3 adrenergic receptors: Molecular, histological, functional and pharmacological approaches. Rom. J. Morphol. Embryol. 2009, 50, 169–179. [Google Scholar] [PubMed]
- Lowell, B.B.; Flier, J.S. Brown adipose tissue, beta 3-adrenergic receptors, and obesity. Annu Rev Med. 1997, 48, 307–316. [Google Scholar] [CrossRef]
- Yasuda, K.; Akanuma, Y.; Kadowaki, T. Beta 3-adrenergic receptors. Nihon. Rinsho. 1997, 55, 468–472. [Google Scholar]
- Galitzky, J.; Carpene, C.; Lafontan, M.; Berlan, M. Specific stimulation of adipose tissue adrenergic beta 3 receptors by octopa-mine. C. R. Acad. Sci. III 1993, 316, 519–523. [Google Scholar] [PubMed]
- Hatakeyama, Y.; Sakata, Y.; Takakura, S.; Manda, T.; Mutoh, S. Acute and chronic effects of FR-149175, a β3-adrenergic receptor agonist, on energy expenditure in Zucker fatty rats. Am. J. Physiol. Integr. Comp. Physiol. 2004, 287, R336–R341. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Uchida, E.; Niiyama, M.; Yoshida, T.; Saito, M. Anti-obesity effects of selective agonists to the BETA.3-adrenergic receptor in dogs. II. Recruitment of thermogenic brown adipocytes and reduction of adiposity after chronic treatment with a BETA.3-adrenergic agonist. J. Veter. Med. Sci. 1998, 60, 465–469. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Duarte, S.F.P.; Francischetti, E.; Genelhu, V.; Cabello, P.H.; Pimentel, M.M.G. LEPR p.Q223R, beta3-AR p.W64R and LEP c.-2548G>A gene variants in obese Brazilian subjects. Genet. Mol. Res. 2007, 6, 1035–1043. [Google Scholar] [PubMed]
- Evans, D.; Minouchehr, S.; Hagemann, G.; Mann, W.A.; Wendt, D.; Wolf, A.; Beisiegel, U. Frequency of and interaction between polymor-phisms in the beta3-adrenergic receptor and in uncoupling proteins 1 and 2 and obesity in Germans. Int. J. Obes. Relat. Metab. Disord. 2000, 24, 1239–1245. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Evans, D.; Wolf, A.; Nellessen, U.; Ahle, S.; Kortner, B.; Kuhlmann, H.; Beisiegel, U. Association between polymorphisms in candidate genes and morbid obesity. Int. J. Obes. 2001, 25, S19–S21. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hao, K.; Peng, S.; Xing, H.; Yu, Y.; Huang, A.; Hong, X.; Wang, Y.; Chen, C.; Wang, B.; Zhang, X. Beta Adrenergic receptor polymorphism and obesity-related phe-notypes in hypertensive patients. Obes. Res. 2004, 12, 125–130. [Google Scholar] [CrossRef]
- O’Dell, S.D.; Bolla, M.K.; Miller, G.J.; Cooper, J.A.; Humphries, S.E.; Day, I.N. W64R mutation in beta-3-adrenergic receptor gene and weight in a large population sample. Int. J. Obes. Relat. Metab. Disord. 1998, 22, 377–379. [Google Scholar] [CrossRef][Green Version]
- Shima, Y.; Tsukada, T.; Nakanishi, K.; Ohta, H. Association of the Trp64Arg mutation of the beta3-adrenergic receptor with fatty liver and mild glucose intolerance in Japanese subjects. Clin. Chim. Acta. 1998, 274, 167–176. [Google Scholar] [CrossRef]
- Verdi, H.; Tulgar Kinik, S.; Yilmaz Yalcin, Y.; Muratoglu Sahin, N.; Yazici, A.C.; Atac, F.B. Beta-3AR W64R Polymorphism and 30-minute post-challenge plasma glucose levels in obese children. J. Clin. Res. Pediatr. Endocrinol. 2015, 7, 7–12. [Google Scholar] [CrossRef]
