
Synthesis, Molecular Docking Analysis and Biological Evaluations of Saccharide-Modified Thiadiazole Sulfonamide Derivatives

 and
and
Abstract

1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Carbonic Anhydrase Inhibition
- (i).
- The IC50 values of intermediates 12a–h against hCA II and IX were 79.1–701.3 and 36.4–65.9 nM, respectively. Among them, 12b showed the strongest inhibitory activity against hCA II, with an IC50 of 79.1 nM. The substitution of halogen or electron withdrawing groups led to a decrease in hCA II inhibitory activity. Intermediate 12h showed the strongest inhibitory activity with hCA IX, with an IC50 of 36.4 nM. The effect of different substitutions on hCA IX inhibitory activity was not obvious. In the in vitro enzyme inhibitory activity test, 12a–h generally showed selectivity for hCA IX, and 12f exhibited the best selectivity for hCA IX; its inhibitory activity for hCA IX was 10 times higher than that for hCA II;
- (ii).
- The IC50 values of saccharide-modified compounds 14a–p against hCA II and IX were 80.3–1403.1 and 29.0–1082.0 nM, respectively; 14b exhibited the strongest inhibitory activity against hCA II, with an IC50 of 80.3 nM, while 14a and 14b had the best inhibitory activity against hCA IX, with an IC50 of 29.0 nM. Compared with 12a–h, the TPSA values of compounds 14a–p were significantly increased, which was beneficial to improving the selectivity through the differences in subcellular localization of isoforms. However, the inhibitory activity against hCA II and IX decreased to varying degrees, and the inhibitory activity against hCA II decreased significantly;
- (iii).
- Among the compounds 14a–p, 14a–b and 14i–j, which did not feature substitution on the benzene ring, were the compounds with the best hCA II inhibitory activities when the aliphatic chain lengths were n = 2 and 3, respectively. Whether the benzene ring was substituted by an electron-donating group or electron-donating group, the inhibitory activity of the compounds against hCA II decreased, especially for the substituents Cl, Br and NO2. The activity data showed that for hCA II, the inhibitory activities of compounds 14a–h with aliphatic chain length n = 3 were generally higher than those of compounds 14i–p with n = 2;
- (iv).
- Compared with hCA II, compounds 14a–p generally had higher inhibitory activity against hCA IX, among which 14a–c, 14g–j and 14p exhibited more desirable IC50 values of less than 100 nM. When the benzene ring was substituted by F or an electron donating group, the inhibitory activity of the compounds toward hCA IX did not change significantly, but when the substitution was for Cl, Br, or -NO2, the inhibitory activity was obviously reduced. Similar to hCA II, the inhibitory activities toward hCA IX for the compounds with aliphatic chain length n = 3 were higher than those of the compounds with n = 2;
- (v).
- Compounds 14a–c, 14g–j, and 14p with good inhibitory activity with hCA IX were tested for their inhibitory activities with hCA XII. The values of IC50 ranged from 40.6 to 140.8 nM. All tested compounds showed strong hCA XII inhibitory activity, of which that of 14j was the most prominent.
2.3. Studies on Docking into the Active Site of hCA IX
2.4. Molecular Dynamics Simulations
2.5. In Vitro Cytotoxicity Studies on Cancer Cells
- (i).
- The inhibitory activities of compounds 14a, 14b, 14h, and 14p on the two tumor cell lines were better than those of the positive control drug AZM;
- (ii).
- Under hypoxic conditions, the inhibitory activities of the tested compounds and AZM on the tumor cell lines were all higher than those under normoxia;
- (iii).
- Compared with the breast adenocarcinoma MDA-MB-231, the tested compounds generally showed higher inhibitory efficacy with the colon cancer cell line HT-29.
