
Inhibition of Carbonic Anhydrase IX Promotes Apoptosis through Intracellular pH Level Alterations in Cervical Cancer Cells

 , , ,
, , , 
Abstract
1. Introduction
2. Results
2.1. Compound E Decreased Viability of Cell Lines
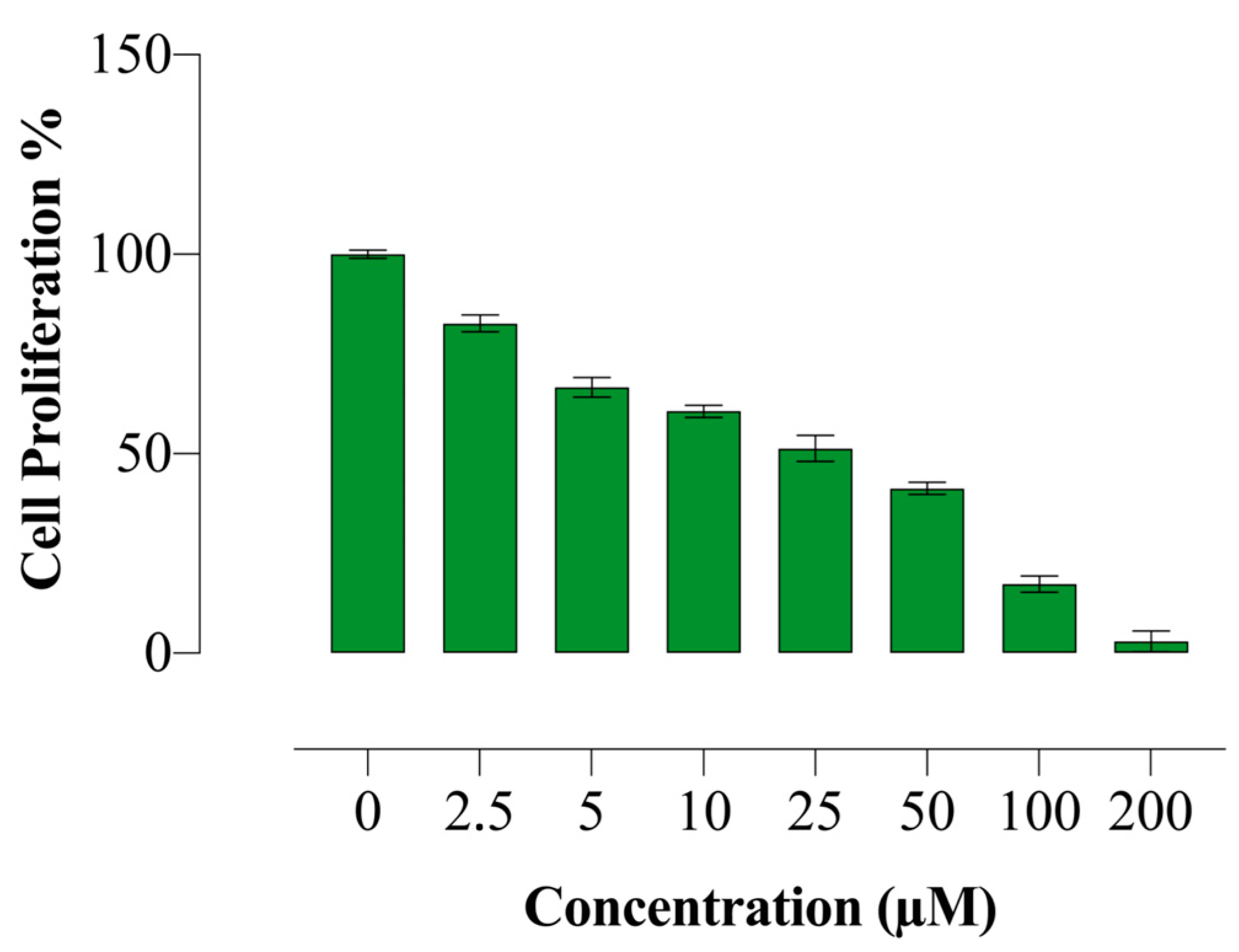
2.2. Compound E Reduced Proliferation of HeLa Cells
2.3. Compound E Triggered Apoptotic Morphology in HeLa Cells
2.4. Compound E Increase Apoptotic Cell Ratio in HeLa Cells
2.5. Compound E Disrupted pH Regulation of HeLa Cells