- Vrydag, W.; Alewijnse, A.E.; Michel, M.C. Do gene polymorphisms alone or in combination affect the function of human be-ta3-adrenoceptors? Br. J. Pharmacol. 2009, 156, 127–134. [Google Scholar] [CrossRef]
- Luo, Z.; Zhang, T.; Wang, S.; He, Y.; Ye, Q.; Cao, W. The Trp64Arg polymorphism in beta3 adrenergic receptor (ADRB3) gene is associated with adipokines and plasma lipids: A systematic review, meta-analysis, and meta-regression. Lipids Health Dis. 2020, 19, 99. [Google Scholar] [CrossRef] [PubMed]
- Piétri-Rouxel, F.; Manning, B.S.J.; Gros, J.; Strosberg, A.D. The biochemical effect of the naturally occurring Trp64-->Arg mutation on human beta3-adrenoceptor activity. JBIC J. Biol. Inorg. Chem. 1997, 247, 1174–1179. [Google Scholar]
- Isogaya, M.; Nagao, T.; Kurose, H. Enhanced cAMP response of naturally occurring mutant of human beta3-adrenergic receptor. Jpn. J. Pharmacol. 2002, 88, 314–318. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Candelore, M.R.; Deng, L.; Tota, L.M.; Kelly, L.J.; Cascieri, M.; Strader, C.D. Pharmacological characterization of a recently described human beta 3-adrenergic receptor mutant. Endocrinology 1996, 137, 2638–2641. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mohammad, S.; Baldini, G.; Granell, S.; Narducci, P.; Martelli, A.M.; Baldini, G. Constitutive Traffic of Melanocortin-4 Receptor in Neuro2A Cells and Immortalized Hypothalamic Neurons. J. Biol. Chem. 2007, 282, 4963–4974. [Google Scholar] [CrossRef]
- Tilley, D.G.; Rockman, H.A. Role of beta-adrenergic receptor signaling and desensitization in heart failure: New concepts and prospects for treatment. Expert Rev. Cardiovasc. Ther. 2006, 4, 417–432. [Google Scholar] [CrossRef]
- Lohse, M.J.; Engelhardt, S.; Danner, S.; Bohm, M. Mechanisms of beta-adrenergic receptor desensitization: From molecular biology to heart failure. Basic Res. Cardiol. 1996, 91, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.J.; Rockman, H.A. Beta-adrenergic receptor desensitization in cardiac hypertrophy and heart failure. Cell Biochem. Biophys. 1999, 31, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Nantel, F.; Bonin, H.; Emorine, L.J.; Zilberfarb, V.; Strosberg, A.D.; Bouvier, M.; Marullo, S. The human beta 3-adrenergic receptor is resistant to short term agonist-promoted desensitization. Mol. Pharmacol. 1993, 43, 548–555. [Google Scholar] [PubMed]
- Zhang, D.; Wang, Y.; Lin, H.; Sun, Y.; Wang, M.; Jia, Y.; Yu, X.; Jiang, H.; Xu, W.; Sun, J.; et al. Function and therapeutic potential of G protein-coupled receptors in epididymis. Br. J. Pharmacol. 2020, 177, 5489–5508. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, M.; Soliman, M. G-Protein Coupled Receptors (GPCRs): A Comprehensive Computational Perspective. Comb. Chem. High Throughput Screen. 2015, 18, 346–364. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.-T.; Sun, W.-Y.; Li, X.-R.; Sun, J.-C.; Du, J.-J.; Wei, W. Emerging roles of G protein-coupled receptors in hepatocellular carcinoma. Int. J. Mol. Sci. 2018, 19, 1366. [Google Scholar] [CrossRef]