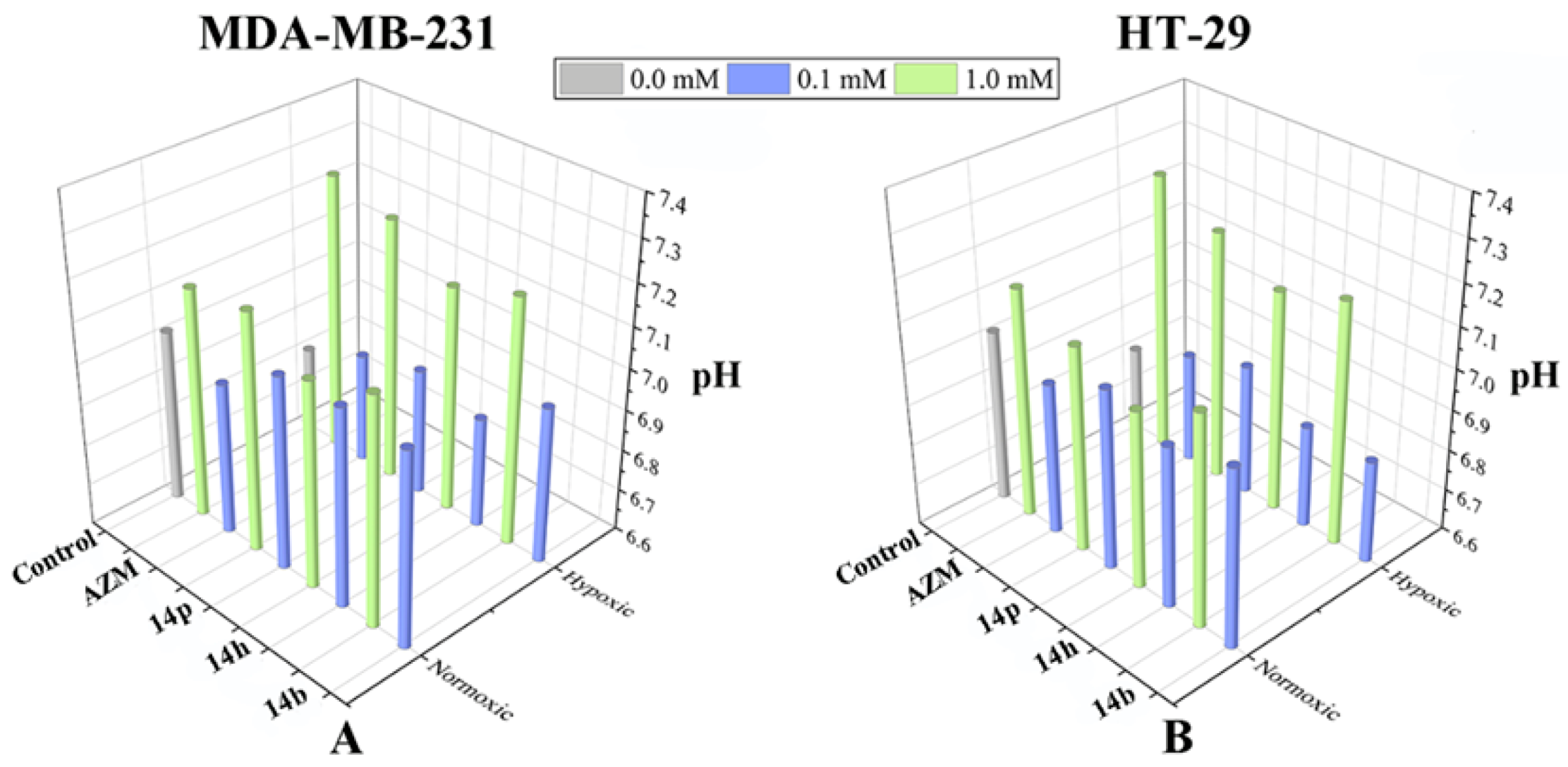
2.6. Extracellular pH Measurement in the Presence of Compound 14b, 14h, and 14p
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of Intermediate 2–4
3.1.2. General Procedure of (((2R,3R,4S,5R,6R)-3,4,5-Tris(benzoyloxy)-6-((benzoyloxy)methyl)tetrahydro-2H-pyran-2-yl)amino) Acid (5a and 5b)
4-Oxo-4-(((2R,3R,4S,5R,6R)-3,4,5-tris(benzoyloxy)-6-((benzoyloxy)methyl)tetrahydro-2H-pyran-2-yl)amino)butanoic Acid (5a)
5-Oxo-5-(((2R,3R,4S,5R,6R)-3,4,5-tris(benzoyloxy)-6-((benzoyloxy)methyl)tetrahydro-2H-pyran-2-yl)amino)pentanoic Acid (5b)
3.1.3. General Procedure of 4-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)benzoic Acid Derivatives (7a–h)
3.1.4. General Procedure of (9H-Fluoren-9-yl)methyl (4-(chlorocarbonyl)phenyl)carbamate Derivatives (8a–h)
3.1.5. Synthesis of 5-Amino-1,3,4-thiadiazole-2-sulfonamide (10)
3.1.6. General Procedure of (9H-Fluoren-9-yl)methyl (4-((5-sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)carbamate Derivatives (11a–h)
3.1.7. General Procedure of 4-Amino-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)benzamide Derivatives (12a–h)
4-Amino-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)benzamide (12a)
3-Amino-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)benzamide (12b)
4-Amino-3-fluoro-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)benzamide (12c)
4-Amino-3-chloro-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)benzamide (12d)
4-Amino-3-bromo-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)benzamide (12e)
4-Amino-3-nitro-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)benzamide (12f)
4-Amino-3-methyl-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)benzamide (12g)
4-Amino-3-methoxy-N-(5-sulfamoyl-1,3,4-thiadiazol-2-yl)benzamide (12h)
3.1.8. General Procedure of (2R,3R,4S,5R,6R)-2-((Benzoyloxy)methyl)-6-(4-oxo-4-((4-((5-sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)amino)butanamido)tetrahydro-2H-pyran-3,4,5-triyl Tribenzoate Derivatives (13a–p)
3.1.9. General Procedure of N1-(4-((5-Sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)-N5-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)glutaramide Derivatives (14a–p)
N1-(4-((5-Sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)-N5-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)glutaramide (14a)
N1-(3-((5-Sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)-N5-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)glutaramide (14b)
N1-(2-Fluoro-4-((5-sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)-N5-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)glutaramide (14c)
N1-(2-Chloro-4-((5-sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)-N5-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)glutaramide (14d)