2.6. Compound E Increased Intracellular ROS Level in HeLa Cells
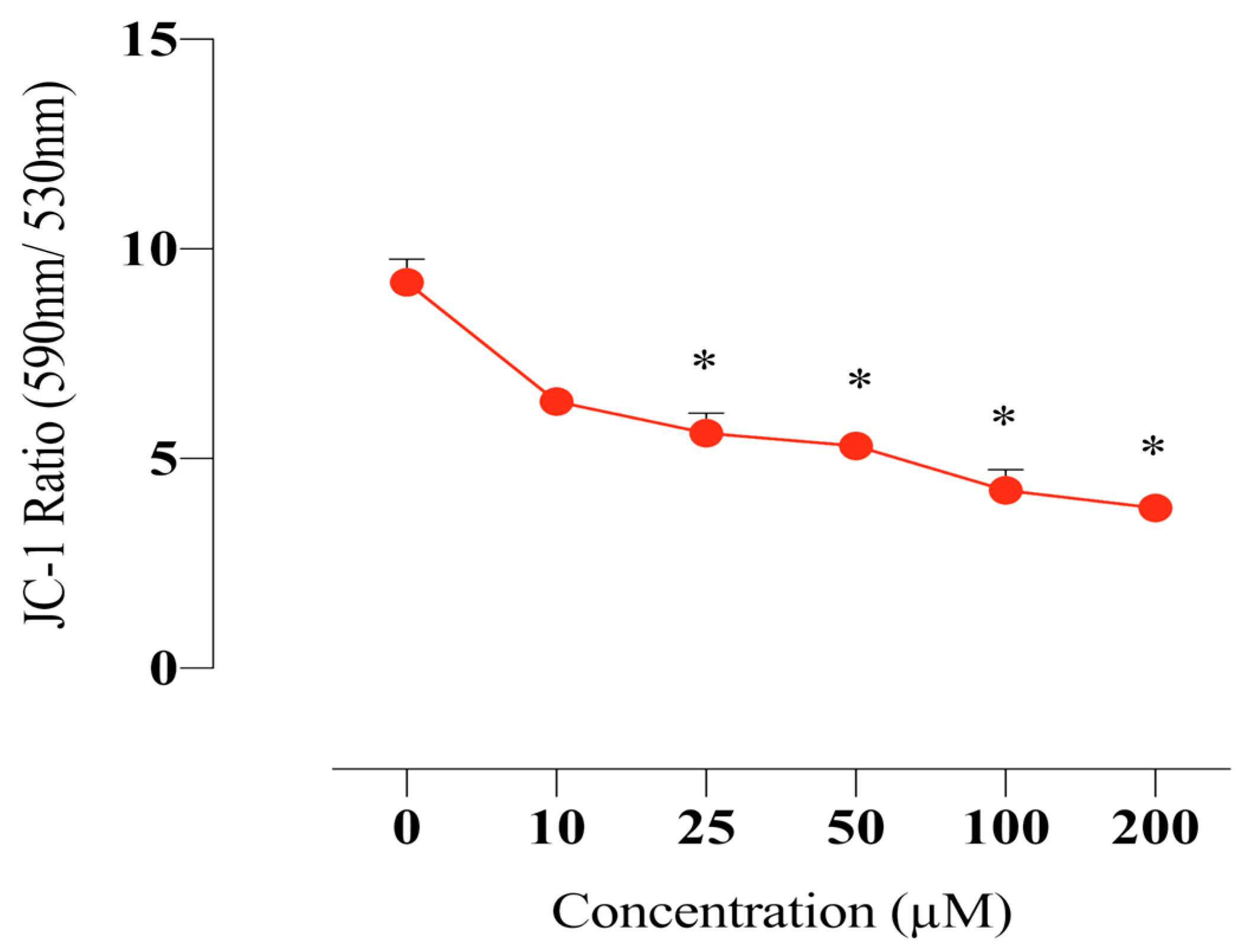
2.7. Compound E Disrupted Mitochondrial Membrane Potential in HeLa Cells
2.8. Sulphonamide E Arrested Cell Cycle in HeLa Cells
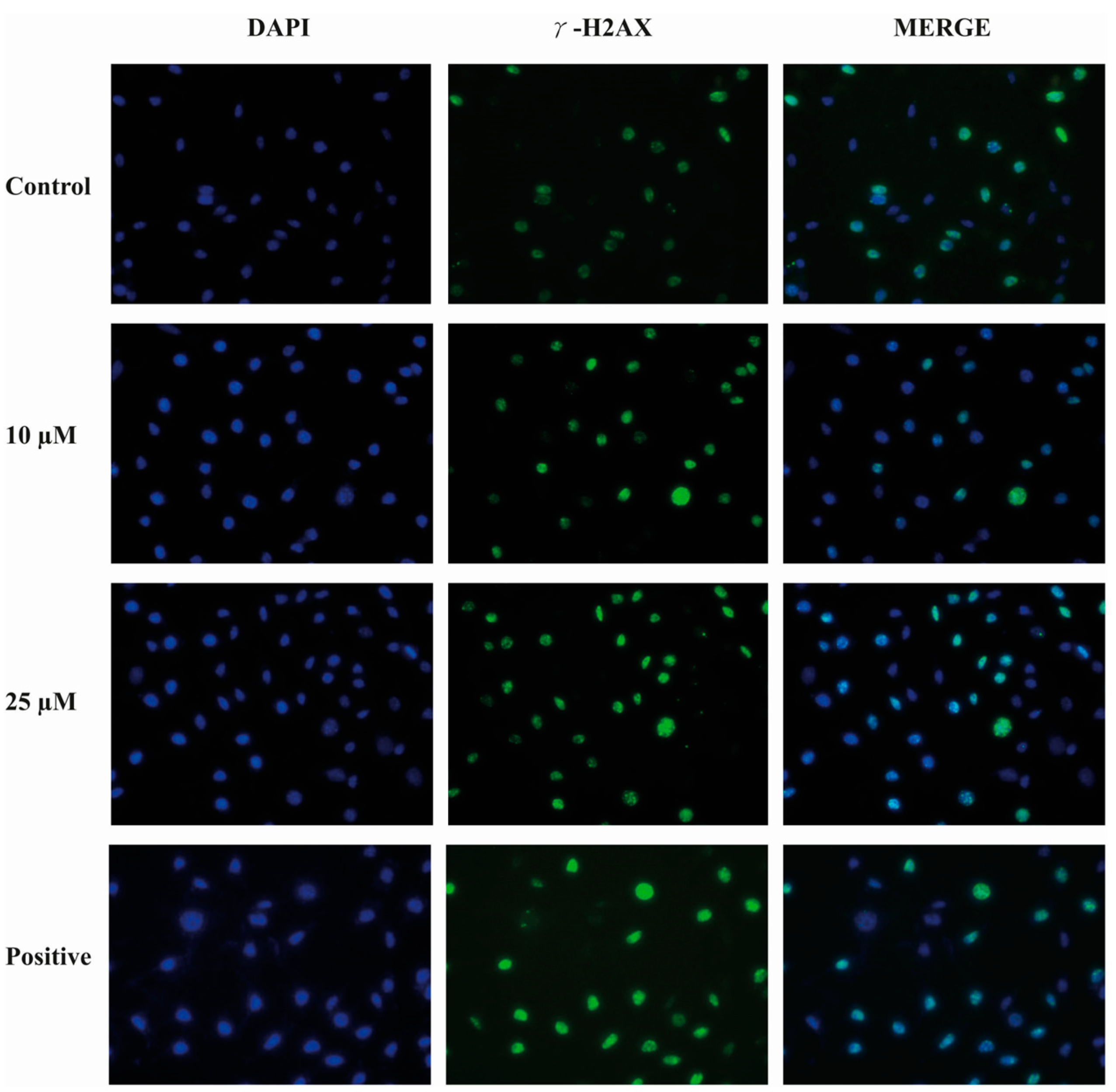
2.9. Compound E Triggered DNA Damage in HeLa Cells
2.10. Sulphonamide E Increased the Expression of Apoptosis-Related Genes in HeLa Cells
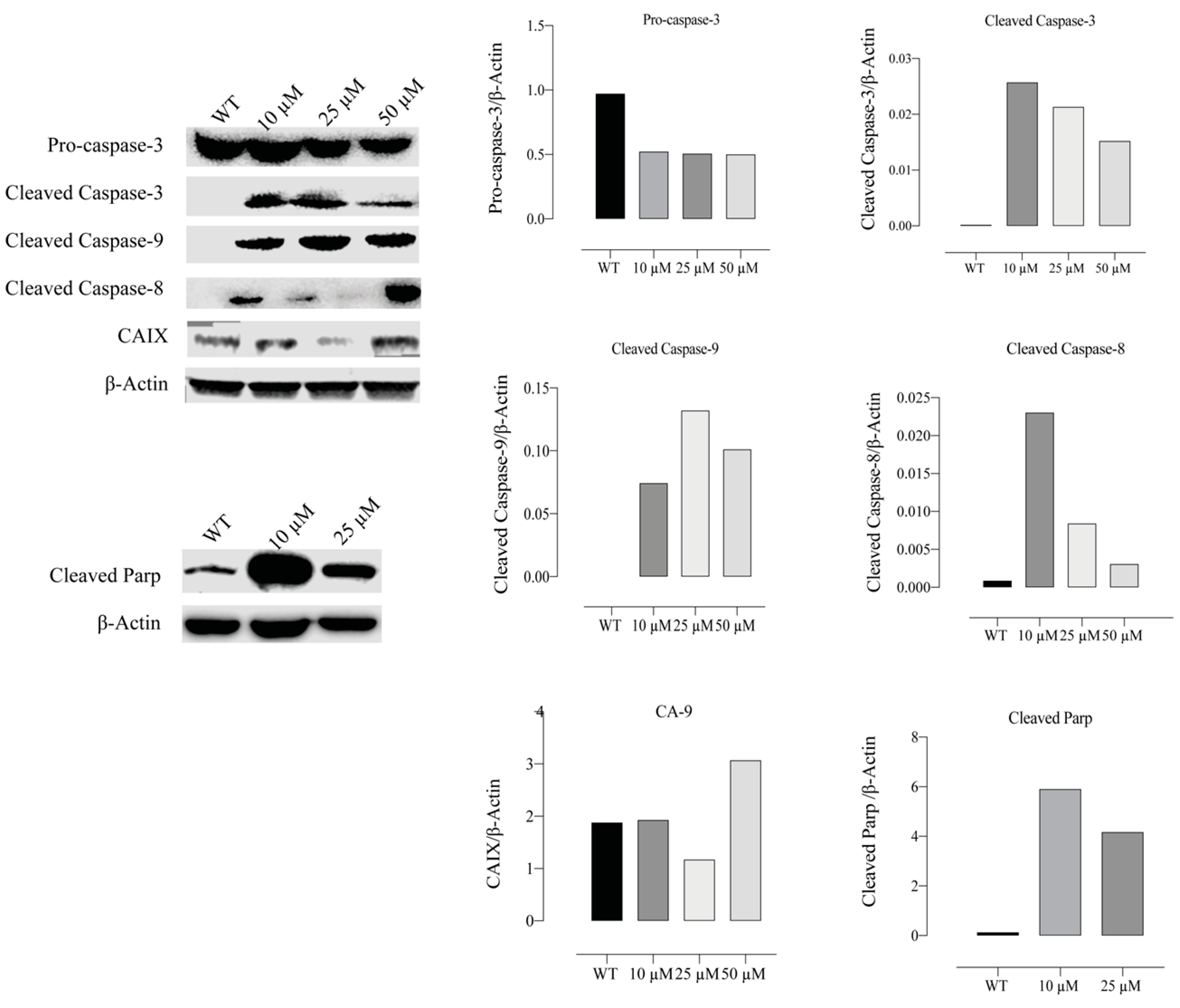
2.11. Compound E Increased the Level of Apoptosis-Related Proteins in HeLa Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Sulphonamide Derivative E
4.4. Cytotoxicity Analysis
4.5. Investigation of Antiproliferative Effects
4.6. Acridine Orange and Ethidium Bromide (AO/EB) Staining Assay
4.7. Apoptosis Detection by Annexin V-PI
4.8. Detection of Intracellular pH
4.9. Detection of Intracellular ROS Production
4.10. Detection of Mitochondrial Membrane Potential (MMP)
4.11. Cell Cycle Assay
4.12. Detection DNA Damage (γ-H2AX) by Immunofluorescence Staining
4.13. RNA Extraction and Real-time Quantitative PCR
4.14. Immunoblot Analysis
4.15. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Pastorekova, S.; Gillies, R.J. The role of carbonic anhydrase IX in cancer development: Links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev. 2019, 38, 65–77. [Google Scholar] [CrossRef]
- Hoyert, D.L. 75 Years of Mortality in the United States, 1935–2010; US Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Health Statistics (NCHS Data Brief): Washington, DC, USA, 2012; Volume 88, pp. 1–8.