- Oh, D.Y.; Olefsky, J.M. G protein-coupled receptors as targets for anti-diabetic therapeutics. Nat. Rev. Drug Discov. 2016, 15, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Mendez, I.; Mendez-Maldonado, K.; Manzo-Francisco, L.A.; Juarez-Hernandez, E.; Uribe, M.; Barbero-Becerra, V.J. G protein-coupled receptors: Key molecules in metabolic associated fatty liver disease development. Nutr. Res. 2021, 87, 70–79. [Google Scholar] [CrossRef]
- Granell, S.; Mohammad, S.; Ramanagoudr-Bhojappa, R.; Baldini, G. Obesity-linked variants of Melanocortin-4 receptor are misfolded in the endoplasmic reticulum and can be rescued to the cell surface by a chemical chaperone. Mol. Endocrinol. 2010, 24, 1805–1821. [Google Scholar] [CrossRef]
- Mohammad, S.; Patel, R.T.; Bruno, J.; Panhwar, M.S.; Wen, J.; McGraw, T.E. A naturally occurring gip receptor variant undergoes enhanced agonist-induced desensitization, which impairs gip control of adipose insulin Sensitivity. Mol. Cell. Biol. 2014, 34, 3618–3629. [Google Scholar] [CrossRef]
- O’Brien, J.B.; Wilkinson, J.C.; Roman, D.L. Regulator of G-protein signaling (RGS) proteins as drug targets: Progress and future potentials. J. Biol. Chem. 2019, 294, 18571–18585. [Google Scholar] [CrossRef]
- McPherson, K.B.; Leff, E.R.; Li, M.-H.; Meurice, C.; Tai, S.; Traynor, J.R.; Ingram, S.L. Regulators of G-Protein Signaling (RGS) proteins promote receptor coupling to G-protein-coupled inwardly rectifying potassium (GIRK) channels. J. Neurosci. 2018, 38, 8737–8744. [Google Scholar] [CrossRef]
- Kach, J.; Sethakorn, N.; Dulin, N.O. A finer tuning of G-protein signaling through regulated control of RGS proteins. Am. J. Physiol. Circ. Physiol. 2012, 303, H19–H35. [Google Scholar] [CrossRef]
- Masuho, I.; Balaji, S.; Muntean, B.S.; Skamangas, N.K.; Chavali, S.; Tesmer, J.J.; Babu, M.M.; Martemyanov, K.A. A Global Map of G Protein Signaling Regulation by RGS Proteins. Cell 2020, 183, 503–521. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, S.; Shenoy, S.K. GPCR desensitization: Acute and prolonged phases. Cell. Signal. 2018, 41, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Haji, E.; Mahri, S.A.; Aloraij, Y.; Malik, S.; Mohammad, S. Single-cell Analysis of beta2-adrenergic receptor dynamics by quan-titative fluorescence microscopy. Curr. Mol. Med. 2020, 20, 488–493. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haji, E.; Al Mahri, S.; Aloraij, Y.; Malik, S.S.; Mohammad, S. Functional Characterization of the Obesity-Linked Variant of the β3-Adrenergic Receptor. Int. J. Mol. Sci. 2021, 22, 5721. https://doi.org/10.3390/ijms22115721
Haji E, Al Mahri S, Aloraij Y, Malik SS, Mohammad S. Functional Characterization of the Obesity-Linked Variant of the β3-Adrenergic Receptor. International Journal of Molecular Sciences. 2021; 22(11):5721. https://doi.org/10.3390/ijms22115721
Chicago/Turabian StyleHaji, Esraa, Saeed Al Mahri, Yumna Aloraij, Shuja Shafi Malik, and Sameer Mohammad. 2021. "Functional Characterization of the Obesity-Linked Variant of the β3-Adrenergic Receptor" International Journal of Molecular Sciences 22, no. 11: 5721. https://doi.org/10.3390/ijms22115721
APA StyleHaji, E., Al Mahri, S., Aloraij, Y., Malik, S. S., & Mohammad, S. (2021). Functional Characterization of the Obesity-Linked Variant of the β3-Adrenergic Receptor. International Journal of Molecular Sciences, 22(11), 5721. https://doi.org/10.3390/ijms22115721