N1-(2-Bromo-4-((5-sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)-N5-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)glutaramide (14e)
N1-(2-Nitro-4-((5-sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)-N5-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)glutaramide (14f)
N1-(2-Methyl-4-((5-sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)-N5-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)glutaramide (14g)
N1-(2-Methoxy-4-((5-sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)-N5-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)glutaramide (14h)
N1-(4-((5-Sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)-N4-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)succinimide (14i)
N1-(3-((5-Sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)-N4-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)succinimide (14j)
N1-(2-Fluoro-4-((5-sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)-N4-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)succinimide (14k)
N1-(2-Chloro-4-((5-sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)-N4-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)succinimide (14l)
N1-(2-Bromo-4-((5-sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)-N4-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)succinimide (14m)
N1-(2-Nitro-4-((5-sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)-N4-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)succinimide (14n)
N1-(2-Methy-4-((5-sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)-N4-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)succinimide (14o)
N1-(2-Methoxy-4-((5-sulfamoyl-1,3,4-thiadiazol-2-yl)carbamoyl)phenyl)-N4-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)succinimide (14p)
3.2. hCA Enzyme Inhibition Assays
3.3. Preparation for Docking Studies and Prediction by TPSA
3.4. Preparation for Molecular Dynamics Simulation
3.5. CCK-8 Assay In Vitro
3.6. Measurement of Extracellular pH
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Supuran, C.T. Experimental carbonic anhydrase inhibitors for the treatment of hypoxic tumors. J. Exp. Pharmacol. 2020, 12, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Angeli, A.; Carta, F.; Nocentini, A.; Winum, J.Y.; Zalubovskis, R.; Akdemir, A.; Onnis, V.; Eldehna, W.M.; Capasso, C.; De Simone, G.; et al. Carbonic Anhydrase Inhibitors Targeting Metabolism and Tumor Microenvironment. Metabolites 2020, 10, 412. [Google Scholar] [CrossRef] [PubMed]
- Winum, J.Y.; Colinas, P. Chapter 21-Carbonic Anhydrases as Esterases and Their Biotechnological Applications. CARBONIC Anhydrases as Biocatalysts; Elsevier: Amsterdam, The Netherlands, 2015; pp. 361–371. [Google Scholar]