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [PubMed]
- Mun, E.J.; Babiker, H.M.; Weinberg, U.; Kirson, E.D.; Von Hoff, D.D. Tumor-treating fields: A fourth modality in cancer treatment. Clin. Cancer Res. 2018, 24, 266–275. [Google Scholar] [CrossRef]
- Vaupel, P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin. Radiat. Oncol. 2004, 14, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O 2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.; Meehan, J.; Gray, M.; Kunkler, I.H.; Langdon, S.P.; Argyle, D.J. Carbonic anhydrase IX (CAIX), cancer, and radiation responsiveness. Metabolites 2018, 8, 13. [Google Scholar] [CrossRef]
- Potter, C.; Harris, A.L. Hypoxia inducible carbonic anhydrase IX, marker of tumour: Hypoxia, survival pathway and therapy target. Cell Cycle 2004, 3, 159–162. [Google Scholar] [CrossRef]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1α-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [PubMed]
- Wykoff, C.C.; Beasley, N.J.; Watson, P.H.; Turner, K.J.; Pastorek, J.; Sibtain, A.; Wilson, G.D.; Turley, H.; Talks, K.L.; Maxwell, P.H. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000, 60, 7075–7083. [Google Scholar] [PubMed]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef]
- Debreova, M.; Csaderova, L.; Burikova, M.; Lukacikova, L.; Kajanova, I.; Sedlakova, O.; Kery, M.; Kopacek, J.; Zatovicova, M.; Bizik, J. CAIX regulates invadopodia formation through both a pH-dependent mechanism and interplay with actin regulatory proteins. Int. J. Mol. Sci. 2019, 20, 2745. [Google Scholar] [CrossRef]
- Meehan, J.; Ward, C.; Turnbull, A.; Bukowski-Wills, J.; Finch, A.J.; Jarman, E.J.; Xintaropoulou, C.; Martinez-Perez, C.; Gray, M.; Pearson, M. Inhibition of pH regulation as a therapeutic strategy in hypoxic human breast cancer cells. Oncotarget 2017, 8, 42857–42875. [Google Scholar] [CrossRef]
- Koyuncu, I.; Gonel, A.; Durgun, M.; Kocyigit, A.; Yuksekdag, O.; Supuran, C.T. Assessment of the antiproliferative and apoptotic roles of sulfonamide carbonic anhydrase IX inhibitors in HeLa cancer cell line. J. Enzym. Inhib. Med. Chem. 2019, 34, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, I.; Gonel, A.; Kocyigit, A.; Temiz, E.; Durgun, M.; Supuran, C.T. Selective inhibition of carbonic anhydrase-IX by sulphonamide derivatives induces pH and reactive oxygen species-mediated apoptosis in cervical cancer HeLa cells. J. Enzym. Inhib. Med. Chem. 2018, 33, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Güttler, A.; Theuerkorn, K.; Riemann, A.; Wichmann, H.; Kessler, J.; Thews, O.; Bache, M.; Vordermark, D. Cellular and radiobiological effects of carbonic anhydrase IX in human breast cancer cells. Oncol. Rep. 2019, 41, 2585–2594. [Google Scholar]
- Hsin, M.; Hsieh, Y.; Hsiao, Y.; Chen, P.; Wang, P.; Yang, S. Carbonic Anhydrase IX Promotes Human Cervical Cancer Cell Motility by Regulating PFKFB4 Expression. Cancers 2021, 13, 1174. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.C.; Chia, S.; Bedard, P.L.; Chu, Q.; Lyle, M.; Tang, L.; Singh, M.; Zhang, Z.; Supuran, C.T.; Renouf, D.J. A phase 1 study of SLC-0111, a novel inhibitor of carbonic anhydrase IX, in patients with advanced solid tumors. Am. J. Clin. Oncol. 2020, 43, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Carta, F.; Monti, S.M.; De Simone, G. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: Three for the price of one. Med. Res. Rev. 2018, 38, 1799–1836. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Experimental Carbonic Anhydrase Inhibitors for the Treatment of Hypoxic Tumors. J. Exp. Pharmacol. 2020, 12, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Emerging role of carbonic anhydrase inhibitors. Clin. Sci. 2021, 135, 1233–1249. [Google Scholar] [CrossRef] [PubMed]