- Akgul, O.; Mannelli, L.C.; Vullo, D.; Angeli, A.; Ghelardini, C.; Bartolucci, G.; Altamimi, A.S.A.; Scozzafava, A.; Supuran, C.T.; Carta, F. Discovery of novel nonsteroidal anti-inflammatory drugs and carbonic anhydrase inhibitors hybrids (NSAIDs-CAIs) for the management of rheumatoid arthritis. J. Med. Chem. 2018, 61, 4961–4977. [Google Scholar] [CrossRef] [PubMed]
- Andring, J.T.; Fouch, M.; Akocak, S.; Angeli, A.; Supuran, C.T.; Ilies, M.A.; McKenna, R. Structural basis of nanomolar inhibition of tumor-associated carbonic anhydrase IX: X-ray crystallographic and inhibition study of lipophilic inhibitors with acetazolamide backbone. J. Med. Chem. 2020, 63, 13064–13075. [Google Scholar] [CrossRef]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple binding modes of inhibitors to carbonic anhydrases: How to design specific drugs targeting 15 different isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef]
- Ali, M.; Bozdag, M.; Farooq, U.; Angeli, A.; Carta, F.; Berto, P.; Zanotti, G.; Supuran, C.T. Benzylaminoethyureido-tailed benzenesulfonamides: Design, synthesis, kinetic and X-ray Investigations on human carbonic anhydrases. Int. J. Mol. Sci. 2020, 21, 2560. [Google Scholar] [CrossRef]
- Zhang, Z.P.; Yin, Z.F.; Li, J.Y.; Wang, Z.P.; Wu, Q.J.; Wang, J.; Liu, Y.; Cheng, M.S. Synthesis, molecular docking analysis, and carbonic anhydrase inhibitory evaluations of benzenesulfonamide derivatives containing thiazolidinone. Molecules 2019, 24, 2418. [Google Scholar] [CrossRef]
- Mam, Y.M.; Brian, P.M.; Robert, M.; Susan, C.F. Carbonic anhydrases: Role in pH control and cancer. Metabolites 2018, 8, 19. [Google Scholar]
- Supuran, C.T. Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017, 7, 48. [Google Scholar] [CrossRef]
- Lee, S.H.; McIntyre, D.; Honess, D.; Hulikova, A.; Pacheco-Torres, J.; Cerdán, S.; Swietach, P.; Harris, A.L.; Griffiths, J.R. Carbonic anhydrase IX is a pH-stat that sets an acidic tumour extracellular pH in vivo. Br. J. Cancer 2018, 119, 622. [Google Scholar] [CrossRef]
- Galati, S.; Yonchev, D.; Rodríguez-Pérez, R.; Vogt, M.; Tuccinardi, T.; Bajorath, J. Predicting isoform-selective carbonic anhydrase inhibitors via machine learning and rationalizing structural features important for selectivity. ACS Omega 2021, 6, 4080–4089. [Google Scholar] [CrossRef] [PubMed]
- Strapcova, S.; Takacova, M.; Csaderova, L.; Martinelli, P.; Lukacikova, L.; Gal, V.; Kopacek, J.; Svastova, E. Clinical and pre-clinical evidence of carbonic anhydrase IX in pancreatic cancer and its high expression in pre-cancerous lesions. Cancers 2020, 12, 2005. [Google Scholar] [CrossRef]
- A Study of SLC-0111 and Gemcitabine for Metastatic Pancreatic Ductal Cancer in Subjects Positive for CAIX. Available online: https://clinicaltrials.gov/ct2/show/NCT03450018 (accessed on 10 April 2020).
- Pacchiano, F.; Carta, F.; McDonald, P.C.; Lou, Y.; Vullo, D.; Scozzafava, A.; Dedhar, S.; Supuran, C.T. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J. Med. Chem. 2011, 54, 1896–1902. [Google Scholar] [CrossRef] [PubMed]
- Bonardi, A.; Nocentini, A.; Bua, S.; Combs, J.; Lomelino, C.; Andring, J.; Lucarini, L.; Sgambellone, S.; Masini, E.; McKenna, R.; et al. Sulfonamide inhibitors of human carbonic anhydrases designed through a three-tails approach: Improving ligand/isoform matching and selectivity of action. J. Med. Chem. 2020, 63, 7422–7444. [Google Scholar] [CrossRef]
- Supuran, C.T. Structure-based drug discovery of carbonic anhydrase inhibitors. J. Enzym. Inhib. Med. Chem. 2012, 27, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Yamashita, M.; Yokota, D.; Hirano, I.; Ono, T.; Fujie, M.; Shibata, K.; Niimi, T.; Suyama, T.; Maddali, K.; et al. Development and pharmacologic characterization of deoxybromophospha sugar derivatives with antileukemic activity. Investig. New Drugs 2010, 28, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, H.S.; Tolan, H.E.M.; El-Bayaa, M.N.; Radwan, M.A.A.; El-Manawaty, M.; El-Sayed, W.A. Synthesis and anticancer activity of new pyridine-thiophene and pyridine-furan hybrid compounds, their sugar hydrazone, and glycosyl derivatives. Russ. J. Gen. Chem. 2020, 90, 1706–1715. [Google Scholar] [CrossRef]
- Nassar, I.F.; El-Farargy, A.F.; Abdelrazek, F.M.; Hamza, Z. Synthesis of new uracil derivatives and their sugar hydrazones with potent antimicrobial, antioxidant and anticancer activities. Nucleos. Nucleot. Nucl. 2020, 39, 991–1010. [Google Scholar] [CrossRef]
- D’Andrea, F.; Sartini, S.; Piano, I.; Franceschi, M.; Quattrini, L.; Guazzelli, L.; Ciccone, L.; Orlandini, E.; Gargini, C.; Motta, C.L.; et al. Oxy-imino saccharidic derivatives as a new structural class of aldose reductase inhibitors endowed with anti-oxidant activity. J. Enzym. Inhib. Med. Chem. 2020, 35, 1194–1205. [Google Scholar] [CrossRef]
- Chen, X.; Gu, H.; Lyu, Z.; Liu, X.; Wang, L.; Chen, H.; Brash, J.L. Sulfonate groups and saccharides as essential structural elements in heparin-mimicking polymers used as surface modifiers: Optimization of relative contents for anti-thrombogenic properties. ACS Appl. Mater. Interfaces 2018, 10, 1440–1449. [Google Scholar] [CrossRef]