- Pastorek, J.; Pastorekova, S. Hypoxia-induced carbonic anhydrase IX as a target for cancer therapy: From biology to clinical use. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2015; Volume 31, pp. 52–64. [Google Scholar]
- Williams, K.J.; Gieling, R.G. Preclinical evaluation of ureidosulfamate carbonic anhydrase IX/XII inhibitors in the treatment of cancers. Int. J. Mol. Sci. 2019, 20, 6080. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, V.; Filippelli, A.; Angeli, A.; Supuran, C.T.; Morbidelli, L. Pharmacological Inhibition of CA-IX Impairs Tumor Cell Proliferation, Migration and Invasiveness. Int. J. Mol. Sci. 2020, 21, 2983. [Google Scholar] [CrossRef]
- Iikuni, S.; Tanimura, K.; Watanabe, H.; Shimizu, Y.; Saji, H.; Ono, M. Development of the 99mTc-Hydroxamamide Complex as a Probe Targeting Carbonic Anhydrase IX. Mol. Pharm. 2019, 16, 1489–1497. [Google Scholar] [CrossRef]
- Tülüce, Y.; Ahmed, B.A.; Koyuncu, İ.; Durgun, M. The cytotoxic, apoptotic and oxidative effects of carbonic anhydrase IX inhibitor on colorectal cancer cells. J. Bioenerg. Biomembr. 2018, 50, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Chafe, S.C.; McDonald, P.C.; Saberi, S.; Nemirovsky, O.; Venkateswaran, G.; Burugu, S.; Gao, D.; Delaidelli, A.; Kyle, A.H.; Baker, J.H. Targeting hypoxia-induced carbonic anhydrase IX enhances immune-checkpoint blockade locally and systemically. Cancer Immunol. Res. 2019, 7, 1064–1078. [Google Scholar] [CrossRef]
- McDonald, P.C.; Winum, J.-Y.; Supuran, C.T.; Dedhar, S. Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget 2012, 3, 84–97. [Google Scholar] [CrossRef]
- Koyuncu, I.; Tülüce, Y.; Slahaddin Qadir, H.; Durgun, M.; Supuran, C.T. Evaluation of the anticancer potential of a sulphonamide carbonic anhydrase IX inhibitor on cervical cancer cells. J. Enzym. Inhib. Med. Chem. 2019, 34, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Andreucci, E.; Peppicelli, S.; Carta, F.; Brisotto, G.; Biscontin, E.; Ruzzolini, J.; Bianchini, F.; Biagioni, A.; Supuran, C.T.; Calorini, L. Carbonic anhydrase IX inhibition affects viability of cancer cells adapted to extracellular acidosis. J. Mol. Med. 2017, 95, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Doyen, J.; Parks, S.K.; Marcié, S.; Pouysségur, J.; Chiche, J. Knock-down of hypoxia-induced carbonic anhydrases IX and XII radiosensitizes tumor cells by increasing intracellular acidosis. Front. Oncol. 2013, 2, 199. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-W.; Li, R.-N.; Wang, H.-R.; Liu, J.-R.; Tang, J.-Y.; Huang, H.-W.; Chan, Y.-H.; Yen, C.-Y. Withaferin A induces oxidative stress-mediated apoptosis and DNA damage in oral cancer cells. Front. Physiol. 2017, 8, 634. [Google Scholar] [CrossRef]
- Chang, Y.-T.; Huang, C.-Y.; Li, K.-T.; Li, R.-N.; Liaw, C.-C.; Wu, S.-H.; Liu, J.-R.; Sheu, J.-H.; Chang, H.-W. Sinuleptolide inhibits proliferation of oral cancer Ca9-22 cells involving apoptosis, oxidative stress, and DNA damage. Arch. Oral Biol. 2016, 66, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Shu, M.; Tang, J.; Chen, L.; Zeng, Q.; Li, C.; Xiao, S.; Jiang, Z.; Liu, J. Tumor microenvironment triple-responsive nanoparticles enable enhanced tumor penetration and synergetic chemo-photodynamic therapy. Biomaterials 2020, 268, 120574. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, İ.; Gönel, A.; Temiz, E.; Karaoğul, E.; Uyar, Z. Pistachio Green Hull Extract Induces Apoptosis Through Multiple Signaling Pathways by Causing Oxidative Stress on Colon Cancer Cells. Anti-Cancer Agents Med. Chem. 2020, 21, 725–737. [Google Scholar] [CrossRef]