- Guler, O.O.; De Sımone, G.; Supuran, C.T. Drug design studies of the novel antitumor targets carbonic anhydrase IX and XII. Curr. Med. Chem. 2010, 17, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Mishra, C.B.; Tiwari, M.; Supuran, C.T. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: Where are we today? Med. Res. Rev. 2020, 40, 1–81. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Lin, B.; Bao, Y.; Yan, H.N.; Zhang, M.; Chang, X.W.; Zhang, X.X.; Wang, Z.J.; Wei, G.F.; Cheng, M.S.; et al. Dual-tail approach to discovery of novel carbonic anhydrase IX inhibitors by simultaneously matching the hydrophobic and hydrophilic halves of the active site. Eur. J. Med. Chem. 2017, 132, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nocentini, A.; Trallori, E.; Singh, S.; Lomelino, C.L.; Bartolucci, G.; Di, C.M.L.; Ghelardini, C.; Mckenna, R.; Gratteri, P.; Supuran, C.T. 4-Hydroxy-3-nitro-5-ureido-benzenesulfonamides selectively target the tumor-associated carbonic anhydrase isoforms IX and XII showing hypoxia-enhanced antiproliferative profiles. J. Med. Chem. 2018, 61, 10860–10874. [Google Scholar] [CrossRef]
- Akocak, S.; Güzel-Akdemir, Ö.; Sanku, R.K.K.; Russom, S.S.; Iorga, B.I.; Supuran, C.T.; Ilies, M.A. Pyridinium derivatives of 3-aminobenzenesulfonamide are nanomolarpotent inhibitors of tumor-expressed carbonic anhydrase isozymes CA IX and CA XII. Bioorg. Chem. 2020, 103, 104204. [Google Scholar] [CrossRef]
- Kumar, D.; Mishra, K.B.; Mishra, B.B.; Mondal, S.; Tiwari, V.K. Click chemistry inspired highly facile synthesis of triazolyl ethisterone glycoconjugates. Steroids 2014, 80, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Li, F.R.; Fan, Z.F.; Qi, S.J.; Wang, Y.S.; Wang, J.; Liu, Y.; Cheng, M.S. Design, synthesis, molecular docking analysis, and carbonic anhydrase IX inhibitory evaluations of novel N-Substituted-β-d-glucosamine derivatives that incorporate benzenesulfonamides. Molecules 2017, 22, 785. [Google Scholar]
- Verpoorte, J.A.; Mehta, S.; Edsall, J.T. Esterase activities of human carbonic anhydrases B and C. J. Boil. Chem. 1967, 242, 4221–4229. [Google Scholar] [CrossRef]
- Leitans, J.; Kazaks, A.; Balode, A.; Ivanova, J.; Zalubovskis, R.; Supuran, C.T.; Tars, K. Efficient expression and crystallization system of cancer-associated carbonic anhydrase isoform IX. J. Med. Chem. 2015, 58, 9004–9009. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Diogo, S.M.; Stefano, F.; Maria, J.R.; Arthur, J.O. AutoDock4Zn: An improved autodock force field for small-molecule docking to zinc metalloproteins. J. Chem. Inf. Model. 2014, 54, 2371–2379. [Google Scholar]
- Discovery Studio User Manual; Accelrys Inc.: San Diego, CA, USA, 2008.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | R | hCA II (IC50, nM) | hCA IX (IC50, nM) | hCA XII (IC50, nM) | TPSA (Å2) |
|---|---|---|---|---|---|
| 12a | R = H | 109.9 ± 6.9 | 44.6 ± 8.3 | n.t. | 140.3 |
| 12b | - | 79.1 ± 5.4 | 63.8 ± 6.3 | n.t. | 140.3 |
| 12c | R = 3-F | 242.4 ± 9.4 | 41.3 ± 6.2 | n.t. | 140.3 |
| 12d | R = 3-Cl | 401.1 ± 12.3 | 36.5 ± 7.6 | n.t. | 140.3 |
| 12e | R = 3-Br | 543.2 ± 10.3 | 65.9 ± 8.3 | n.t. | 140.3 |