- Temiz, E.; Koyuncu, İ.; Sahin, E. CCT3 Suppression Prompts Apoptotic Machinery Through Oxidative Stress and Energy Deprivation in Breast and Prostate Cancers. Free Radic. Biol. Med. 2021, 165, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Ziech, D.; Franco, R.; Georgakilas, A.G.; Georgakila, S.; Malamou-Mitsi, V.; Schoneveld, O.; Pappa, A.; Panayiotidis, M.I. The role of reactive oxygen species and oxidative stress in environmental carcinogenesis and biomarker development. Chem. Biol. Interact. 2010, 188, 334–339. [Google Scholar] [CrossRef]
- Sadowski-Debbing, K.; Coy, J.F.; Mier, W.; Hug, H.; Los, M.J. Caspases-their role in apoptosis and other physiological processes as revealed by knock-out studies. Arch. Immunol. Ther. Exp. 2002, 50, 19–34. [Google Scholar]
- Simon, H.-U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Wang, J.-P.; Hsieh, C.-H.; Liu, C.-Y.; Lin, K.-H.; Wu, P.-T.; Chen, K.-M.; Fang, K. Reactive oxygen species-driven mitochondrial injury induces apoptosis by teroxirone in human non-small cell lung cancer cells. Oncol. Lett. 2017, 14, 3503–3509. [Google Scholar] [CrossRef] [PubMed]
- Akkafa, F.; Koyuncu, İ.; Temiz, E.; Dagli, H.; Dïlmec, F.; Akbas, H. miRNA-mediated apoptosis activation through TMEM 48 inhibition in A549 cell line. Biochem. Biophys. Res. Commun. 2018, 503, 323–329. [Google Scholar] [CrossRef]
- Bekeschus, S.; Schütz, C.S.; Nießner, F.; Wende, K.; Weltmann, K.-D.; Gelbrich, N.; von Woedtke, T.; Schmidt, A.; Stope, M.B. Elevated H2AX phosphorylation observed with kINPen plasma treatment is not caused by ROS-mediated DNA damage but is the consequence of apoptosis. Oxidative Med. Cell. Longev. 2019, 2019, 8535163. [Google Scholar] [CrossRef] [PubMed]
- Brodská, B.; Holoubek, A. Generation of reactive oxygen species during apoptosis induced by DNA-damaging agents and/or histone deacetylase inhibitors. Oxidative Med. Cell. Longev. 2011, 2011, 253529. [Google Scholar] [CrossRef] [PubMed]
- Blanca, H.; Álvarez, A.M.; Sanchez, A.; Fernandez, M.; Roncero, C.; Benıto, M.; Fabregat, I. Reactive oxygen species (ROS) mediates the mitochondrial-dependent apoptosis induced by transforming growth factor ß in fetal hepatocytes. FASEB J. 2001, 15, 741–751. [Google Scholar]
- Durgun, M.; Turkmen, H.; Ceruso, M.; Supuran, C.T. Synthesis of Schiff base derivatives of 4-(2-aminoethyl)-benzenesulfonamide with inhibitory activity against carbonic anhydrase isoforms I, II, IX and XII. Bioorg. Med. Chem. Lett. 2015, 25, 2377–2381. [Google Scholar] [CrossRef] [PubMed]
- Durgun, M.; Turkmen, H.; Ceruso, M.; Supuran, C.T. Synthesis of 4-sulfamoylphenyl-benzylamine derivatives with inhibitory activity against human carbonic anhydrase isoforms I, II, IX and XII. Bioorg. Med. Chem. 2016, 24, 982–988. [Google Scholar] [CrossRef]
- Nocentini, A.; Angeli, A.; Carta, F.; Winum, J.Y.; Zalubovskis, R.; Carradori, S.; Capasso, C.; Donald, W.A.; Supuran, C.T. Reconsidering anion inhibitors in the general context of drug design studies of modulators of activity of the classical enzyme carbonic anhydrase. J. Enzym. Inhib. Med. Chem. 2021, 36, 561–580. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µM) a | SI (µM) | ||||||
|---|---|---|---|---|---|---|---|
| Compound E | 24 h | 48 h | 72 h | 24 h | 48 h | 72 h | |
| HEK-293 | 152.0 ± 3.44 | 296.4 ± 4.67 | 76.1 ± 2.1 | HEK-293/HELA | >3.2 | >3.4 | >3.7 |