| 12f | R = 3-NO2 | 701.3 ± 13.2 | 55.1 ± 11.5 | n.t. | 183.1 |
| 12g | R = 3-CH3 | 119.7 ± 14.8 | 43.8 ± 6.6 | n.t. | 140.3 |
| 12h | R = 3-OCH3 | 147.2 ± 15.3 | 36.4 ± 8.3 | n.t. | 149.2 |
| 14a | n = 3, R = H | 140.9 ± 12.9 | 29.0 ± 4.2 | 140.8 ± 5.5 | 266.2 |
| 14b | n = 3 | 80.3 ± 11.5 | 29.0 ± 1.5 | 56.0 ± 8.5 | 266.2 |
| 14c | n = 3, R = 3-F | 215.0 ± 9.5 | 76.6 ± 6.2 | 81.8 ± 11.1 | 266.2 |
| 14d | n = 3, R = 3-Cl | 765.4 ± 15.1 | 279.8 ± 10.2 | n.t. | 266.2 |
| 14e | n = 3, R = 3-Br | 885.6 ± 13.7 | 1082.0 ± 8.5 | n.t. | 266.2 |
| 14f | n = 3, R = 3-NO2 | 995.3 ± 10.6 | 117.1 ± 6.9 | n.t. | 309.0 |
| 14g | n = 3, R = 3-CH3 | 165.2 ± 9.1 | 53.4 ± 6.2 | 70.5 ± 10.9 | 266.2 |
| 14h | n = 3, R = 3-OCH3 | 271.7 ± 9.0 | 68.8 ± 10.2 | 84.9 ± 7.6 | 275.1 |
| 14i | n = 2, R = H | 197.4 ± 7.9 | 54.9 ± 6.2 | 52.8 ± 11.4 | 266.2 |
| 14j | n = 2 | 260.3 ± 12.6 | 69.4 ± 5.7 | 40.6 ± 6.9 | 266.2 |
| 14k | n = 2, R = 3-F | 474.4 ± 7.1 | 141.1 ± 8.1 | n.t. | 266.2 |
| 14l | n = 2, R = 3-Cl | 612.0 ± 8.5 | 255.9 ± 7.9 | n.t. | 266.2 |
| 14m | n = 2, R = 3-Br | 823.7 ± 11.1 | 865.0 ± 10.7 | n.t. | 266.2 |
| 14n | n = 2, R = 3-NO2 | 1403.1 ± 24.6 | 290.9 ± 10.9 | n.t. | 309.0 |
| 14o | n = 2, R = 3-CH3 | 269.8 ± 13.9 | 135.0 ± 7.0 | n.t. | 266.2 |
| 14p | n = 2, R = 3-OCH3 | 332.1 ± 16.8 | 47.9 ± 7.1 | 42.3 ± 9.4 | 275.1 |
| AZM | 16.7 ± 4.7 | 50.2 ± 12.8 | 38.2 ± 10.3 | 113.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.-P.; Zhong, Y.; Han, Z.-B.; Zhou, L.; Su, H.-S.; Wang, J.; Liu, Y.; Cheng, M.-S. Synthesis, Molecular Docking Analysis and Biological Evaluations of Saccharide-Modified Thiadiazole Sulfonamide Derivatives. Int. J. Mol. Sci. 2021, 22, 5482. https://doi.org/10.3390/ijms22115482
Zhang Z-P, Zhong Y, Han Z-B, Zhou L, Su H-S, Wang J, Liu Y, Cheng M-S. Synthesis, Molecular Docking Analysis and Biological Evaluations of Saccharide-Modified Thiadiazole Sulfonamide Derivatives. International Journal of Molecular Sciences. 2021; 22(11):5482. https://doi.org/10.3390/ijms22115482
Chicago/Turabian StyleZhang, Zuo-Peng, Ye Zhong, Zhen-Bin Han, Lin Zhou, Hua-Sheng Su, Jian Wang, Yang Liu, and Mao-Sheng Cheng. 2021. "Synthesis, Molecular Docking Analysis and Biological Evaluations of Saccharide-Modified Thiadiazole Sulfonamide Derivatives" International Journal of Molecular Sciences 22, no. 11: 5482. https://doi.org/10.3390/ijms22115482
APA StyleZhang, Z.-P., Zhong, Y., Han, Z.-B., Zhou, L., Su, H.-S., Wang, J., Liu, Y., & Cheng, M.-S. (2021). Synthesis, Molecular Docking Analysis and Biological Evaluations of Saccharide-Modified Thiadiazole Sulfonamide Derivatives. International Journal of Molecular Sciences, 22(11), 5482. https://doi.org/10.3390/ijms22115482