| PNT1-A | 214.1 ± 9.3 | 167.3 ± 11.4 | 398.3 ± 10.4 | HEK-293/HT-29 | >1.7 | >6.9 | 0.9 |
| MDA-MB-231 | 1948.4 ± 4.65 | 205.3 ± 3.55 | 62.9 ± 10.3 | HEK-293/MDA-MB-231 | 0.07 | 1.3 | >3.7 |
| HT-29 | 89.3 ± 12.2 | 42.9 ± 16.3 | 83.3 ± 17.4 | ||||
| HeELA | 46.4 ± 4.3 | 86.8 ± 2.3 | 20.1 ± 4.2 | ||||
| 5-FU | 24 h | 48 h | 72 h | 24 h | 48 h | 72 h | |
| HEK-293 | 44.6 ± 7.2 | 19.8 ± 2.3 | 14.5 ± 3.2 | HEK-293/HELA | >1.1 | 0.6 | 0.9 |
| PNT1-A | 23.1 ± 4.55 | 21.2 ± 2.2 | 17.5 ± 7.6 | HEK-293/HT-29 | 0.9 | 0.4 | 0.7 |
| MDA-MB-231 | 42.6 ± 2.56 | 37.9 ± 12.3 | 21.0 ± 4.3 | HEK-293/MDA-MB-231 | 1.0 | 0.5 | 0.6 |
| HT-29 | 47.3 ± 1.2 | 23.4 ± 2.1 | 19.3 ± 1.2 | ||||
| HELA | 37.8 ± 4.3 | 31.2 ± 2.3 | 14.7 ± 0.6 | ||||
| Primer | Forward (5′-3′) | Reverse (3′-5′) |
|---|---|---|
| CASPASE 3 | GAGCACTGGAATGTCATCTCGCTCTG | TACAGGAAGTCAGCCTCCACCGGTATC |
| CASPASE 8 | CATCCAGTCACTTTGCCAGA | GCATCTGTTTCCCCATGTTT |
| CASPASE 9 | ATTCCTTTCCAGGCTCCATC | CACTCACCTTGTCCCTCCAG |
| CASPASE 12 | GCCATGGCTGATGAGAAACCA | TCGCATCCCCAAAAGGTCAA |
| CA9 | AGTCATTGGCGCTATGGAGG | TCTGAGCCTTCCTCAGCGAT |
| NRF-2 | TTCGGCTACGTTTCAGTCAC | TCACTGTCAACTGGTTGGGG |
| BAX | TCCATTCAGGTTCTCTTGACC | GCCAAACATCCAAACACAGA |
| BCL-2 | ATCGTCGCCTTCTTCGAGTT | ATCGTCGCCTTCTTCGAGTT |
| LC3 | ATCATCGAGCGCTACAAGGG | AGAAGCCGAAGGTTTCCTGG |
| BECLIN-1 | CGACTGGAGCAGGAAGAAG | TCTGAGCATAACGCATCTGG |
| GAPDH | GGAAGGACTCATGACCACAGT | GGATGATGTTCTGGAGAGCCC |
| Antibody | Brand | Dilution Ratio | Rabbit/Mouse | Time |
|---|---|---|---|---|
| Pro-Caspase 3 | ST John’s Laboratory | 1/1000 | Rabbit | Overnight +4 °C |
| Cleaved Caspase 3 | ST John’s Laboratory | 1/1000 | Rabbit | Overnight +4 °C |
| Cleaved Caspase 8 | ST John’s Laboratory | 1/1000 | Rabbit | Overnight +4 °C |
| Cleaved Caspase 9 | ST John’s Laboratory | 1/1000 | Rabbit | Overnight +4 °C |
| Cleaved PARP | Milipore | 1/1000 | Rabbit | Overnight +4 °C |
| CAIX | Cell Signaling | 1/1000 | Rabbit | Overnight +4 °C |
| B-actin | Cell Signaling | 1/50,000 | Mouse | 1–2 h +4 °C |
| AntiRabbit IgG HRP | Cell signaling | 1/10,000 | Rabbit | 1–2 h +4 °C |
| AntiMouse IgG HRP | Santa cruz | 1/10,000 | Mouse | 1–2 h +4 °C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Temiz, E.; Koyuncu, I.; Durgun, M.; Caglayan, M.; Gonel, A.; Güler, E.M.; Kocyigit, A.; Supuran, C.T. Inhibition of Carbonic Anhydrase IX Promotes Apoptosis through Intracellular pH Level Alterations in Cervical Cancer Cells. Int. J. Mol. Sci. 2021, 22, 6098. https://doi.org/10.3390/ijms22116098
Temiz E, Koyuncu I, Durgun M, Caglayan M, Gonel A, Güler EM, Kocyigit A, Supuran CT. Inhibition of Carbonic Anhydrase IX Promotes Apoptosis through Intracellular pH Level Alterations in Cervical Cancer Cells. International Journal of Molecular Sciences. 2021; 22(11):6098. https://doi.org/10.3390/ijms22116098
Chicago/Turabian StyleTemiz, Ebru, Ismail Koyuncu, Mustafa Durgun, Murat Caglayan, Ataman Gonel, Eray Metin Güler, Abdurrahim Kocyigit, and Claudiu T. Supuran. 2021. "Inhibition of Carbonic Anhydrase IX Promotes Apoptosis through Intracellular pH Level Alterations in Cervical Cancer Cells" International Journal of Molecular Sciences 22, no. 11: 6098. https://doi.org/10.3390/ijms22116098
APA StyleTemiz, E., Koyuncu, I., Durgun, M., Caglayan, M., Gonel, A., Güler, E. M., Kocyigit, A., & Supuran, C. T. (2021). Inhibition of Carbonic Anhydrase IX Promotes Apoptosis through Intracellular pH Level Alterations in Cervical Cancer Cells. International Journal of Molecular Sciences, 22(11), 6098. https://doi.org/10.3390/ijms22116098